

Abstract
Cyclooxygenase-2 (COX-2) and 5-lipoxygenase (5-LO) play a role in inflammation and carcinogenesis. Biomarkers that reflect tobacco smoke-induced tissue injury are needed. In this study, levels of urinary prostaglandin E metabolite (PGE-M) and leukotriene E4 (LTE4), biomarkers of the COX and 5-LO pathways, were compared in never smokers, former smokers and current smokers. The effects of celecoxib, a selective COX-2 inhibitor, on levels of PGE-M and LTE4 were determined. Baseline levels of PGE-M and LTE4 were positively associated with smoking status; levels of PGE-M and LTE4 were higher in current vs. never smokers. Treatment with celecoxib 200 mg bid for 6 ± 1 days led to a reduction in urinary PGE-M levels in all groups, but exhibited the greatest effect among subjects with high baseline PGE-M levels. Thus, high baseline PGE-M levels in smokers reflected increased COX-2 activity. In individuals with high baseline PGE-M levels, treatment with celecoxib led to a significant increase in levels of urinary LTE4, an effect that was not found in individuals with low baseline PGE-M levels. In conclusion, increased levels of urinary PGE-M and LTE4 were found in human smokers, a result that may reflect subclinical lung inflammation. In individuals with high baseline levels of PGE-M (elevated COX-2 activity), celecoxib administration shunted arachidonic acid into the pro-inflammatory 5-LO pathway. Because 5-LO activity and LTE4 have been suggested to play a role in cardiovascular disease, these results may help to explain the link between use of COX-2 inhibitors and cardiovascular complications.
Keywords: Smoking, Biomarker, Celecoxib, 5-lipoxygenase, cardiovascular toxicity
Introduction
Cigarette smoking is the leading preventable cause of death in the U.S., and is the primary cause of chronic obstructive pulmonary disease (COPD) and lung cancer. Smoking is responsible for approximately 440,000 deaths per year in the U.S. (1). Several studies have shown that the presence of moderate to severe COPD increases the risk of developing lung cancer (2,3). The risk of dying from smoking-related diseases including COPD and lung cancer remains increased in former smokers compared with never smokers (4). Host responses to cigarette smoke are heterogeneous (5). To identify individuals at greatest risk for developing smoking-related lung disease, noninvasive biomarkers of tissue injury are needed. Given the link between smoking and lung inflammation, measurements of inflammatory mediators could be of value in identifying high-risk individuals.
This report focuses on two pro-inflammatory pathways involving the enzymes cyclooxygenase (COX) and 5-lipoxygenase (5-LO) (Fig. 1). COX-1, a constitutively expressed enzyme (6), and COX-2, an enzyme rapidly induced by stimuli including cytokines, growth factors and tobacco carcinogens, catalyze the conversion of arachidonic acid to PGH2 (7-10). PGH2 is converted by distal synthases to a variety of prostaglandins (PGs) including PGE2 (11). PGs have long been implicated as playing a key role in inflammation and malignancy, and PGE2 in particular has been shown to inhibit apoptosis, promote cell proliferation, induce angiogenesis, and suppress immune surveillance (12). Catabolism of PGE2 is initiated by 15-PG dehydrogenase and results in a stable end metabolite, 11-α-hydroxy-9, 15-dioxo-2,3,4,5-tetranor-prostane-1, 20-dioic acid (PGE-M) that is excreted in the urine (Fig. 1) and used as an index of systemic PGE2 production (13-15).
Fig. 1.

Formation of PGE-M and LTE4 via COX and LOX pathways
Arachidonic acid can also be converted by 5-LO and its molecular partner, 5-LO-activating protein (FLAP), to the unstable intermediate, leukotriene A4 (LTA4) (Fig. 1) (16). LTA4 is then conjugated to reduced glutathione by leukotriene C4 synthase, forming LTC4. LTC4 is converted to LTD4, and then to the terminal product LTE4 by a dipeptidase. The final biologically active metabolites of the 5-LOX pathway include LTB4 and the cysteinyl-LTs (cys-LTs), LTC4, LTD4 and LTE4. The cys-LTs are potent inflammatory mediators that increase vascular permeability and stimulate smooth muscle contraction (17,18). The cys-LTs, LTC4 and LTD4 are metabolized to the end product LTE4, which is excreted in the urine without further modification (19,20).
Previously, we reported results from a Phase II biomarker trial of urinary PGE-M in squamous cell carcinoma of the head and neck (21). Although PGE-M was unable to discriminate between squamous cell carcinoma of the head and neck cases and controls, PGE-M was positively associated with cigarette smoke exposure, with statistically significant differences between never and ever smokers. In the current study, we had four objectives: 1) to confirm that levels of urinary PGE-M are increased in smokers, a secondary finding in our original study; 2) to determine whether increased levels of PGEM reflected enhanced COX-2 activity; 3) to evaluate whether smoking increased levels of urinary LTE4, a product of the 5-LO pathway; and 4) to investigate whether celecoxib, a selective COX-2 inhibitor, could shunt arachidonic acid into the 5-LO pathway. Products of the 5-LO pathway including cys-LTs have been suggested to play a role in inducing cardiovascular disease (16,17,22). Use of selective COX-2 inhibitors has been associated with an increased risk of cardiovascular complications (23,24). Our findings suggest that levels of urinary PGE-M and LTE4 are promising biomarkers of smoking-induced tissue injury. Moreover, celecoxib shunted arachidonic acid into the 5-LO pathway, a result that may help explain the cardiovascular toxicity of selective COX-2 inhibitors.
Materials and Methods
Study Design
This was an open-label, non-randomized trial of 200 mg twice daily oral treatment of celecoxib (Celebrex, Pfizer, New York, NY) for 6 ± 1 days in 3 groups of healthy subjects with varying smoke exposure according to the following definitions: never smokers (<100 cigarettes per lifetime); former smokers (>1 year post cessation and > 10 pack year exposure); and current smokers (active smokers with > 10 pack year exposure). The protocol was approved by the Weill Cornell Medical College Institutional Review Board and Clinical and Translational Science Center, and conducted in accordance with an assurance filed with and approved by the Department of Health and Human Services. All subjects provided written informed consent for participation.
Participant Selection
Eligible subjects were healthy volunteers, aged 18-80 years, recruited from the community and hospital. Exclusion criteria included presence of a chronic inflammatory condition, ongoing or active infection (e.g. HIV) or a history of nonsteroidal anti-inflammatory drug (NSAID) use, including aspirin or selective COX-2 inhibitors, within the previous week. Subjects with active cardiac disease, or a history of myocardial infarction or angina within the past six months were ineligible. Subjects who were breast feeding, or presented with liver or kidney failure, a bleeding history or sulfa allergy were also ineligible. Subjects were age- and gender-matched according to the following a priori strata: male >=55 years, male < 55 years, female >=55 years, female <55 years.
Study Schema, Treatment and Study Assessments
After signing informed consent, participants underwent a baseline evaluation including smoking assessment and questionnaire (demographics and medical history), eligibility was confirmed and single void urine specimens collected. Subjects received drug and began taking oral celecoxib 200 mg twice daily for 6 ± 1 day. This is the maximum recommended dose for the treatment of arthritis. The length of treatment was chosen to be certain steady-state levels of drug were achieved. At day 6 (± 1), urine and blood were collected and pill counts were performed to assess compliance. Toxicity was monitored according to the NCI Common Toxicity Criteria. Urine specimens were aliquoted into 5 × 2-mL cryovials and stored at -80°C. Blood samples were centrifuged at 3,000 RPM for 15 min and stored at -80°C.
Study Endpoints
Urine was analyzed for PGE-M and LTE4. Post-treatment plasma specimens were analyzed for cotinine levels as a biological measure of tobacco smoke exposure and celecoxib levels as a measure of drug compliance. All measurements were carried out in a blinded manner.
Urinary PGE-M
Urinary PGE-M levels were measured by mass spectrometry as previously described (15,21). Briefly, 1 mL of urine was acidified to pH 3 with 1 mol/L HCl, and endogenous PGE-M was then converted to the O-methyloxime derivative by treatment with 0.5 mL of 16% (w/v) methyloxime HCl in 1.5 mol/L sodium acetate buffer (pH 5). Following a 1 h incubation, the methoximated PGE-M was extracted with 10 mL water adjusted to pH 3, and the aqueous sample was applied to a C-18 Sep-Pak (Waters, Milford, MA) that had been preconditioned with 5 mL methanol and 5 mL water (pH 3). The Sep-Pak was washed with 20 mL water (pH 3) and 10 mL heptane. PGE-M was then eluted from the Sep-Pak with 5 mL ethyl acetate, and any residual aqueous material was removed from the eluate by aspiration. The [2H6]O-methyloxime PGE-M internal standard (6.2 ng in 10 μL ethanol) was then added, and the eluate was evaporated under a continuous stream of nitrogen at 37°C. The dried residue was resuspended in 50 μL mobile phase A and was filtered through a 0.2-μm Spin-X filter (Corning, Corning, NY). This was followed by liquid chromatography-tandem mass spectrometry as described previously (15,21). Liquid chromatography was done on a 2.1 × 50-mm, 3-μm particle Luna C-18 column (Phenomenex, Torrance, CA) attached to a Surveyor MS Pump (ThermoFinnigan, San Jose, CA). Precursor ions (m/z 385 and 391 for unlabeled PGE-M and the [2H6]-PGE-M internal standard, respectively) were collisionally activated at 22eV under 1.5mT argon gas. For endogenous PGE-M, the predominant product ion m/z 336 representing [M-(OCH3 + H2O)]- and the analogous ion m/z 339 [M-(OC[2H3] + H2O)]- for the deuterated internal standard were monitored using multiple reaction monitoring (MRM). Quantification of endogenous PGE-M used the ratio of the mass chromatogram peak areas of the m/z 336 and 339 ions. Data acquisition and analysis were performed using XCaliber software, version 2.0.
Urinary LTE4 measurement
LTE4 and 20,20,20-2H3-LTE4 were purchased from BIOMOL International, L.P. (Plymouth Meeting, PA). Empore SD C-18 extraction cartridges (3M, St. Paul, MN) were obtained from VWR International (West Chester, PA) and Thermo Fisher Scientific (Waltham, MA). All organic reagents were of HPLC quality and purchased from EM Sciences (Gibbstown, NJ).
Purification and analysis of urinary LTE4
Urine (5-7.5mL) was acidified to pH 3 with 1 mol/L HCl. To the acidified urine was added the internal standard, [2H3]-LTE4 (1 ng). The sample was then applied to an Empore C-18 solid phase extraction column (standard density, 6 mL capacity, 3M, St. Paul, MN) that had been pre-washed with methanol (6 mL) and water (pH 3) (6 mL). The column was subsequently washed with water (pH 3) (6 mL), methanol:water (50:50, v/v) (2 mL), and heptane (6 mL). The analyte was then eluted with methanol (1 mL). The eluate was evaporated under a continuous stream of dry nitrogen. The sample was then dissolved in 100 μL methanol and filtered using a 0.2-μm Spin-X filter (Corning, Corning, NY). The sample was dried under a stream of nitrogen and then dissolved in 25μL methanol: water (50:50, v/v) for analysis by UPLC/ESI-MS/MS.
Analysis of urinary LTE4 by Ultra Pressure Liquid Chromatography/Mass Spectrometry
UPLC was performed on a 1.0 × 100-mm, 1.7-μm particle Acquity UPLC BEH C-18 column (Waters Corporation, Milford, Massachusetts, USA) attached to an Acquity UPLC (Waters Corporation, Milford, Massachusetts, USA). Mobile phase A is 8.3 mmol/L ammonium acetate:acetic acid (100:0.1, pH 5.7), and mobile phase B is 90:10 (v/v/v) acetonitrile:mobile phase A. Samples were separated using a gradient of 30-100% of mobile phase B over 3 min at a flow rate of 150μL/min prior to delivery to a ThermoFinnigan TSQ Quantum Ultra triple quadrupole mass spectrometer operating in the negative ion mode using MRM. Precursor ions (m/z 438 and 441 for unlabeled LTE4 and the [2H3]-LTE4 internal standard, respectively) were collisionally activated at 21eV under 1.5mT argon gas. For endogenous LTE4, the predominant product ion, m/z 333, and the analogous ion for the deuterated internal standard, m/z 336, were monitored. Quantification of endogenous LTE4 uses the ratio of the mass chromatogram peak areas of the m/z 333 and m/z 336 ions. Data acquisition and analysis were performed using XCaliber software, version 2.0.
Plasma celecoxib assay
Celecoxib levels were measured in plasma by liquid chromatography-tandem mass spectrometry as previously described (25). Briefly, 100 μL of plasma were diluted with an equal volume of 10 mmol/L ammonium acetate (pH 8.5). To this solution, 4 mL hexane/ethyl acetate (1:1, v/v) were added; the mixture was vortex mixed for 5 min and then centrifuged at 4,000 rpm at 5°C for 5 min. The extraction was repeated twice, and the upper organic layer was collected, pooled, and evaporated to dryness under a stream of nitrogen at room temperature under reduced room light conditions to limit the possibility of photooxidation. The sample was then reconstituted in 200 μL of methanol/10 mmol/L ammonium acetate (pH 8.5, 1:1, v/v). The celecoxib level in the samples was determined by liquid chromatography-tandem mass spectrometry. Ten μL of the sample were injected on a Luna 3-μm phenyl-hexyl 2 × 150 mm analytic column (Phenomenex, Torrance, CA). Celecoxib was detected and quantified by operating the mass spectrometer in electrospray negative ion mode and monitoring the transition m/z 380.2 > 316.1. Quantification was done by comparing the sample peak areas to a standard curve constructed from peak areas of extracted plasma sample added to known amounts of celecoxib.
Cotinine assay
The cotinine assay kit was obtained from OraSure Technologies, Inc. (Bethlehem, PA). This is a competitive micro-plate immunoassay, and the test relies on the competitive binding between free cotinine in the sample and that bound to enzyme conjugate for antibody coated on a polystyrene plate. Excess enzyme is washed away, substrate is added, and the measured absorbance is inversely proportional to the amount of cotinine present in the sample, calibrator or control. Cotinine levels were used to verify smoking history. Subjects with self-reported histories of being never or former smokers with cotinine levels greater than 2 ng/ml were excluded from the analysis.
Statistical Analysis
Demographic and smoking characteristics of the subjects were compared across groups using methods appropriate for the type of data. For age, ANOVA and Kruskal-Wallis methods were used to compare the means and medians, respectively. For the categorical variables, Fisher's exact test was used to compare the differences in proportions.
PGE-M and LTE4 values were analyzed primarily using non-parametric tests and reported in terms of median (range). Differences in baseline levels of PGE-M and LTE4 across different groups were examined using the Kruskal-Wallis test. Pair-wise comparison was carried out using the Wilcoxon rank sum test. P-values were adjusted for multiple comparison using Bonferroni method. Pre/post change in PGE-M and LTE4 following celecoxib treatment for subjects in a specific group was evaluated using Wilcoxon signed rank test. Magnitude of change across groups was compared using Kruskal-Wallis test.
Consistent results were obtained for log-transformed data when corresponding parametric methods were used. Further analyses adjusting for age, gender and race were carried out using multiple regression for log-transformed PGE-M and LTE4 data. Age, gender and race had no effect on any of the reported results.
Results
A total of 95 subjects (31 never smokers, 30 former smokers and 34 current smokers) were enrolled in the trial. In accordance with exclusion and biological endpoint criteria, 8 asthmatics (2 never smokers and 6 current smokers) and 5 subjects (1 never smoker and 4 former smokers) with plasma cotinine levels indicative of passive or current smoke exposure were excluded from the analysis. Hence, baseline analyses were carried out on information available from a total of 82 subjects. A description of the characteristics of the 82 subjects is presented in Table 1. Although participants were ageand gender-matched according to prespecified criteria, there was a non-statistically significant trend toward former smokers being older than never and current smokers. There was an even distribution of male and female participants across all three groups. Current smokers had the greatest percentage of black participants. The majority of former and current smokers reported a greater than 20 pack year history of smoking.
Table 1.
Distribution of demographic and smoking characteristics
| Characteristic | Never (n=28) | Former (n=26) | Current (n=28) | p |
|---|---|---|---|---|
| Age (years) | ||||
| Mean ± SD | 46.9 ± 14.8 | 56.6 ± 16.7 | 51.5 ± 12.0 | 0.06 |
| Median (range) | 48.5 (26, 70) | 56.5 (26, 79) | 49.5 (23, 75) | 0.09 |
| Gender, n (%) | ||||
| Male | 14 (50) | 15 (58) | 16 (57) | 0.85 |
| Female | 14 (50) | 11 (42) | 12 (43) | |
| Race, n (%) | ||||
| Caucasian | 15 (54) | 16 (60) | 13 (46) | 0.03 |
| Black | 4 (14) | 3 (12) | 13 (46) | |
| Hispanic | 6 (21) | 3 (12) | 2 (7) | |
| Asian | 2 (7) | 1 (4) | 0 (0) | |
| Other | 1 (4) | 3 (12) | 0 (0) | |
| Pack-year, n (%) | ||||
| < 20 | n/a | 6 (24) | 10 (36) | 0.66 |
| 21-40 | n/a | 9 (36) | 9 (32) | |
| > 40 | n/a | 10 (40) | 9 (32) |
Seventy-six of the 82 subjects (93%) completed the trial. The 6 subjects (1 never smoker and 5 current smokers) who failed to complete the study withdrew for personal reasons unrelated to the study. Celecoxib was well tolerated with no reports of serious adverse events. Notably, celecoxib was undetectable in the plasma in 5 of the 76 subjects (7%, including 2 never smokers, 1 former smoker and 2 current smokers) who completed the trial. This indicated noncompliance with the study medication and led to exclusion of these 5 subjects in the analysis of treatment effect. Hence, data from 71 subjects (25 never smokers, 25 former smokers, 21 current smokers) who completed the treatment phase of the study and had measurable celecoxib levels in plasma were included in the treatment effect analyses.
Smoking is associated with increased levels of PGE-M and LTE4
Baseline levels of urinary PGE-M and LTE4 were positively associated with smoking status, with statistically significant increases in median levels of PGE-M (p=0.02; Fig. 2A) and LTE4 (p=0.03; Fig. 2B) from never to former to current smokers. Between group comparisons showed that median baseline PGE-M and LTE4 values were statistically significantly higher in current versus never smokers (p=0.045 and p=0.018, respectively).
Fig. 2.

Baseline levels of urinary PGE-M (ng/mg creatinine) and LTE4 (pg/mg creatinine) are associated with smoking status. A, A statistically significant increase in baseline urinary PGE-M levels [median (range)] was observed from never [6.9 (0.2, 27.5)] to former [12.7 (2.7, 42.5)] to current [13.7 (1.2, 63.0)] smokers (p=0.02). B, Baseline levels [median (range)] of urinary LTE4 were statistically significantly increased from never [67 (10, 351)] to former [74 (10, 1414)] to current [103 (10, 333)] smokers (p=0.03).
Elevated PGE-M is due to increased COX-2 activity
Next we evaluated whether elevated urinary PGE-M levels reflected increased COX-2 activity. To address the role of COX-2, celecoxib, the selective COX-2 inhibitor, was used as a pharmacological probe. Levels of celecoxib were determined in the plasma of subjects who completed the trial. Median values were: never smokers (1.15 μM), former smokers (1.54 μM) and current smokers (1.37 μM). These differences were not statistically significant. Treatment with celecoxib at 200 mg bid led to a statistically significant reduction in median urinary PGE-M levels in all groups (Fig. 3A), demonstrating that COX-2 activity contributes to the production of urinary PGE-M even in never smokers. Next we wished to determine whether high baseline PGE-M values reflected increased COX-2 activity. To evaluate this possibility, the magnitude of celecoxib-induced decrease in PGE-M was compared in subjects with high vs. low baseline PGE-M levels. Individuals with baseline values above the median (11.2 ng/mg Cr) were defined as “high PGE-M” whereas those subjects with baseline values below or equal to the median were labeled “low PGE-M”. Regardless of smoking status, treatment with celecoxib led to significantly lower PGE-M values in both those with high baseline PGE-M levels and those with low baseline PGEM levels, with the median (range) decrease being -11.7 (-44.8, 5.5) (p<0.001) and -1.9 (-6.9, 19.2) (p=0.002), respectively. Importantly, the magnitude of the decrease induced by treatment with the COX-2 inhibitor was significantly greater in those with high baseline PGE-M levels compared with those with low PGE-M levels (p<0.001, Fig. 3B). These results suggest that the elevated level of PGE-M detected at baseline in smokers reflects increased COX-2 activity. The increased levels of urinary PGE-M found in smokers (Fig. 2A) could reflect a greater proportion of subjects with high COX-2 activity. To evaluate this possibility, we compared the proportion of current smokers, former smokers and never smokers with high baseline PGE-M. As shown in Fig. 3C, a greater proportion of former (64%) and current smokers (60%) had high baseline PGE-M levels compared with never smokers (28%) (p=0.03).
Fig. 3.

Elevated baseline PGE-M in ever smokers reflects increased COX-2 activity. A, Treatment with celecoxib 200 mg twice a day led to a statistically significant reduction in urinary PGE-M levels [median (range)] among never [pre PGE-M = 6.9 (0.2, 26.1) vs. post PGE-M = 3.0 (0.2, 15.2); p=0.001], former [pre PGE-M=13.0 (2.7, 42.5) vs. post PGE-M=6.3 (0.6, 38.4); p<0.001] and current [pre PGE-M=13.7 (1.2, 62.3) vs. post PGE-M=6.2 (0.7, 17.5); p<0.001] smokers. B, Treatment with celecoxib led to a significant reduction in levels of urinary PGE-M among subjects with high baseline PGEM [-11.7 ng/mg Cr (-44.8, 5.5); p<0.001] and among subjects with low baseline PGE-M values [-1.9 ng/mg Cr (-6.9, 19.2); p=0.002]. The reduction in urinary PGE-M levels was significantly greater among subjects with high baseline PGE-M compared to those with low baseline PGE-M levels, p<0.001. C, A greater proportion of ever smokers have high baseline PGE-M values compared with never smokers, p=0.03.
Celecoxib stimulates the formation of urinary LTE4 in individuals with high baseline levels of PGE-M
Arachidonic acid is a substrate for both the COX and 5-LO pathways (Fig. 1). As noted above, high baseline levels of PGE-M appears to reflect increased COX-2 activity. In theory, celecoxib might shunt arachidonic acid into the 5-LO pathway in a subset of individuals with high baseline levels of COX-2 activity. To evaluate this possibility, we determined the effect of celecoxib on the synthesis of LTE4. Interestingly, in individuals with high baseline levels of PGE-M, celecoxib treatment caused a significant increase in levels of urinary LTE4 (Fig. 4, p=0.03). By contrast, in individuals with low baseline PGE-M levels, celecoxib did not cause a significant increase in levels of urinary LTE4.
Fig. 4.
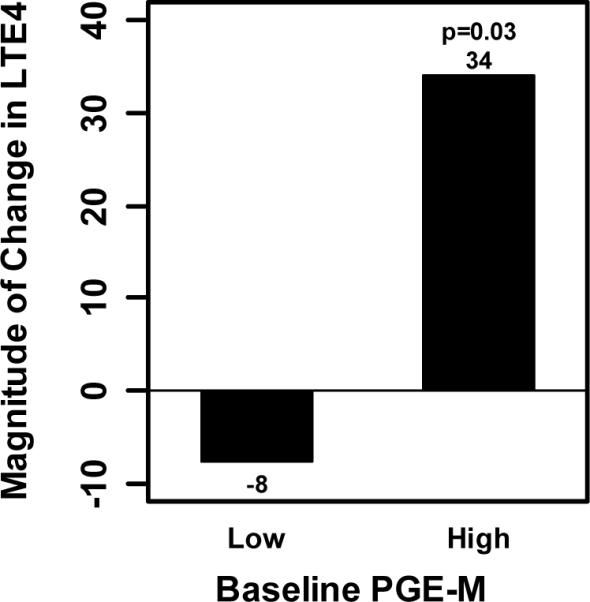
Treatment with celecoxib led to a significant increase in urinary LTE4 levels among subjects with high baseline PGE-M levels. For individuals with high PGE-M, the median (range) magnitude of change induced by celecoxib was 34 pg/mg Cr (-208, 4673), p=0.03. For individuals with low PGE-M, the median (range) magnitude of change induced by celecoxib was -8 pg/mg Cr (-244, 229), p=0.96.
Discussion
The current results confirm our original finding that levels of urinary PGE-M are increased in smokers vs. never smokers (21) (Fig. 2A). Furthermore, we show for the first time that levels of urinary LTE4 are also increased in active smokers (Fig. 2B). Notably, there was considerable overlap in levels of PGE-M and LTE4 between groups, with most but not all current smokers exhibiting higher levels of these urinary metabolites compared to never smokers. The fact that levels of the two pro-inflammatory biomarkers weren't uniformly increased in smokers is consistent with known inter-individual variability in host responses to smoking (5). Additional studies are warranted to determine whether healthy appearing smokers with elevated levels of urinary PGE-M and LTE4 are at increased risk of developing COPD or lung cancer. If so, these biomarkers could also prove useful in evaluating therapies that aim to reduce the risk of smoking-induced lung disease.
Cyclooxygenase-2, the inducible form of COX, can be rate-limiting for PGE2 synthesis (12). We next determined whether COX-2 activity contributed to either baseline PGE-M levels in never smokers or increased PGE-M levels in smokers. Celecoxib, the selective COX-2 inhibitor, was used as a pharmacological tool to evaluate these possibilities. Treatment with celecoxib led to a significant reduction in PGE-M levels in never, former and current smokers (Fig. 3A). Consistent with previous reports (15,26), our results suggest that COX-2 contributes to basal production of PGs even in healthy never smoking subjects. Celecoxib caused a greater reduction in levels of urinary PGE-M in individuals with high vs. low baseline PGE-M levels (Fig. 3B). Thus, individuals with high baseline PGE-M levels have higher COX-2 activity than individuals with low baseline PGE-M levels. Importantly, a greater percentage of current and former smokers had high baseline levels of PGE-M/COX-2 activity than never smokers (Fig. 3C). Collectively, these results imply that the increased urinary PGE-M in smokers vs. never smokers reflects increased COX-2 activity.
Another important question concerns the source of increased COX-2 activity and enhanced PGE2 production in smokers. The lung is the most likely source of increased COX-2 expression and PGE2 synthesis because of its vast surface area and the known link between tobacco smoke exposure and lung inflammation. Several findings support this notion. Increased levels of COX-2 have been found in airway cells from patients with COPD compared with unaffected control subjects (27). Higher concentrations of PGE2 have been reported in the sputum of both smokers and patients with COPD compared with non-smoking controls (28). Finally, increased levels of exhaled PGE2 have been detected in patients with COPD vs. healthy controls (29). Previous studies have suggested that 5-LO may play a role in smoking-induced COPD (30,31). Elevated levels of urinary LTE4 have been observed in asthmatics (18,32). Although the evidence is circumstantial, it seems likely that the observed increase in urinary LTE4 in smokers is of pulmonary origin. Additional studies will be needed to further evaluate this possibility.
The levels of arachidonic acid available for eicosanoid synthesis can be affected by both dietary intake and phospholipase A2 activity (33,34). Both COX-2 and 5-LO use arachidonic acid as the substrate for eicosanoid synthesis (Fig. 1). Hence, changes in amounts of free arachidonic acid or the activities of enzymes that metabolize it can potentially alter PG and leukotriene production. Previously, Mao et al. showed that treatment of active smokers with celecoxib led to increased levels of LTB4 in bronchoalveolar lavage fluid, which suggested a drug-induced shunt of arachidonic acid into the 5-LO pathway (35). In our study, treatment with celecoxib led to a marked increase in urinary LTE4 levels in individuals with elevated COX-2 activity manifested by high baseline PGE-M levels (Fig. 4). Importantly, this effect of celecoxib was not found in individuals with low baseline PGE-M levels suggesting that significant levels of COX-2 needed to be expressed for the drug effect to occur. The potential implications of celecoxib shunting arachidonic acid into the 5-LO pathway need to be considered. 5-LO has been implicated in both inflammation and carcinogenesis (16,18,36-38). Consequently, the anti-inflammatory and chemopreventive properties of celecoxib may be compromised in cells and tissues in which arachidonic acid is shunted into the 5-LO pathway. Co-administration of an agent, e.g., inhibitor of 5-LO or FLAP, which inhibits the production of leukotrienes may increase the utility of a selective COX-2 inhibitor in conditions in which this type of shunt occurs. Our findings may also provide new insights into the mechanisms underlying the cardiovascular complications associated with use of selective COX-2 inhibitors. Both COX-2 and 5-LO are expressed in atherosclerotic plaques (39-41). Based on the current findings, we speculate that a selective COX-2 inhibitor will shunt arachidonic acid into the 5-LO pathway leading to the production of multiple bioactive lipids in atherosclerotic plaques. This is a potentially important idea because of evidence that 5-LO plays a role in cardiovascular disease. In addition to the 5-LO pathway being abundantly expressed in arterial walls of patients with various stages of atherosclerosis (41), 5-LO has been causally linked to atherosclerosis in some mouse models (42). Moreover, two human genetic studies have correlated polymorphisms of the 5-LO pathway with relative risk for myocardial infarction, stroke and atherosclerosis (43,44). In fact, therapies are being developed to target the 5-LO pathway to reduce the risk of myocardial infarction (22,45). Additional studies are needed to determine whether shunting of arachidonic acid into the 5-LO pathway contributes to the cardiovascular complications associated with use of COX inhibitors. If so, risk might be minimized by co-administering an agent that targets the 5-LO pathway or using dual COX/5-LO inhibitors rather than COX inhibitors. Given the potential of dietary lipid intake to modulate arachidonic acid levels, it's possible that changes in diet may also affect an individual's risk. Other potential mechanisms have been suggested to explain the cardiovascular toxicity of selective COX-2 inhibitors. It has been suggested, for example, that selective COX-2 inhibitors block the production of cardioprotective PGI2 by vascular endothelium, without inhibiting COX-1-dependent platelet thromboxane A2 synthesis, supporting a pro-thrombotic mechanism (46,47). Whether one or more mechanisms contribute to the cardiovascular complications associated with use of COX inhibitors remains to be determined. Certainly, the results of the current study suggest that other potential mechanisms need to be strongly considered.
Acknowledgments
This paper is dedicated to the memory of Dr. Jason Morrow whose untimely passing is a great loss to all of us. We are grateful to Drs. Chaya Moskowitz and Neil Gross for their assistance in study design, and Helen Leung and Donielle Sliwa who contributed to data collection.
Grant Support: FAMRI (Dannenberg, Duffield-Lillico), Pfizer Inc. (Dannenberg), Memorial Sloan-Kettering Cancer Center Prevention Control and Population Research Program Pilot Project Award (Duffield-Lillico), NCI P01 CA77839 (Milne and Dannenberg) and CTSC UL1-RR024996
References
- 1.CDC Annual Smoking-Attributable Mortality, Years of Potential Life Lost, and Productivity Losses - United States, 1997-2001. MMWR. 2005;54:625–8. [PubMed] [Google Scholar]
- 2.Tockman MS, Anthonisen NR, Wright EC, Donithan MG. Airways obstruction and the risk for lung cancer. Ann Intern Med. 1987;106:512–8. doi: 10.7326/0003-4819-106-4-512. [DOI] [PubMed] [Google Scholar]
- 3.Skillrud DM, Offord KP, Miller RD. Higher risk of lung cancer in chronic obstructive pulmonary disease. A prospective, matched, controlled study. Ann Intern Med. 1986;105:503–7. doi: 10.7326/0003-4819-105-4-503. [DOI] [PubMed] [Google Scholar]
- 4.Halpern MT, Gillespie BW, Warner KE. Patterns of absolute risk of lung cancer mortality in former smokers. J Natl Cancer Inst. 1993;85:457–64. doi: 10.1093/jnci/85.6.457. [DOI] [PubMed] [Google Scholar]
- 5.Engels EA, Wu X, Gu J, Dong Q, Liu J, Spitz MR. Systematic evaluation of genetic variants in the inflammation pathway and risk of lung cancer. Cancer Res. 2007;67:6520–7. doi: 10.1158/0008-5472.CAN-07-0370. [DOI] [PubMed] [Google Scholar]
- 6.Smith WL, DeWitt DL. Biochemistry of prostaglandin endoperoxide H synthase-1 and synthase-2 and their differential susceptibility to nonsteroidal anti-inflammatory drugs. Semin Nephrol. 1995;15:179–94. [PubMed] [Google Scholar]
- 7.DuBois RN, Awad J, Morrow J, Roberts LJ, Bishop PR. Regulation of eicosanoid production and mitogenesis in rat intestinal epithelial cells by transforming growth factor-α and phorbol ester. J Clin Invest. 1994;93:493–8. doi: 10.1172/JCI116998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Liu W, Reinmuth N, Stoeltzing O, et al. Cyclooxygenase-2 is up-regulated by interleukin-1β in human colorectal cancer cells via multiple signaling pathways. Cancer Res. 2003;63:3632–36. [PubMed] [Google Scholar]
- 9.Moraitis D, Du B, De Lorenzo MS, et al. Levels of cyclooxygense-2 are increased in the oral mucosa of smokers: evidence for the role of epidermal growth factor and its ligands. Cancer Res. 2005;65:664–70. [PubMed] [Google Scholar]
- 10.Smith WL, DeWitt DL, Garavito RM. Cyclooxygenases: structural, cellular, and molecular biology. Annu Rev Biochem. 2000;69:145–82. doi: 10.1146/annurev.biochem.69.1.145. [DOI] [PubMed] [Google Scholar]
- 11.Jakobsson P-J, Thoren S, Morgenstern R, Samuelsson B. Identification of human prostaglandin E synthase: a microsomal glutathione-dependent, inducible enzyme constituting a potential novel drug target. Proc. Natl. Acad. Sci. USA. 1999;96:7220–25. doi: 10.1073/pnas.96.13.7220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dannenberg AJ, Subbaramaiah K. Targeting cyclooxygenase-2 in human neoplasia: rationale and promise. Cancer Cell. 2003;4:431–6. doi: 10.1016/s1535-6108(03)00310-6. [DOI] [PubMed] [Google Scholar]
- 13.Hamberg M, Samuelsson B. On the metabolism of prostaglandins E 1 and E 2 in man. J Biol Chem. 1971;246:6713–21. [PubMed] [Google Scholar]
- 14.Seyberth HW, Sweetman BJ, Frolich JC, Oates JA. Quantifications of the major urinary metabolite of the E prostaglandins by mass spectrometry: evaluation of the method's application to clinical studies. Prostaglandins. 1976;11:381–97. doi: 10.1016/0090-6980(76)90160-x. [DOI] [PubMed] [Google Scholar]
- 15.Murphey LJ, Williams MK, Sanchez SC, et al. Quantification of the major urinary metabolite of PGE2 by a liquid chromatographic/mass spectrometric assay: determination of cyclooxygenase-specific PGE2 synthesis in healthy humans and those with lung cancer. Anal Biochem. 2004;334:266–75. doi: 10.1016/j.ab.2004.08.019. [DOI] [PubMed] [Google Scholar]
- 16.Peters-Golden M, Henderson WR. Leukotrienes. N Engl J Med. 2007;357:1841–54. doi: 10.1056/NEJMra071371. [DOI] [PubMed] [Google Scholar]
- 17.De Caterina R, Zampolli A. From asthma to atherosclerosis-5-lipoxygenase, leukotrienes, and inflammation. N Engl J Med. 2004;350:4–7. doi: 10.1056/NEJMp038190. [DOI] [PubMed] [Google Scholar]
- 18.Capra V, Thompson MD, Sala A, Cole DE, Folco G, Rovati GE. Cysteinylleukotrienes and their receptors in asthma and other inflammatory diseases: critical update and emerging trends. Med Res Rev. 2007;27:469–527. doi: 10.1002/med.20071. [DOI] [PubMed] [Google Scholar]
- 19.Westcott JY, Voelkel NF, Jones K, Wenzel SE. Inactivation of leukotriene C4 in the airways and subsequent urinary leukotriene E4 excretion in normal and asthmatic subjects. Am Rev Respir Dis. 1993;148:1244–51. doi: 10.1164/ajrccm/148.5.1244. [DOI] [PubMed] [Google Scholar]
- 20.Kumlin M. Measurement of leukotrienes in humans. Am J Respir Crit Care Med. 2000;161:S102–6. doi: 10.1164/ajrccm.161.supplement_1.ltta-20. [DOI] [PubMed] [Google Scholar]
- 21.Gross ND, Boyle JO, Morrow JD, et al. Levels of prostaglandin E metabolite, the major urinary metabolite of prostaglandin E2, are increased in smokers. Clin Cancer Res. 2005;11:6087–93. doi: 10.1158/1078-0432.CCR-05-0733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Funk CD. Leukotriene modifiers as potential therapeutics for cardiovascular disease. Nat Rev Drug Discov. 2005;4:664–672. doi: 10.1038/nrd1796. [DOI] [PubMed] [Google Scholar]
- 23.Solomon SD, McMurray JJ, Pfeffer MA, et al. Cardiovascular risk associated with celecoxib in a clinical trial for colorectal adenoma prevention. N Engl J Med. 2005;352:1071–1080. doi: 10.1056/NEJMoa050405. [DOI] [PubMed] [Google Scholar]
- 24.Solomon SC, Wittes J, Finn PV, et al. Cardiovascular risk of celecoxib in 6 randomized placebo-controlled trials. Circulation. 2008;117:2104–2113. doi: 10.1161/CIRCULATIONAHA.108.764530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Patel MI, Subbaramaiah K, Du B, et al. Celecoxib inhibits prostate cancer growth: evidence of a cyclooxygenase-2-independent mechanism. Clin Cancer Res. 2005;11:1999–2007. doi: 10.1158/1078-0432.CCR-04-1877. [DOI] [PubMed] [Google Scholar]
- 26.McAdam BF, Catella-Lawson F, Mardini IA, Kapoor S, Lawson JA, FitzGerald GA. Systemic biosynthesis of prostacyclin by cyclooxygenase (COX)-2: the human pharmacology of a selective inhibitor of COX-2. Proc Natl Acad Sci U S A. 1999;96:272–277. doi: 10.1073/pnas.96.1.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Taha R, Olivenstein R, Utsumi T, et al. Prostaglandin H synthase 2 expression in airway cells from patients with asthma and chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2000;161:636–640. doi: 10.1164/ajrccm.161.2.9811063. [DOI] [PubMed] [Google Scholar]
- 28.Chen Y, Chen P, Hanaoka M, Droma Y, Kubo K. Enhanced levels of prostaglandin E2 and matrix metalloproteinase-2 correlate with severity of airflow limitation in stable COPD. Respirology. 2008;13:1014–1021. doi: 10.1111/j.1440-1843.2008.01365.x. [DOI] [PubMed] [Google Scholar]
- 29.Montuschi P, Kharitonov SA, Ciabattoni G, Barnes PJ. Exhanled leukotrienes and prostaglandins in COPD. Thorax. 2003;58:585–588. doi: 10.1136/thorax.58.7.585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kilfeather S. 5-lipoxygenase inhibitors for the treatment of COPD. Chest. 2002;121:197S–200S. doi: 10.1378/chest.121.5_suppl.197s. [DOI] [PubMed] [Google Scholar]
- 31.Santus P, Sola A, Carlucci P, et al. Lipid peroxidation and 5-lipoxygenase activity in chronic obstructive pulmonary disease. Am J Respir Crit Care Med. 2005;171:838–843. doi: 10.1164/rccm.200404-558OC. [DOI] [PubMed] [Google Scholar]
- 32.Green SA, Malice MP, Tanaka W, Tozzi CA, Reiss TF. Increase in urinary leukotriene LTE4 levels in acute asthma: correlation with airflow limitation. Thorax. 2004;59:100–104. doi: 10.1136/thorax.2003.006825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lands WE. Biochemistry and physiology of n-3 fatty acids. FASEB J. 1992;6:2530–2536. doi: 10.1096/fasebj.6.8.1592205. [DOI] [PubMed] [Google Scholar]
- 34.Schaloske RH, Dennis EA. The phospholipase A2 superfamily and its group numbering system. Biochim Biophys Acta. 2006;1761:1246–59. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 35.Mao JT, Tsu IH, Dubinett SM, et al. Modulation of pulmonary leukotriene B4 production by cyclooxygenase-2 inhibitors and lipopolysaccharide. Clin Cancer Res. 2004;10:6872–8. doi: 10.1158/1078-0432.CCR-04-0945. [DOI] [PubMed] [Google Scholar]
- 36.Stanke-Labesque F, Pofelski J, Moreau-Gaudry A, Bessard G, Bonaz B. Urinary leukotriene E4 excretion: a biomarker of inflammatory bowel disease activity. Inflamm Bowel Dis. 2008;14:769–774. doi: 10.1002/ibd.20403. [DOI] [PubMed] [Google Scholar]
- 37.Avis IM, Jett M, Boyle T, et al. Growth control of lung cancer by interruption of 5-lipoxygenase-mediated growth factor signaling. J Clin Invest. 1996;97:806–813. doi: 10.1172/JCI118480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sun Z, Sood S, Li N, et al. Involvement of the 5-lipoxygenase/leukotriene A4 hydrolase pathway in 7,12-dimethylbenz[a]anthracene (DMBA)-induced oral carcinogenesis in hamster cheek pouch, and inhibition of carcinogenesis by its inhibitors. Carcinogenesis. 2006;27:1902–1908. doi: 10.1093/carcin/bgl039. [DOI] [PubMed] [Google Scholar]
- 39.Schonbeck U, Sukhova GK, Graber P, Coulter S, Libby P. Augmented expression of cyclooxygenase-2 in human atherosclerotic lesions. Am J Pathol. 1999;155:1281–1291. doi: 10.1016/S0002-9440(10)65230-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Cipollone F, Prontera C, Pini B, et al. Overexpression of functionally coupled cyclooxygenase-2 and prostaglandin E synthase in symptomatic atherosclerotic plaques as a basis of prostaglandin E2-dependent plaque instability. Circulation. 2001;104:921–927. doi: 10.1161/hc3401.093152. [DOI] [PubMed] [Google Scholar]
- 41.Spanbroek R, Grabner R, Lotzer K, et al. Expanding expression of the 5-lipoxygenase pathway with the arterial wall during human atherogenesis. Proc Natl Acad Sci USA. 2003;100:1238–1243. doi: 10.1073/pnas.242716099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mehrabian M, Allayee H, Wong J, et al. Identification of 5-lipoxygenase as a major gene contributing to atherosclerosis susceptibility in mice. Circ Res. 2002;91:120–126. doi: 10.1161/01.res.0000028008.99774.7f. [DOI] [PubMed] [Google Scholar]
- 43.Dwyer JH, Allayee H, Dwyer KM, et al. Arachidonate 5-lipoxygenase promoter genotype, dietary arachidonic acid, and atherosclerosis. N Engl J Med. 2004;350:29–37. doi: 10.1056/NEJMoa025079. [DOI] [PubMed] [Google Scholar]
- 44.Helgadottir A, Manolescu A, Thorleifsson G, et al. The gene encoding 5-lipoxygenase activating protein confers risk of myocardial infarction and stroke. Nat Genet. 2004;36:233–239. doi: 10.1038/ng1311. [DOI] [PubMed] [Google Scholar]
- 45.Hakonarson H, Thorvaldsson S, Helgadottir A, et al. Effects of a 5-lipoxygenase-activating protein inhibitor on biomarkers associated with risk of myocardial infarction. JAMA. 2005;293:2245–2256. doi: 10.1001/jama.293.18.2245. [DOI] [PubMed] [Google Scholar]
- 46.Fitzgerald GA. Coxibs and cardiovascular disease. N Engl J Med. 2004;351:1709–1711. doi: 10.1056/NEJMp048288. [DOI] [PubMed] [Google Scholar]
- 47.Grosser T, Fries S, Fitzgerald GA. Biologic basis for the cardiovascular consequences of COX-2 inhibition: therapeutic challenges and opportunities. J Clin Invest. 2006;116:4–15. doi: 10.1172/JCI27291. [DOI] [PMC free article] [PubMed] [Google Scholar]