

Abstract
A behavior-related deficit in the release of ascorbate (AA), an antioxidant vitamin, occurs in the striatum of R6/2 mice expressing the human mutation for Huntington’s disease (HD), a dominantly inherited condition characterized by striatal dysfunction. To determine the role of corticostriatal fibers in AA release, we combined slow-scan voltammetry with electrical stimulation of cortical afferents to measure evoked fluctuations in extracellular AA in wild-type (WT) and R6/2 striatum. Although cortical stimulation evoked a rapid increase in AA release in both groups, the R6/2 response had a significantly shorter duration and smaller magnitude than WT. To determine if corticostriatal dysfunction also underlies the behavior-related AA deficit in R6/2s, we measured striatal AA release in separate groups of mice treated with d-amphetamine (5 mg/kg), a psychomotor stimulant known to release AA from corticostriatal terminals independently of dopamine. Relative to WT, both AA release and behavioral activation were diminished in R6/2 mice. Collectively, our results show that the corticostriatal pathway is directly involved in AA release and that this system is dysfunctional in HD. Moreover, because AA release requires glutamate uptake, a failure of striatal AA release in HD is consistent with an overactive glutamate system and diminished glutamate transport, both of which are thought to be central to HD pathogenesis.
Keywords: ascorbate, cerebral cortex, Huntington’s disease, striatum, voltammetry
1. Introduction
As the deprotonated form of vitamin C, ascorbate (AA) protects against oxidative stress and excitotoxicity, which commonly occur in neurodegenerative disease (Browne and Beal, 2006; Rice, 2000; Zeron et al., 2001). In striatum, a major target of the neuropathology associated with Huntington’s disease (HD), the level of extracellular AA is significantly diminished in HD mouse models (Dorner et al., 2007; Rebec et al., 2002). Although the antioxidant role of AA may influence striatal function at a variety of levels -- from receptor binding (Majewska et al., 1990) to neuronal excitability (Cortright and Rebec, 2006; Kiyatkin and Rebec, 1998) -- it is especially interesting that striatal AA release appears to exert a strong influence on behavioral output. Not only is the level of behavioral activation in rodents correlated with striatal AA release (O’Neill and Fillenz, 1985), but depletion of AA from striatal extracellular fluid impairs locomotion and other forms of motor activity (Rebec and Wang, 2001). Behavioral activation also promotes striatal glutamate release (Sandstrom and Rebec, 2007), and cortical afferents, which supply the striatum with the bulk of its glutamate input (Groves, 1983), are essential for the release of AA. Cortical ablations, for example, lower extracellular AA levels by >70% (Basse-Tomusk and Rebec, 1990). It appears, therefore, that cortical activation increases striatal AA release. In HD, a condition characterized by progressive loss of motor control and cognitive decline, this process is likely to be impaired owing to a dysregulation of corticostriatal glutamate transmission (for review see Cepeda et al., 2007; Cepeda et al., 2003).
To determine if cortical activation promotes striatal AA release, we combined slow-scan voltammetry, which provides a distinct and selective signal for AA (Gonon et al., 1981; Rebec, 2007), with electrical stimulation of overlying motor cortex (M1). Evoked AA release was monitored in R6/2 mice, the most widely used HD model, and in age-matched, wild-type (WT) controls. All mice were anesthetized to avoid the behavioral complications of M1 stimulation. To assess corticostriatal involvement in AA release under naturally occurring conditions, separate groups of behaving R6/2 and WT mice received amphetamine, a psychomotor stimulant known to promote striatal AA release via the corticostriatal pathway independently of dopamine (Basse-Tomusk and Rebec, 1990; Gonon et al., 1981; Kamata et al., 1986; Pierce et al., 1992). Collectively, our results suggest that a dysfunctional corticostriatal pathway underlies the AA release deficit in HD mice.
2. Results
2.1 Histology
Histological analysis confirmed placement of stimulating electrodes in M1 cortex and working voltammetry electrodes in striatum (Fig. 1A, B). Data from mice with lesions not located in striatum were not included in analyses.
Fig. 1.
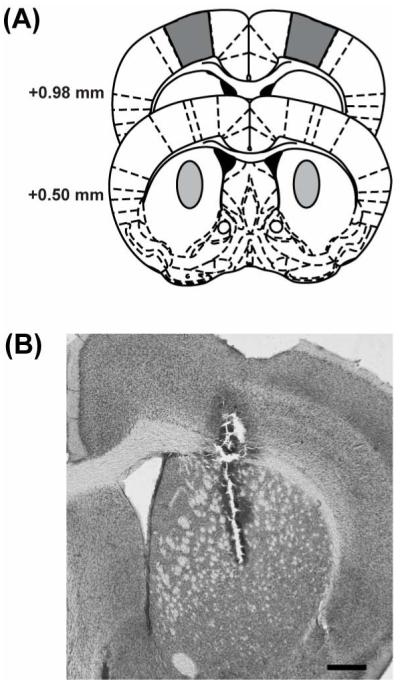
Locations of electrode placement. (A) Diagram of coronal sections showing locations of voltammetry electrode placement in striatum (0.5 mm anterior, 2 mm lateral to bregma and 3.2 to 3.6 mm ventral from skull surface; light gray) and stim electrode in cortex (M1; 1.0 mm anterior, 1.5 mm lateral from bregma, 0.5 mm ventral from skull surface; dark gray). (B) Photomicrograph of coronal section from lesioned R6/2 mouse shows voltammetry electrode tract and proper placement in striatum.
2.2 Cortically evoked striatal AA release
Although cortical stimulation elevated striatal AA release in both WT and R6/2 mice, the magnitude of the evoked AA response at each post-stimulation scan was smaller (p < 0.05) in R6/2 than WT mice (Fig. 2A, B). In addition, the basal extracellular concentration of AA was lower in R6/2 mice relative to WT both before and after cortical stimulation (Table 1). In fact, while WT AA remained 10-20% above baseline as late as 50 s following stimulation, R6/2 levels dropped below baseline at 20 s after stimulation and remained there for the remainder of the 50 s recording period (Fig. 2B). We found no significant difference in the percent change in AA from the first to the last pre-stimulation baseline in either group (p > 0.05), indicating that our results cannot be explained by a progressive loss of AA over the course of multiple stimulations in the R6/2 line. Collectively, our results support evidence for a direct link between cortical activation and striatal AA release and suggest that this link is dysfunctional in HD.
Fig. 2.
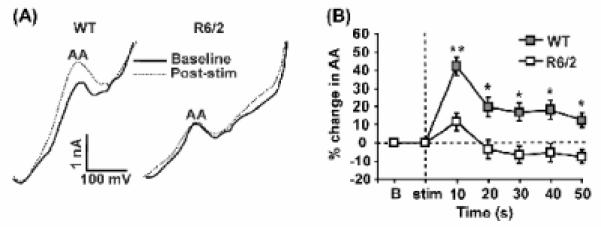
Striatal ascorbate (AA) release in response to cortical stimulation (stim). (A) Sample voltammograms obtained from a WT mouse and a R6/2 mouse. Baseline AA peak (solid line) represents a single scan taken immediately before stim. The post stim peak (dashed line) represents the first single scan taken 10 s after cortical stim of 10 s duration. The AA oxidation peak occurred between -100 and -200 mV versus reference. Note that the AA peak increases after cortical stim in the WT mouse but does not in the R6/2 mouse. (B) Mean percent change in striatal AA release across time. Baseline (b) represents the last baseline scan before stim. Dashed lines denote baseline on y-axis and stim on x-axis. Single scans at each time point were compared to baseline. (** p < 0.001, * p < 0.01) n = 23 sessions in 5 WT mice and 25 sessions in 5 R6/2 mice.
Table 1.
Ascorbate Concentrations Before and After Cortical Stimulation
| Genotype | n | Pre-stim (AA μM) | Post-stim (AA μM) |
|---|---|---|---|
| WT | 17(3) | 97 ± 10**† | 124 ± 11†† |
| R6/2 | 22(4) | 60 ± 9* | 66 ± 10 |
Mean ± SEM ascorbate concentrations (μM) estimated from re-testing electrode in vitro.
p < 0.001 compared to WT post-stim.
p < 0.05 compared to R6/2 post-stim
p < 0.001 compared to R6/2 post-stim
p < 0.01 compared to R6/2 pre-stim
n = number of sessions (number of mice)
2.3 Amphetamine-induced striatal AA release
To assess corticostriatal AA release under behavioral conditions, separate groups of R6/2 and WT mice received either amphetamine or vehicle in the home cage. As expected (Rebec et al., 2006; Rebec et al., 2002), vehicle treatment revealed lower extracellular AA (p < 0.05) in R6/2 than WT mice (Fig. 3A). Amphetamine made this AA difference even more pronounced. WT mice, for example, responded to amphetamine with a frank and prolonged increase in AA release relative to baseline (p < 0.05), whereas R6/2 AA failed to increase and, in fact, tended to fall below baseline (Fig. 3B,C). No significant differences were found between measurements taken on the right versus left hemispheres or first versus second sessions.
Fig. 3.

Striatal ascorbate (AA) release in behaving mice. (A) Overall mean percent change in vehicle (VEH)-treated animals. R6/2 mice exhibited a diminished release of AA compared to wild-type (WT) mice (* p < 0.05 compared to R6/2 mice; n = 13 sessions in 11 WT mice and 15 sessions in 11 R6/2 mice). (B) Overall mean percent change in striatal AA release in amphetamine (AMPH)-treated animals. WT mice showed an increase in striatal AA release in response to AMPH that was not observed in R6/2 mice (* p < 0.05 compared to R6/2 mice; n = 14 WT mice and 12 R6/2 mice). (C) Mean percent change in striatal AA over time from baseline taken 5 min before injection of AMPH. WT mice increased AA release in response to AMPH, while R6/2 mice did not. (* p < 0.05 compared to R6/2 mice; n = 14 WT mice and 12 R6/2 mice).
2.4 Amphetamine-evoked behavioral responses
Behavioral analyses paralleled our AA results. Whereas WT mice responded to amphetamine with an increase in percent time spent in locomotor activity relative to baseline and to vehicle treatment (p < 0.01), the R6/2 locomotor response was not different from that observed after vehicle (Fig 4A). In fact, the amphetamine-induced increase in locomotor time was greater in WT than R6/2 mice (p < 0.05). Moreover, although cage scratching occurred in all animals, only WT mice treated with amphetamine increased the percent time spent cage scratching over that recorded after vehicle treatment (p < 0.001); R6/2 mice showed no such change (Fig. 4B). Genotypic differences also emerged when we assessed the number of times specific amphetamine-induced behaviors occurred. WT mice, for example, showed significantly more instances of cage scratching, rearing, circling and head bobbing (p < 0.001 in all cases) in response to amphetamine than R6/2 mice. Total occurrences of each behavior along with corresponding X2 analyses are shown in Fig. 4C-F.
Fig. 4.

Effects of amphetamine (AMPH) on behavior of WT and R6/2 mice. Behavior was coded for a 3-min period during the pre-amphetamine baseline and at 45 min after injection around the time of peak motor activity. (A) Percent of time spent in locomotor activity. AMPH significantly increased locomotor activity in WT but not R6/2 mice (**p < 0.01, *p < 0.05). (B) Percent of time spent scratching. AMPH significantly increased time spent scratching in WT but not R6/2 mice (***p < 0.001, **p < 0.01). (C) Instances of scratching. AMPH significantly increased instances of scratching in WT but not R6/2 mice (X2 = 123.33, p < 0.001). Please note that instances of specific behaviors occurred minimally in R6/2 mice. Thus X2 analyses were used and standard error is not applicable for this and the following figures. (D) Number of rears. The number of rears significantly increased in response to AMPH in WT but not R6/2 mice (X2 = 82.78, p < 0.001). (E) Number of circles. AMPH increased the number of circles in WT mice (X2 = 103.22, p < 0.001). No circling behavior was observed in R6/2 mice. (F) Instances of head bobbing. Instances of head bobbing significantly increased in response to AMPH in WT but not R6/2 mice (X2 = 58.00, p < 0.001). n = 17 sessions in 15 WT mice treated with vehicle (VEH), 15 sessions in 15 WT mice treated with AMPH, 14 sessions in 13 R6/2 mice treated with VEH, 14 sessions in 14 R6/2 mice treated with AMPH.
3. Discussion
Our results not only support previous evidence showing diminished AA release in striatum of HD mice (Dorner et al., 2007; Rebec et al., 2006; Rebec et al., 2002), but also implicate corticostriatal dysfunction as an underlying mechanism. Lesion studies indicate that the corticostriatal pathway is the major source of AA in striatal extracellular fluid, accounting for most of the AA present under resting or baseline conditions as well as the increase in AA that occurs during spontaneous behavioral activation (O’Neill et al., 1983), after an infusion of ethanol (Liu et al., 1999) or following an injection of amphetamine (Basse-Tomusk and Rebec, 1990). We now report that both cortical stimulation and amphetamine administration resulted in diminished release of striatal AA in R6/2 relative to WT mice, suggesting that corticostriatal activity is impaired in HD.
Striatal AA release appears to require activation of glutamate receptors and/or glutamate uptake. Support for the former comes from research showing that ethanol-induced AA release in striatum can be diminished by amantadine, a non-selective NMDA receptor antagonist that facilitates dopamine release, and significantly antagonized by high doses of MK-801, a non-competitive NMDA receptor antagonist (Liu et al., 1999). Furthermore, research on the retina shows that AMPA receptors, either activated directly by glutamate or in response to glutamate-induced activation of NMDA receptors, promote AA release (Portugal et al., 2009). Although AMPA and/or NMDA receptors may modulate striatal AA release, a postsynaptic mechanism for release is unlikely. In striatum, kainic acid lesions fail to alter extracellular AA (Pierce et al., 1992), ruling out both medium spiny neurons, which account for >90% of the striatal neuronal population, and interneurons as the source of extracellular AA. In contrast, the involvement of glutamate uptake in AA release is supported by ample in vitro and in vivo evidence. For example, intra-striatal infusions of L-glutamate, the naturally occurring isomer, promote AA release (Ghasemzedah et al., 1991; Grünewald and Fillenz,1984; O’Neill et al., 1984), whereas D-glutamate, which has no affinity for transport, does not (Grünewald and Fillenz,1984). In fact, large rapid changes in AA in response to infusions as low as 100 μM of L-glutamate in vivo reflect glutamate release and uptake (Ghasemzedah et al., 1991). Pharmacological blockade of glutamate uptake, moreover, blocks L-glutamate-evoked AA release (Grünewald and Fillenz,1984), whereas large increases in extracellular AA are accompanied by similarly large decreases in extracellular glutamate (Yusa, 2001), suggesting that AA release occurs by hetero-exchange with glutamate uptake. It also is interesting that adding a high concentration of AA to striatal extracellular fluid slows the uptake of glutamate released by corticostriatal terminals (Rebec et al., 2005), indicating not only that AA is released down a concentration gradient but that glutamate uptake itself is modulated by the level of extracellular AA. This AA-glutamate interaction may involve either direct release of AA by the glutamate transporter or an indirect process in which glutamate uptake, which primarily occurs on glial cells, triggers a series of events that promote AA release from corticostriatal terminals (Fillenz et al., 1986; Rebec and Pierce, 1994). In either case, a failure to release AA in R6/2 mice suggests a failure of glutamate uptake, and that is exactly what no-net-flux microdialysis of R6/2 striatum indicates (Miller et al., 2008). This finding also is in line with decreased expression of GLT1, the glial protein responsible for the removal of most extracellular glutamate, in aged R6/2 mice (Behrens et al., 2002; Lievens et al., 2001).
One reason for AA release in conjunction with corticostriatal activation is antioxidant protection, specifically, protection against glutamate excitotoxicity (Qiu et al., 2007; Rice, 2000). In fact, the failure to release AA in the face of heightened glutamate transmission is a likely contributor to the oxidative stress and neuronal pathology evident in HD striatum (Browne & Beal, 2006; Rice, 2000; Zeron et al., 2001). A high level of extracellular AA also can prevent the hyperactivity of striatal neurons in symptomatic R6/2 mice. These animals, for example, have a significantly higher proportion of fast-firing neurons than WT, but this difference disappears when R6/2 mice receive multiple injections of AA to restore the extracellular concentration of striatal AA to WT levels (Rebec et al., 2006). Interestingly, AA treatment also improves the R6/2 behavioral phenotype (Rebec et al., 2003). In rats, intra-striatal infusion of ascorbate oxidase, which lowers extracellular AA by >50%, impairs a wide range of behavioral responses (Rebec and Wang, 2001). Thus, low extracellular AA in R6/2 striatum could allow glutamate to disrupt neuronal processing and impair behavioral output.
Neurons accumulate AA via the sodium-dependent vitamin C transporter (Tsukaguchi et al., 1999), which is highly expressed in cerebral cortex (Mun et al., 2006). Although glia also accumulate AA by active uptake and SVCT2 in culture (Siushansian and Wilson, 1995; Wilson, 1989), SVCT2 is not usually expressed in glia in vivo (Berger and Hediger, 2000). Thus, although the mechanism is not well understood, a failure of AA release may involve a failure of neurons, glia, or both to store and/or subsequently release adequate amounts of AA. Because the intracellular concentration of AA is at least 10-times higher in neurons than glia (Rice, 1999), the axon terminals of cortical afferents are the most likely source of extracellular AA in striatum. Striatal neurons themselves are not an AA source as selective loss of these cells fails to alter AA release in striatum (Pierce et al., 1992). A higher tissue level of AA in R6/2 mice (Tkac et al., 2007) indicates that cellular AA may not be adequately released into extracellular fluid.
The results of our cortical stimulations also show that the evoked release of AA is not monotonic. In WT mice, the sharp rise in response to cortical stimulation is quickly followed by a rapid decline that stabilizes at a level above the pre-stimulation baseline for the remainder of the post-stimulation recording period. Both the initial peak and the more prolonged subsequent AA response may simply reflect the level of glutamate uptake. Cortical stimulation, for example, evokes a glutamate response in rat striatum that nicely parallels the time-course of the AA response in WT mice: a sharp initial peak followed by a glutamate level that remains above baseline for many seconds thereafter (Rebec et al., 2005).
The AA concentrations reported here (Table 1) are lower than previously reported in awake mice (Rebec et al., 2002) and in rats (Miele and Fillenz, 1996). This difference is likely due to the need for general anesthesia during cortical stimulation. In fact, when used by itself, chloral hydrate, which is a component of our chloropent anesthesia, can block electrically stimulated release of AA (O’Neill et al., 1984). Our results show that chloropent did not block this effect in either R6/2 or WT mice, indicating some level of corticostriatal activation with this anesthetic, and R6/2s showed a clear AA release deficit. To assess corticostriatal AA release under behavioral conditions, we tested the effects of amphetamine in a separate group of animals.
Although drugs that enhance dopamine transmission, including amphetamine, promote striatal AA release (Mueller and Haskett, 1987; Pierce and Rebec, 1990; Zetterstrom et al., 1991), near-total loss of the nigro-striatal dopamine projection does not significantly attenuate amphetamine-induced striatal AA release in rats (Gonon et al., 1981; Pierce and Rebec, 1992), but cortical ablations do (Basse-Tomusk et al., 1990). In fact, amphetamine is capable of activating cortex via noradrenergic and other non-dopamine pathways (Carey et al., 2008; Pierce et al., 1994; Steketee, 2003). Thus, intact corticostriatal afferents are necessary for amphetamine-induced AA release, and the diminished AA response in R6/2 mice further supports corticostriatal dysfunction in HD.
Our behavioral assessment also showed a diminished R6/2 response to amphetamine compared to WT. Other drugs that interfere with dopamine transport, such as methamphetamine and cocaine, elicit similar behavioral deficits in HD models (Hickey et al., 2002; Johnson et al., 2006). Corticostriatal dysfunction, including a decline in AA release, could account for these results. Cortical ablations, for example, decrease AA levels and AA oxidation in striatum and attenuate the behavioral effects associated with scopolamine and apomorphine, drugs known to facilitate striatal AA release (Miele et al., 1995). Cortical ablations also attenuate the ability of amphetamine to elicit behavior-related changes in striatal neuronal activity (Ryan et al., 1989; Tschanz et al., 1991), and depletion of striatal extracellular AA significantly impairs spontaneous as well as goal-directed movement (Rebec and Wang, 2001). These results, however, do not rule out a role for dopamine in the blunted behavioral response of R6/2s. In fact, a reduction in the density and activity of both D1 and D2 dopamine receptors is prominent in HD patients (Joyce et al., 1988; Richfield et al., 1991; Turjanski et al., 1995) and R6/2 mice (Ariano et al., 2002; Cha et al., 1998). Moreover, although dopamine uptake is unimpaired, the dopamine response to other psychomotor stimulants such as cocaine and methamphetamine is diminished in R6/2 mice as early as 6-weeks of age when behavioral signs are just beginning to emerge (Hickey et al., 2002; Johnson et al., 2006). Interestingly, however, tetrabenazine, which depletes dopamine by interfering with vesicular storage, has beneficial effects on the R6/2 behavioral phenotype and protects against striatal cell loss (Tang et al., 2007). Thus, although dopamine transmission is likely to be altered in HD, it is unclear how this alteration contributes to the behavioral phenotype.
Our results show that cortical activation promotes striatal AA release and that this process is deficient in the R6/2 model of HD. A likely mechanism is a problem with glutamate transmission, either at the level of receptor activation or removal from the synapse. An increase in glutamate, diminished function of the GLT1 transporter and deficient GLU uptake without a subsequent rise in extracellular AA could contribute to striatal dysfunction and eventual degeneration. Diminished corticostriatal dependent AA release also may underlie the decrease in R6/2 behavioral activation induced by amphetamine. Further research on the relationship between glutamate transmission and striatal AA release is likely to suggest new therapeutic targets in HD treatment.
4. Experimental procedure
4.1 Animal Care
Male transgenic R6/2 mice (B6CBA-TgN[HDexon1]62Gpb) and WT controls were obtained from The Jackson Laboratory (Bar Harbor, ME) at 6 weeks of age. The R6/2 line is characterized by a rapidly progressive behavioral phenotype leading to death in ∼ 14 weeks (Mangiarini et al., 1996). Data were collected from mice at 7-9 weeks of age, when motor symptoms are clearly present (Carter et al., 1999; Rebec et al., 2002). All mice were housed individually in the departmental animal colony under standard conditions (12 hr light/dark cycle with lights on at 07:30) with access to food and water ad libitum. Both the housing and experimental use of animals followed the National Institutes of Health guidelines and were approved by the Institutional Animal Care and Use Committee.
4.2 Genotyping and Sequencing
To confirm genotype and determine CAG repeat length, genomic DNA was extracted from tail tissue samples in 250 μL cell lysis buffer (50 mM Tris, pH 8.0; 10 mM EDTA; 2% SDS) and proteinase K (10 mg/ml; 30 μg/reaction) at 55°C for 16 h. The lysate was then incubated with 5 unit/sample RNase T1 and 10 units/sample RNase A at 37°C for 1 h. Protein was removed by a double extraction with 250 μL phenol:chloroform:iosamyl alcohol (25:24:1) followed by centrifugation at 10,000x g for 10 min at 4°C. The aqueous layer was removed to a clean microcentrifuge tube after each extraction. DNA was then concentrated with 250 μL 7.5 M ammonium acetate and precipitated by layering 1 mL cold 95% ethanol onto the sample. The DNA was pelleted at 10,000x g for 10 min at 4°C, the supernatant discarded, and the pellet washed twice with 700 μL cold 70% ethanol. The pellet was then rehydrated in 35 μL sterile distilled water and stored at 4 °C.
PCR reactions consisted of 2.0 μl DNA template (20 to 50 ng/μL), 0.4 μl each primer (20 μM stock), 7.2 μl filter-sterilized distilled water, and 10.0 μl 2x Biomix® Red (Bioline USA Inc., Taunton, MA) for a 20 μl total volume. The forward primer sequence is 5′-GCGACCCTGGAAAAGCTGATG-3′, and the 3′ end is 27 nucleotides upstream of the CAG repeat region. The reverse primer is 5′-GGCGGCTGAGGAAGCTGAGGA-3′, and its 3′ end is 35 nucleotides downstream of the CAG region. Cycling conditions were 94°C for 90 s, followed by 30 cycles of 94°C for 30 s, 62°C for 45 s, 72°C for 90 s, with a final elongation at 72°C for 10 min. Electrophoresis of samples was performed in 4.0% NuSieve® 3:1 analytical agarose (Lonza, Rockland, ME) in 1x TAE with 0.2 μg/mL ethidium bromide at 5V/cm for 180 min using a 100bp DNA ladder as standard (New England Biolabs, Ipswich, MA) .
Gels were evaluated with Kodak Image Station 4000R and Kodak Molecular Imaging software (Carestream Molecular Imaging, New Haven, CT) to confirm genotype and determine CAG repeat length. Using Clone Manager software (Sci-Ed Software, Cary, NC) primers were aligned to the htt gene sequence acquired from the National Center for Biotechnology Information (www.ncbi.nlm.nih.gov). Alignment of primers to template showed the DNA fragment amplified by PCR is 104 bp longer than the CAG repeat region.
Dye-terminator sequencing of PCR products was also employed to confirm electrophoretically determined CAG repeat length. Eight identical amplication reactions from a single genomic template were pooled, and the PCR product was purified using the High Pure PCR Product Purification Kit (Roche Diagnostics, Indianapolis, IN). This purified PCR product was then used as the template for the dye-terminator sequencing reaction. Each reaction contained 2 μL sterile distilled water, 3 μL 5 mM MgCl2, 2 μL forward primer (2 μM stock), 2 μL template (10 to 20 ng/μL), and 1 μL BigDye® Terminator v3.1 Reaction Mix (Applied Biosystems, Foster City, CA). Cycling conditions were 98°C for 30 s, followed by 20 cycles of 98°C for 15 s, 51°C for 30 s, 72°C for 90 s, with a final 10 min elongation period at 72°C. The samples were taken to the institutional sequencing facility where the ABI3730 automated sequencer (Applied Biosystems, Foster City, CA) provided electropherograms of sequenced templates. Subsequent analysis using CodonCode Aligner (Dedham, MA) allowed visual confirmation of the exact number of CAG repeats for each of 8 R6/2 mice. The mean CAG repeat number was 115.6 with a range of 112 to 118.
4.3 Surgery and Voltammetry
Mice were anesthetized with chloropent (0.4 ml/100 g, i.p.) and mounted in a stereotaxic frame. The skull was exposed and bilateral holes were drilled over striatum (anterior 0.5 mm and lateral 2.0 mm from bregma). For cortical stimulation, holes were extended to include the area over M1 (anterior 1.0 mm and lateral 1.5 mm from bregma). These mice remained under anesthesia for voltammetric testing of electrically evoked AA release in striatum and were not used again. For assessment of amphetamine effects in behaving mice, a plastic cylindrical hub (∼3.1 mm in diameter) sealed with a plastic cap was positioned at a 5° angle over each striatal hole and was sealed to the skull with dental cement. A week of post-surgical recovery was allowed before testing with amphetamine or vehicle. Thus, separate groups of mice were used for the stimulation- and amphetamine-evoked AA recordings.
For all experiments, a locally constructed, glass-insulated carbon fiber (10 μm in diameter) served as the working electrode. The active surface area of the carbon fiber extended ∼150 μm beyond the pulled-glass tip. Electrochemical pretreatment separated the oxidation signal for AA from other easily oxidized compounds such as 3,4-dihydroxyphenylacetic acid (DOPAC) (Gonon et al., 1981; Rebec, 2007). Subsequent tests in citrate phosphate buffer containing 100 μM AA and 20 μM DOPAC ensured adequate sensitivity and peak separation. Storage of sampled current and generation of waveforms for slow-scan staircase voltammetry was carried out by a computer interfaced to a locally constructed potentiostat operating in two-electrode mode.
The working electrode was lowered into left or right striatum, 3.2 to 3.5 mm ventral from skull surface (Fig. 1A); a Ag/AgCl reference was positioned on the surface of the brain through the contralateral hole. A potential was applied in 6 mV steps from -350 to +450 mV versus the reference to ensure AA oxidation. In roughly half of our recordings, the working electrode was removed after the session ended and was retested for sensitivity in citrate phosphate buffer containing 100 μM AA and 20 μM DOPAC; the calculated AA concentration was used as a basis for estimating the concentration recorded in vivo.
4.4 Cortical Stimulation
A bipolar Polyamide® coated stainless steel stimulating electrode (0.2 mm diameter; Plastics One, Inc., Roanoke, VA) was lowered 0.5 mm into M1 (Fig. 1A). Scans were obtained every 5 min until the carbon fiber electrode detected stable oxidation peaks. For each cortical stimulation (10 s, 500 μA at 60Hz), the scan rate was set at 100 mV/s to achieve the temporal resolution necessary to track stimulation-evoked changes in AA release. We made 5 pre-stimulation scans, which were obtained at 10-s intervals and served as baseline, followed by 5 post-stimulation scans. Each animal underwent 5 to 7 stimulations, with roughly half applied to each hemisphere. Inter-stimulation intervals were ∼5 min. Mice were perfused immediately following the experiment and histology was performed as described below.
4.5 Amphetamine Administration
On the day of recording, the working and reference electrodes were fitted into separate microdrives, and each mouse was lightly anesthetized (chloropent, 0.2ml/100g) to allow for insertion of the microdrives into the head-mounted hubs and subsequent lowering of the electrodes to their respective target sites (Rebec et al., 2002). A cable attached to each microdrive was connected to the potentiostat via a commutator, which allowed the mouse to move freely. Individual scans (20 mV/s) were obtained every 5 min beginning ∼20 min after chloropent injection. This scan rate is sufficient for associating AA release with behavior (Pierce and Rebec, 1990). When the mice showed signs of waking (∼40 to 60 min later), d-amphetamine sulfate (Sigma-Aldrich, St. Louis, MO) or saline vehicle was injected subcutaneously at a dose (5 mg/kg, salt) known to increase behavioral activity in mice (Fukushiro et al., 2007). Scans continued at 5-min intervals until 85 min post-amphetamine. Mice were returned to the colony for 1 to 2 weeks before a second assessment in which the working electrode was placed into the hemisphere opposite of that for the first session to ensure recording from a fresh striatal site. Amphetamine and vehicle injections also were switched to prevent multiple drug injections; injection order was counterbalanced.
4.6 Behavioral Assessments
For voltammetry, mice were placed in their home cage (27.5L × 16W × 12H cm) of Plexiglas® and standard bedding and videotaped for subsequent behavioral assessment. Video recording began immediately before amphetamine injection and continued for 45 min. Videotapes were analyzed by an independent observer, blind to genotype, who recorded the occurrence of several different behaviors, including: locomotion (running or walking), rearing (forelimbs off the ground), cage scratching (forelimbs scratching against the side of the cage), head bobbing (repetitive up and down movements of the head), and circling (locomotion in a circular pattern). Behavior was coded for a 3-min period during the pre-amphetamine baseline and at 45 min after injection around the time of peak motor activity induced by amphetamine. We assessed the percent time engaged in a specific behavior and the number of instances the behavior occurred.
4.7 Histology
After the last day of recording in behaving animals or after cortical stimulation experiments, mice were anesthetized with chloropent (2 times the surgical dose), and current was passed through the working electrode to mark the recording site. The animals were transcardially perfused with saline in 10% formalin. Brains were removed, frozen, sectioned, stained with cresyl violet and examined under a light microscope to confirm recording location (Paxinos and Franklin, 2001).
4.8 Data Analysis
Mean peak height (apex of the curve) of each voltammetric scan was measured. Mean percent change in striatal AA release was determined by comparing mean peak height of scans taken before stimulation or amphetamine to scans taken after stimulation or amphetamine (baseline), respectively. Stimulation sessions were averaged for each animal to provide mean percent change in AA. Student’s t-tests were used to compare the percent change in striatal AA release in WT and R6/2 mice, to compare right versus left hemispheres, and first session versus second session. To evaluate the effects of amphetamine, a repeated-measures analysis of variance (ANOVA) was used to compare the mean percent change in striatal AA from baseline taken 5 min before injection to the mean of 10 scans taken at 45 min and again at 85 min following injection. Separate, two-way ANOVAs were used to measure percent time engaged in each assessed behavior. Separate X2 analyses were used to evaluate instances of each behavior. In all cases, statistically significant data were reported (p < 0.05). Sphericity was assumed for reporting degrees of freedom; p-values were adjusted using the Huynh-Feldt test. Contrasts on means using the Bonferroni adjustment were used to evaluate interaction effects.
Acknowledgments
This work was supported by grants from the National Institute of Neurological Disorders and Stroke: R01 NS-35663 (GVR), F31 NS-060218-01 (JLD) and F31 NS-064791 (BRM); the National Science Foundation Graduate Research Fellowship Program (JLD); the Neuroscience Node of the Indiana University Metabolomics and Cytomics (METACyt) Initiative funded, in part, by a grant from the Lilly Endowment. The Indiana University Science Technology and Research Scholars Program, and the Center for the Integrative Study of Animal Behavior Research Experience for Undergraduates supported undergraduate students involved in this project. Faye Caylor and Paul Langley provided administrative and technical support.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Classification Terms:
Section: Disease-Related Neuroscience
REFERENCES
- Ariano MA, Aronin N, Difiglia M, Tagle DA, Sibley DR, Leavitt BR, Hayden MR, Levine MS. Striatal neurochemical changes in transgenic models of Huntington’s disease. J Neurosci Res. 2002;68:716–29. doi: 10.1002/jnr.10272. [DOI] [PubMed] [Google Scholar]
- Basse-Tomusk A, Rebec GV. Corticostriatal and thalamic regulation of amphetamine-induced ascorbate release in the neostriatum. Pharmacol Biochem Behav. 1990;35:55–60. doi: 10.1016/0091-3057(90)90204-u. [DOI] [PubMed] [Google Scholar]
- Behrens PF, Franz P, Woodman B, Lindenberg KS, Landwehrmeyer GB. Impaired glutamate transport and glutamate-glutamine cycling: downstream effects of the Huntington mutation. Brain. 2002;125:1908–22. doi: 10.1093/brain/awf180. [DOI] [PubMed] [Google Scholar]
- Berger UV, Hediger MA. The vitamin C transporter is expressed by astrocytes in culture but not in situ. 2000;11:1395–9. doi: 10.1097/00001756-200005150-00009. [DOI] [PubMed] [Google Scholar]
- Browne SE, Beal MF. Oxidative damage in Huntington’s disease pathogenesis. Antioxid Redox Signal. 2006;8:2061–73. doi: 10.1089/ars.2006.8.2061. [DOI] [PubMed] [Google Scholar]
- Carey RJ, DePalma G, Shanahan A, Damianopoulos EN, Muller CP, Huston JP. Effects on spontaneous and cocaine-induced behavior of pharmacological inhibition of noradrenergic and serotonergic systems. Pharmacol Biochem Behav. 2008;89:54–63. doi: 10.1016/j.pbb.2007.11.003. [DOI] [PubMed] [Google Scholar]
- Carter RJ, Lione LA, Humby T, Mangiarini L, Mahal A, Bates GP, Dunnett SB, Morton AJ. Characterization of progressive motor deficits in mice transgenic for the human Huntington’s disease mutation. J Neurosci. 1999;19:3248–57. doi: 10.1523/JNEUROSCI.19-08-03248.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Hurst RS, Clvert CR, Hernandez EC, Heagaray E, Nguyen OK, Jocoy E, Christian LJ, Ariano MA, Levine MS. Transient and progressive electrophysiological alterations in the corticostriatal pathway in a mouse model of Huntington’s disease. J Neurosci. 2003;23:961–69. doi: 10.1523/JNEUROSCI.23-03-00961.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cepeda C, Wu N, Andre VM, Cummings DM, Levine MS. The corticostriatal pathway in Huntington’s disease. Prog Neurobiol. 2007;81:253–71. doi: 10.1016/j.pneurobio.2006.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cha JH, Kosinski CM, Kerner JA, Alsdorf SA, Mangiarini L, Davies SW, Penney JB, Bates GP, Young AB. Altered brain neurotransmitter receptors in transgenic mice expressing a portion of an abnormal human huntington disease gene. Proc Natl Acad Sci U S A. 1998;95:6480–5. doi: 10.1073/pnas.95.11.6480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortright JJ, Rebec GV. Ascorbate modulation of sensorimotor processing in striatum of freely moving rats. Brain Res. 2006;1092:108–16. doi: 10.1016/j.brainres.2006.03.079. [DOI] [PubMed] [Google Scholar]
- Dorner JL, Miller BR, Barton SJ, Brock TJ, Rebec GV. Sex differences in behavior and striatal ascorbate release in the 140 CAG knock-in mouse model of Huntington’s disease. Behav Brain Res. 2007;178:90–7. doi: 10.1016/j.bbr.2006.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fillenz M, O’Neill RD, Grünewald RA. Changes in extracellular brain ascorbate concentration as an index of excitatory aminoacid release. In: Joseph MH, Fillenz M, MacDonald IA, Marsden CA, editors. Monitoring Neurotransmitter Release During Behaviour. Ellis Norwood; Chichester: 1986. pp. 144–163. [Google Scholar]
- Fukushiro DF, Calzavara MB, Trombin TF, Lopez GB, Abilio VC, Andersen ML, Tufik S, Frussa-Filho R. Effects of environmental enrichment and paradoxical sleep deprivation on open-field behavior of amphetamine-treated mice. Physiol Behav. 2007;92:773–9. doi: 10.1016/j.physbeh.2007.06.002. [DOI] [PubMed] [Google Scholar]
- Ghasemzedah B, Cammack J, Adams RN. Dynamic changes in extracellular fluid ascorbic acid monitored by in vivo electrochemistry. Brain Res. 1991;547:162–166. [PubMed] [Google Scholar]
- Gonon F, Buda M, Cespuglio R, Jouvet M, Pujol JF. Voltammetry in the striatum of chronic freely moving rats: detection of catechols and ascorbic acid. Brain Res. 1981;223:69–80. doi: 10.1016/0006-8993(81)90807-6. [DOI] [PubMed] [Google Scholar]
- Groves PM. A theory of the functional organization of the neostriatum and the neostriatal control of voluntary movement. Brain Res. 1983;286:109–32. doi: 10.1016/0165-0173(83)90011-5. [DOI] [PubMed] [Google Scholar]
- Grünewald RA, Fillenz M. Release of ascorbate from synaptosomal fraction of rat brain. Neurochem Int. 1984;6:491–500. doi: 10.1016/0197-0186(84)90120-7. [DOI] [PubMed] [Google Scholar]
- Hickey MA, Reynolds GP, Morton AJ. The role of dopamine in motor symptoms in the R6/2 transgenic mouse model of Huntington’s disease. J Neurochem. 2002;81:46–59. doi: 10.1046/j.1471-4159.2002.00804.x. [DOI] [PubMed] [Google Scholar]
- Johnson MA, Rajan V, Miller CE, Wightman RM. Dopamine release is severely compromised in the R6/2 mouse model of Huntington’s disease. J Neurochem. 2006;97:737–46. doi: 10.1111/j.1471-4159.2006.03762.x. [DOI] [PubMed] [Google Scholar]
- Joyce JN, Lexow N, Bird E, Winokur A. Organization of dopamine D1 and D2 receptors in human striatum: receptor autoradiographic studies in Huntington’s disease and schizophrenia. Synapse. 1988;2:546–57. doi: 10.1002/syn.890020511. [DOI] [PubMed] [Google Scholar]
- Kamata K, Wilson RL, Alloway RD, Rebec GV. Multiple amphetamine injections reduce the release of ascorbic acid in the neostriatum of the rat. Brain Res. 1986;362:331–8. doi: 10.1016/0006-8993(86)90458-0. [DOI] [PubMed] [Google Scholar]
- Kiyatkin EA, Rebec GV. Ascorbate modulates glutamate-induced excitations of striatal neurons. Brain Res. 1998;812:14–22. doi: 10.1016/s0006-8993(98)00814-2. [DOI] [PubMed] [Google Scholar]
- Lievens JC, Woodman B, Mahal A, Spasic-Boscovic O, Samuel D, Kerkerian-Le Goff L, Bates GP. Impaired glutamate uptake in the R6 Huntington’s disease transgenic mice. Neurobiol Dis. 2001;8:807–21. doi: 10.1006/nbdi.2001.0430. [DOI] [PubMed] [Google Scholar]
- Liu J, Wu C, Liu W, Zhang H, Li C. Involvement of the corticostriatal glutamatergic pathway in ethanol-induced ascorbic acid release in rat striatum. Addict Biol. 1999;4:273–281. doi: 10.1080/13556219971489. [DOI] [PubMed] [Google Scholar]
- Mangiarini L, Sathasivam K, Seller M, Cozens B, Harper A, Hetherington C, Lawton M, Trottier Y, Lehrach H, Davies SW, Bates GP. Exon 1 of the HD gene with an expanded CAG repeat is sufficient to cause a progressive neurological phenotype in transgenic mice. Cell. 1996;87:493–506. doi: 10.1016/s0092-8674(00)81369-0. [DOI] [PubMed] [Google Scholar]
- Miele M, Enrico P, Esposito G, Fresu L, Migheli R, De Natale G, Desole MS. Cortical ablation and drug-induced changes in striatal ascorbic acid oxidation and behavior in the rat. Pharm Biochem Behav. 1995;50:1–7. doi: 10.1016/0091-3057(94)00209-2. [DOI] [PubMed] [Google Scholar]
- Miele M, Fillenz M. In vivo determination of extracellular brain ascorbate. J. Neurosci. Meth. 1996;70:15–19. doi: 10.1016/S0165-0270(96)00094-5. [DOI] [PubMed] [Google Scholar]
- Miller BR, Dorner JL, Shou M, Sari Y, Barton SJ, Sengelaub DR, Kennedy RT, Rebec GV. Up-regulation of GLT1 expression increases glutamate uptake and attenuates the Huntington’s disease phenotype in the R6/2 mouse. Neuroscience. 2008;153:329–37. doi: 10.1016/j.neuroscience.2008.02.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mueller K, Haskett C. Effects of haloperidol on amphetamine-induced increases in ascorbic acid as determined by voltammetry in vivo. Pharmacol Biochem Behav. 1987;27:231–234. doi: 10.1016/0091-3057(87)90563-6. [DOI] [PubMed] [Google Scholar]
- Mun GH, Kim MJ, Lee JH, Kim HJ, Chung YH, Chung YB, Kang JS, Hwang YI, Oh SH, Kim JG, Hwang DH, Shin DH, Lee WJ. Immunohistochemical study of the distribution of sodium-dependent vitamin C transporters in adult rat brain. J Neurosci Res. 2006;83:919–28. doi: 10.1002/jnr.20751. [DOI] [PubMed] [Google Scholar]
- O’Neill RD, Grunewald RA, Fillenz M, Albery WJ. The effect of unilateral cortical lesions on the circadian changes in rat striatal ascorbate and homovanillic acid levels measured in vivo using voltammetry. Neurosci Lett. 1983;42:105–10. doi: 10.1016/0304-3940(83)90430-5. [DOI] [PubMed] [Google Scholar]
- O’Neill RD, Fillenz M, Sundstrom L, Rawlins JNP. Voltammetrically monitored brain ascorbate as an index of excitatory amino acid release in the unrestrained rat. Neurosci Lett. 1984;52:227–233. doi: 10.1016/0304-3940(84)90166-6. [DOI] [PubMed] [Google Scholar]
- O’Neill RD, Fillenz M. Circadian changes in extracellular ascorbate in rat cortex, accumbens, striatum and hippocampus: correlations with motor activity. Neurosci Lett. 1985;60:331–6. doi: 10.1016/0304-3940(85)90599-3. [DOI] [PubMed] [Google Scholar]
- Paxinos G, Franklin KBJ. The mouse brain in stereotaxic coordinates. 2nd ed. Academic Press; San Diego: 2001. [Google Scholar]
- Pierce RC, Rebec GV. Stimulation of both D1 and D2 dopamine receptors increases behavioral activation and ascorbate release in the neostriatum of freely moving rats. Eur J Pharmacol. 1990;191:295–302. doi: 10.1016/0014-2999(90)94161-p. [DOI] [PubMed] [Google Scholar]
- Pierce RC, Miller DW, Reising DB, Rebec GV. Unilateral neostriatal kainate, but not 6-OHDA, lesions block dopamine agonist-induced ascorbate release in the neostriatum of freely moving rats. Brain Res. 1992;597:138–43. doi: 10.1016/0006-8993(92)91515-g. [DOI] [PubMed] [Google Scholar]
- Pierce RC, Clemens AJ, Grabner CP, Rebec GV. Amphetamine promotes neostriatal ascorbate release via a nigro-thalamo-cortico-neostriatal loop. J Neurochem. 1994;63:1499–507. doi: 10.1046/j.1471-4159.1994.63041499.x. [DOI] [PubMed] [Google Scholar]
- Portugal CC, Miya VS, Calaza Kda C, Santos RA, Paes-de-Carvalho R. Glutamate receptors modulate sodium-dependent and calcium-independent vitamin C bedirectional transport in cultured avian retinal cells. J Neurochem. 2009;108:507–20. doi: 10.1111/j.1471-4159.2008.05786.x. [DOI] [PubMed] [Google Scholar]
- Qiu S, Li L, Weeber EJ, May JM. Ascorbate transport by primary cultured neurons and its role in neuronal function and protection against excitotoxicity. J Neurosci Res. 2007;85:1046–56. doi: 10.1002/jnr.21204. [DOI] [PubMed] [Google Scholar]
- Rebec GV, Wang Z. Behavioral activation in rats requires endogenous ascorbate release in striatum. J Neurosci. 2001;21:668–75. doi: 10.1523/JNEUROSCI.21-02-00668.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebec GV, Barton SJ, Ennis MD. Dysregulation of ascorbate release in the striatum of behaving mice expressing the Huntington’s disease gene. J Neurosci. 2002;22:RC202. doi: 10.1523/JNEUROSCI.22-02-j0006.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rebec GV, Barton SJ, Marseilles AM, Collins K. Ascorbate treatment attenuates the Huntington behavioral phenotype in mice. Neuroreport. 2003;14:1263–5. doi: 10.1097/00001756-200307010-00015. [DOI] [PubMed] [Google Scholar]
- Rebec GV, Witowski SR, Sandstrom MI, Rostand RD, Kennedy RT. Extracellular ascorbate modulates cortically evoked glutamate dynamics in rat striatum. Neurosci Lett. 2005;378:166–70. doi: 10.1016/j.neulet.2004.12.027. [DOI] [PubMed] [Google Scholar]
- Rebec GV, Conroy SK, Barton SJ. Hyperactive striatal neurons in symptomatic Huntington R6/2 mice: variations with behavioral state and repeated ascorbate treatment. Neuroscience. 2006;137:327–36. doi: 10.1016/j.neuroscience.2005.08.062. [DOI] [PubMed] [Google Scholar]
- Rebec GV. From interferant anion to neuromodulator: Ascorbate oxidizes its way to respectability. In: Michael AC, Borland LM, editors. Electrochemical Methods for Neuroscience. CRC Press; Boca Raton, FL: 2007. pp. 149–165. [PubMed] [Google Scholar]
- Rice ME. Ascorbate compartmentalization in the CNS. Neurotox Res. 1999;1:81–90. doi: 10.1007/BF03033272. [DOI] [PubMed] [Google Scholar]
- Rice ME. Ascorbate and its neuroprotective role in the brain. TINS. 2000;23:209–216. doi: 10.1016/s0166-2236(99)01543-x. [DOI] [PubMed] [Google Scholar]
- Richfield EK, O’Brien CF, Eskin T, Shoulson I. Heterogeneous dopamine receptor changes in early and late Huntington’s disease. Neurosci Lett. 1991;132:121–6. doi: 10.1016/0304-3940(91)90448-3. [DOI] [PubMed] [Google Scholar]
- Robinson MB. The family of sodium-dependent glutamate transporters: a focus on the GLT-1/EAAT2 subtype. Neurochem Int. 1998;33:479–91. doi: 10.1016/s0197-0186(98)00055-2. [DOI] [PubMed] [Google Scholar]
- Ryan LJ, Young SJ, Segal DS, Groves PM. Antidromically identified striatonigral projection neurons in the chronically implanted behaving rat: relations of cell firing to amphetamine-induced behaviors. Behav Neurosci. 1989;103:3–14. doi: 10.1037//0735-7044.103.1.3. [DOI] [PubMed] [Google Scholar]
- Sandstrom MI, Rebec GV. Extracellular ascorbate modulates glutamate dynamics: role of behavioral activation. BMC Neurosci. 2007;8:32. doi: 10.1186/1471-2202-8-32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siushansian R, Wilson JX. Ascorbate transport and intracellular concentration in cerebral astrocytes. J Neurochem. 1995;65:41–9. doi: 10.1046/j.1471-4159.1995.65010041.x. [DOI] [PubMed] [Google Scholar]
- Steketee JD. Neurotransmitter systems of the medial prefrontal cortex: potential role in sensitization to psychostimulants. Brain Res Brain Res Rev. 2003;41:203–28. doi: 10.1016/s0165-0173(02)00233-3. [DOI] [PubMed] [Google Scholar]
- Tang TS, Chen X, Liu J, Bezprozvanny I. Dopaminergic signaling and striatal neurodegeneration in Huntington’s disease. J Neurosci. 2007;27:7899–910. doi: 10.1523/JNEUROSCI.1396-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tkac I, Dubinsky JM, Keene D, Gruetter R, Low WC. Neurochemical changes in Huntington R6/2 mouse striatum detected by in vivo 1H NMR spectroscopy. J Neurochem. 2007;100:1397–406. doi: 10.1111/j.1471-4159.2006.04323.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tschanz JT, Haracz JL, Griffith KE, Rebec GV. Bilateral cortical ablations attenuate amphetamine-induced excitations of neostriatal motor-related neurons in freely moving rats. Neurosci Lett. 1991;134:127–30. doi: 10.1016/0304-3940(91)90523-v. [DOI] [PubMed] [Google Scholar]
- Tsukaguchi H, Tokui T, Mackenzie B, Berger UV, Chen XZ, Wang Y, Brubaker RF, Hediger MA. A family of mammalian Na+-dependent L-ascorbic acid transporters. Nature. 1999;399:70–5. doi: 10.1038/19986. [DOI] [PubMed] [Google Scholar]
- Turjanski N, Weeks R, Dolan R, Harding AE, Brooks DJ. Striatal D1 and D2 receptor binding in patients with Huntington’s disease and other choreas. A PET study. Brain. 1995;118(Pt 3):689–96. doi: 10.1093/brain/118.3.689. [DOI] [PubMed] [Google Scholar]
- Wilson JX. Ascorbic acid uptake by a high-affinity sodium-dependent mechanism in cultured rat astrocytes. J Neurochem. 1989;53:1064–71. doi: 10.1111/j.1471-4159.1989.tb07396.x. [DOI] [PubMed] [Google Scholar]
- Yusa T. Increased extracellular ascorbate release reflects glutamate re-uptake during the early stage of reperfusion after forebrain ischemia in rats. Brain Res. 2001;897:104–13. doi: 10.1016/s0006-8993(01)02099-6. [DOI] [PubMed] [Google Scholar]
- Zeron MM, Chen N, Moshaver A, Lee AT, Wellington CL, Hayden MR, Raymond LA. Mutant huntingtin enhances excitotoxic cell death. Mol Cell Neurosci. 2001;17:41–53. doi: 10.1006/mcne.2000.0909. [DOI] [PubMed] [Google Scholar]
- Zetterstrom T, Wheeler DB, Boutelle MG, Fillenz M. Striatal ascorbate and its relationship to dopamine receptor stimulation and motor activity. Eur J Neurosci. 1991;3:940–946. doi: 10.1111/j.1460-9568.1991.tb00029.x. [DOI] [PubMed] [Google Scholar]