

Summary
Background
The large von Willebrand factor (VWF) propeptide (VWFpp) plays a critical role in the multimerization and regulated storage of the mature VWF protein. Although our laboratory and others have identified mutations in von Willebrand disease patients that disrupt VWF multimerization, little is known about the affect of mutations on the regulated storage of VWF.
Patients/Methods
We identified a heterozygous 18 base pair, in-frame deletion in exon 12 of the VWF gene in a patient with an unusual, dimer-intense multimer pattern. This deletion results in loss of amino acids 436–442 of VWFpp, which include one cysteine.
Results
Through expression studies, we demonstrate reduced secretion, loss of VWF multimerization, and defective regulated storage of the variant VWF. The loss of VWF storage is secondary to loss of propeptide storage resulting from an apparently defective sorting signal on VWFpp. Suprisingly, coexpressed wild-type VWF or VWFpp functioned in trans to partially restore multimerization of VWF from the variant allele.
Conclusions
The deletion of six amino acids in VWFpp results in defects in VWF processing, regulated storage, and function. Although VWFpp may usually function in a homotypic fashion, acting on its own mature VWF subunit, VWFpp may retain the ability to function in trans on VWF expressed from the variant allele.
Keywords: von Willebrand disease, von Willebrand factor, Weibel–Palade body
von Willebrand factor (VWF) is a multimeric glycoprotein that mediates platelet adhesion to vascular endothelium at the site of vessel injury [1]. A second function of VWF is to serve as a carrier protein for coagulation factor VIII. Defects in VWF function or decreased levels of VWF cause von Willebrand disease (VWD) [1]. Type 3 VWD is a recessive disorder characterized by a complete deficiency of VWF, a secondary deficiency of FVIII, and defective platelet adhesion and fibrin formation leading to severe bleeding. Type 2 VWD includes qualitative deficiencies classified as types 2A, 2B, 2M, and 2N. A partial quantitative deficiency of VWF with normal VWF multimer distribution is characteristic of type 1 VWD [1].
VWF is synthesized in endothelial cells and megakaryocytes as pre-pro-VWF, consisting of a 22 amino acid signal peptide, a 741 amino acid propeptide, and a 2050 amino acid mature VWF molecule [2]. The propeptide (VWFpp) contains two homologous D domains, D1 and D2, and mature VWF contains the following domains: D′–D3–A1–A2–A3–D4–B–C1–C2 [2]. VWF undergoes extensive intracellular processing, including glycosylation, sulfation, C-terminal dimerization, carbohydrate processing, N-terminal multimerization, and propeptide cleavage [3]. VWF and VWFpp are stored together in regulated secretory vesicles, Weibel–Palade bodies in endothelial cells, and α-granules in megakaryocytes and platelets [3].
Several functions of VWF have been mapped specifically to individual domains, such as FVIII binding (D′–D3), collagen binding (A1 and A3), N-terminal multimerization (D3), platelet glycoprotein Ibα binding (A1), and C-terminal dimerization (C2) [3]. Our laboratory and others have shown that granular storage of VWF is independent of multimerization [4–7]. The large propeptide is required for both multimerization and regulated storage of VWF [4,5,8]. We have shown that VWF is routed to storage granules in a VWFpp-dependent manner [5,9]. Although naturally occurring mutations in VWF that abolish multimerization but do not affect regulated storage have been reported, there have been no reports of VWF mutations that result in loss of regulated storage [6,7]. In this study, we report the characterization of a naturally occurring mutation in VWFpp. The patient studied has mild VWD and an aberrant VWF multimer pattern. A mutation was identified in the VWFpp D2 domain that results in decreased VWF secretion, complete loss of VWF multimerization, and loss of regulated storage of VWF.
Materials and methods
Patients
The patient was studied after we received written informed consent. The research protocol was approved by the Institutional Review Boards of the Children's Hospital of Wisconsin, the General Clinical Research Center of the Medical college of Wisconsin, and BloodCenter of Wisconsin's Blood Research Institute. Plasma VWF:Ag, VWF:RCo, VWF multimers and FVIII levels were determined by the Hemostasis Reference Laboratory at BloodCenter of Wisconsin, Milwaukee, WI, USA.
Isolation of genomic DNA and sequencing
Total genomic DNA was prepared from leukocytes isolated from 50 mL of peripheral blood as previously described [6]. Genomic DNA polymerase chain reaction amplification and direct sequencing was performed on each VWF exon, 5′/3′-untranslated regions, and intron/exon boundries at the Partners Healthcare Center for Genetics and Genomics, Harvard Medical School, Boston, MA, USA. The VWF mutation was also identified in a family member.
Expression plasmids
The human VWF, Y87S-VWF, VWFpp and propeptide-deleted VWF (Δpro) expression vectors were as previously described [5,6]. The patient deletion (Δ437–442) was introduced into the full-length VWF expression vector and the VWFpp vector to create pCIneo-VWF-Δ437–442 and pCIneo-VWFpp-Δ437–442, respectively. A pCIneo-P-selectin expression vector was used. For some experiments, a myc-tagged VWF, pCDNA3 VWF-myc-his or green fluorescent protein (GFP)-tagged VWF expression vector (pEGFP-VWF-WT or pEG-FP-VWF-Δ437–442) were used to differentially identify wild-type and variant VWF proteins. See Supporting Information for details on expression vectors.
Antibodies and reagents
Botrocetin was prepared from Bothrops jararaca venom (Sigma Chemical Co., St Louis, MO, USA) and purified as previously described [10]. Anti-VWF antibodies AVW-1, AVW-5 and 105.4, anti-VWFpp antibodies 239.1–239.11, 242.4, and 242.5 and anti-cmyc 9E10 were produced by our laboratory. Purchased antibodies include: rabbit anti-human VWF (DAKO, Carpinteria, CA, USA), rabbit anti-P-selectin (BD Bioscience, San Jose, CA, USA), rabbit-anti-GRP78 (Affinity Bioreagents, Golden, CO, USA), rabbit-anti-adrenocorticotrophic hormone (ACTH) (DAKO), rabbit anti-GFP (Santa Cruz Biotechnology, Santa Cruz, CA, USA), non-immune mouse and rabbit IgG (Jackson Immunoresearch, West Grove, PA, USA), and goat anti-rabbit and anti-mouse IgG (H+L) [F(Ab′)2] fragments conjugated with either AlexaFluor-488 or AlexaFluor-568 (Invitrogen, Carlsbad, CA, USA).
Mammalian cell culture and transfection
Human embryonic kidney cells (HEK293T; D. Ginsburg, University of Michigan, Ann Arbor, MI, USA) and mouse pituitary tumor cells (AtT-20/D16v-F2, CRL 1795; American Type Culture Collection) were cultured and transfected as previously described [5]. Conditioned media were harvested from cells, centrifuged, and frozen at − 80 °C. Transfected HEK293T cells were washed with phosphate-buffered saline and lysed in Reporter Lysis Buffer (Promega, Madison, WI, USA). Transfected AtT-20 cells were either fixed for intracellular staining or used for secretion studies. To assess stimulated release, cells were incubated for 30 min with either 5 mmol L−1 8-Br-cAMP diluted in OptiMEM or OptiMEM (control), and releasates were harvested. VWF:Ag and VWFpp levels were determined by antigen-capture enzyme-linked immunosorbent assay [11].
VWF multimer analysis
VWF in the conditioned medium of transfected cells was analyzed by electrophoresis through a 0.8% (w/v) HGT(P) agarose (FMC Bioproducts, Rockland, ME, USA) stacking gel and 2% or 3% (w/v) HGT(P) agarose running gel containing 0.1% sodium dodecylsulfate for 16 h at 40 V using the Laemmli buffer system and western blotting as previously described [5].
Assays of VWF activity
VWF platelet binding was determined using an assay described by our laboratory [6]. Conditioned media from transfected cells were diluted to equal amounts of VWF and incubated for 60 min with 125I-labeled AVW-1. Formalin-fixed human platelets were added in the presence or absence of ristocetin or botrocetin under non-stirring conditions. After 30 min, reactions were centrifuged at 10 000 ×g for 10 min. Platelet pellets and supernatants were counted to quantitate VWF bound to the platelets relative to a pooled normal plasma standard curve. Binding of expressed VWF to collagen type III from human placenta was determined in a microtiter plate assay [6]. Binding of FVIII to expressed VWF was measured by chromogenic assay using the Chromogenix Coatest FVIII:C/4 kit (diaPharma, West Chester, OH, USA) [6].
Confocal immunofluorescence microscopy
Cells were analyzed for the intracellular location of VWF, VWFpp and other proteins by immunofluorescence antibody staining and confocal laser scanning microscopy in the Imaging Core of the Medical College of Wisconsin, using a Leica TCS SP2 confocal laser imaging system [9]. Transfected cells were fixed using 3.7% (v/v) buffered formalin, permeabilized in 1% Triton X-100, and blocked with 2% normal goat serum. Cells were incubated at 4 °C overnight in 2–5 μg mL−1 primary antibodies, and then washed. Cells were incubated in secondary antibodies diluted 1 : 1000 for 30 min at room temperature, washed, and mounted using Vectashield (Vector Labs, Burlingame, CA, USA).
Results
Characterization of a patient with VWD
We studied a 68-year-old woman diagnosed with VWD. She had a mild bleeding history including bruising, menorrhagia, and bleeding after dental extraction. She had a VWF:Ag level of 74 IU dL−1, an abnormally low VWF:RCo level (45 IU dL−1), and a normal FVIII level (94 IU dL−1). Multimer analysis revealed an overabundance of dimeric VWF and a small decrease in high molecular mass multimers (Fig. 1A). A similar multimer profile in a patient with a Y87S-VWF mutation was reported by our laboratory, but with a more pronounced dimer band (Fig. 1A, right) [6].
Fig. 1.
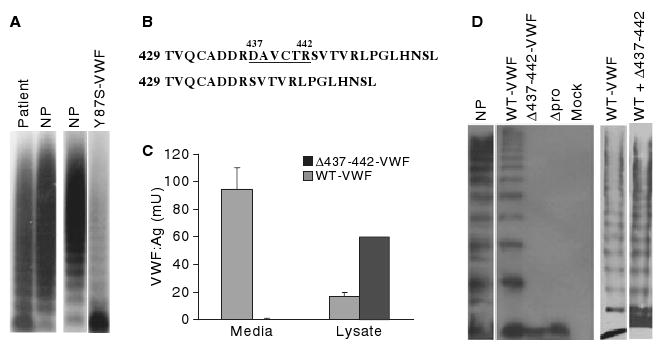
Plasma von Willebrand factor (VWF) multimer analysis, mutation identification, and expression of variant VWF. (A) Patient plasma (lane 1), normal human plasma (NP) and plasma from a previously reported VWD patient were analyzed by sodium dodecylsulfate (SDS)--agarose gel electrophoresis and western blotting [6]. (B) A mutation identified in the D2 domain of VWF propeptide (VWFpp) predicts deletion of amino acids 437--442. (C) VWF concentration in conditioned media and cell lysates from HEK293T cells expressing wild-type (WT) VWF (gray bars) and Δ437--442-VWF (black bars) from three separate transfections with each vector expressed in duplicate. Results shown are mean VWF levels, with error bars indicating standard deviation. (D) Multimeric composition of recombinant VWF proteins (2% SDS--agarose): normal human plasma (lane 1), wild-type VWF (lanes 2 and 6), Δ437--442-VWF (lane 3), propeptide-deleted VWF (Δpro, lane 4), mock-transfected cells (lane 5), coexpressed wild-type VWF and Δ437--442-VWF (lane 7). The Δ437--442-VWF mutation results in decreased VWF secretion and defective multimerization.
Identification and expression of mutant VWF
Each of the VWF exons, intron/exon boundries and 5′/3′-untranslated nucleotides was sequenced. A deletion of nucleotides 1309–1326 was identified in exon 12, predicting the deletion of amino acids 437–442 (Fig. 1B). The mutation was introduced into a VWF expression vector (Δ437–442-VWF) and expressed in HEK293T cells. We observed decreased secretion and increased intracellular retention of homozygous Δ437–442-VWF (Fig. 1C). Multimer analysis revealed a complete loss of Δ437–442-VWF multimers, similar to what was seen with propeptide-deleted (Δpro) VWF (Fig. 1D). Heterozygous expression of wild-type VWF and Δ437–442-VWF demonstrated increased low molecular mass VWF subunits among a full spectrum of multimers, similar to the patient's multimer profile (Fig. 1D).
Functional assessment of recombinant Δ437–442-VWF
Several assays of VWF activity were performed to determine the effect of the Δ437–442 mutation on VWF function as compared with wild-type VWF and dimeric Y87S-VWF (Table 1) [6]. The mutant Δ437–442-VWF did not bind to platelets in the presence of ristocetin, similar to Y87S-VWF. Dimeric Δ437–442-VWF showed reduced botrocetin-induced platelet binding (52% of that of the wild type), whereas Y87S-VWF binding was comparable to that of wild-type VWF. The collagen-binding function of the dimeric Δ437–442-VWF was abolished, whereas Y87S-VWF retained a reduced collagen-binding ability. The Δ437–442-VWF bound FVIII at a reduced level. Together, these data indicate that the Δ437–442-VWF mutation results in a functionally defective VWF protein.
Table 1.
Functional assessment of recombinant von Willebrand factor (VWF)
| Platelet binding assay (% of AVW-1 bound) | Collagen binding | FVIII binding | |||||||
|---|---|---|---|---|---|---|---|---|---|
| Expressed VWF | Ristocetin | % WT | Botrocetin | % WT | No agonist | Amt bound (mU mL−1) | % WT | FVIII binding (mU mL−1) | % WT |
| Wild-type VWF | 60.1 | 100 | 68.7 | 100 | 2.9 | 5.18 | 100 | 4.06 | 100 |
| Δ437–442-VWF | 12.3 | 20.5 | 35.7 | 52.0 | 1.6 | 0.07 | 1.4 | 0.93 | 22.0 |
| Y87S-VWF | 10.1 | 16.8 | 66.8 | 97.2 | 2.8 | 1.17 | 22.6 | 0.8 | 19.7 |
| Mock | 6.4 | 10.6 | 7.7 | 11.2 | 1.9 | 0.07 | 1.4 | <0.1 | < 2 |
WT, wild type.
Intracellular localization of Δ437–442-VWF
VWF is stored in Weibel–Palade bodies in endothelial cells and α-granules in megakaryocytes and platelets [3]. When expressed in AtT-20 cells, VWF trafficks to storage granules [4,5,12]. AtT-20 cells expressing wild-type VWF showed a granular VWFpp and VWF distribution, whereas cells expressing Δ437–442-VWF demonstrated only diffuse staining (Fig. 2A,B). Weibel–Palade bodies contain P-selectin in addition to VWF, and in model cell lines the recruitment of coexpressed P-selectin is used to validate the formation of pseudo-Weibel–Palade bodies [7,12]. Cotransfected P-selectin was recruited to storage granules formed by expression of wild-type VWF, but Δ437–442-VWF did not form granules that recruited P-selectin (Fig. 2C,D). Δ437–442-VWF colocalized with the endoplasmic reticulum (ER) protein GRP78 (Fig. 2E).
Fig. 2.
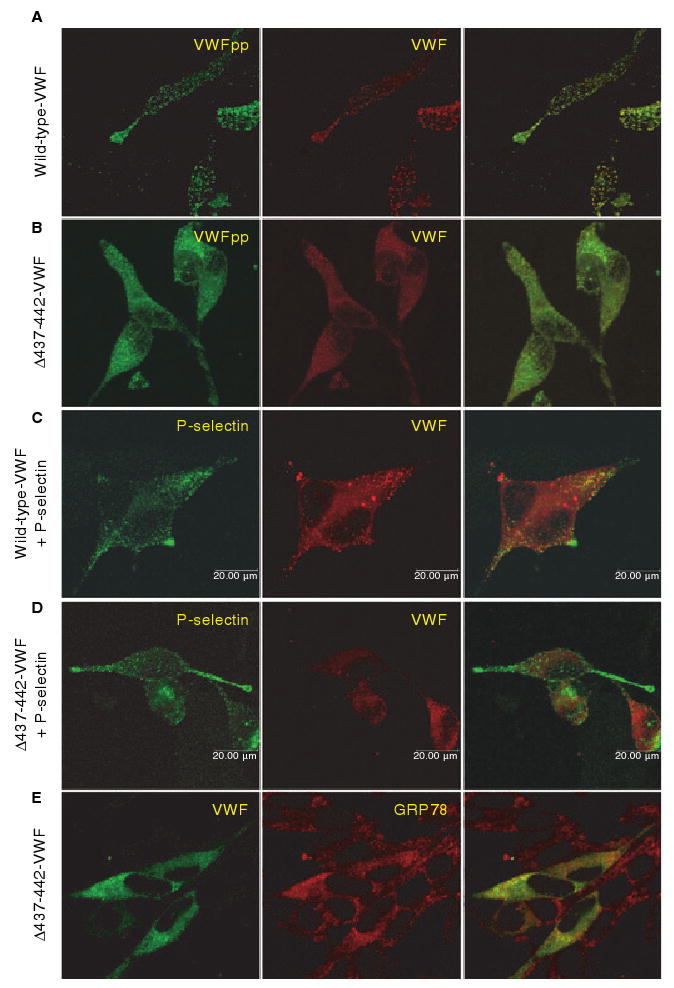
Intracellular localization of Δ437--442-von Willebrand factor (VWF). AtT-20 cells were transfected with wild-type (WT) VWF (A, C) or Δ437--442-VWF (B, D, E), or cotransfected with P-selectin (C, D). Cells were dual-stained for VWF propeptide (VWFpp) and VWF (A, B), P-selectin and VWF (C, D), or VWF and GRP78 (E). Intracellular localization was examined by confocal microscopy. The merged image is shown in the last column, with colocalization shown in yellow. Δ437--442-VWF does not form granules or colocalize with coexpressed P-selectin, but is instead localized to the endoplasmic reticulum.
VWF-regulated storage is a propeptide-dependent event: VWFpp independently trafficks to regulated storage and cotrafficks VWF multimers through a non-covalent association [5]. To determine whether the loss of storage is due to defective VWFpp trafficking, Δ437–442-VWFpp was expressed in AtT-20 cells. Cells expressing wild-type VWFpp demonstrated colocalization of VWFpp with endogenous ACTH granules, but cells expressing Δ437–442-VWFpp showed only diffuse staining [Fig. 3A(1,2)]. Δ437–442-VWFpp colocalized with the ER marker GRP78 [Fig. 3A(3)]. Δ437–442-VWFpp was not released in response to the secretagog 8-Br-cAMP (118% of basal), whereas a > 2-fold increase in wild-type VWFpp was observed (Fig. 3B). Δ437–442-VWFpp is not trafficked to the regulated storage pathway.
Fig. 3.
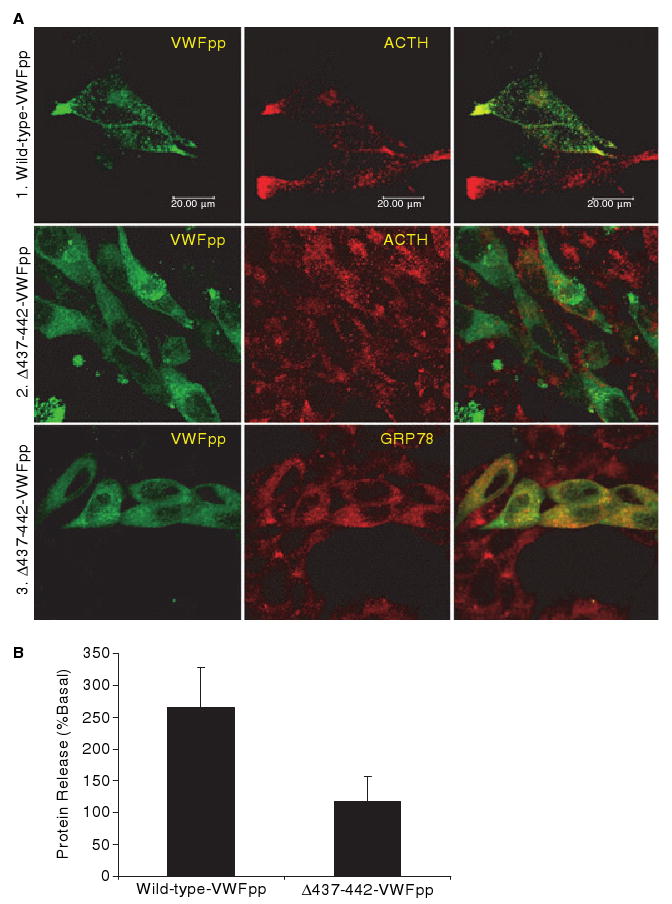
Intracellular localization and regulated secretion of wild-type VWF propeptide (VWFpp) and Δ437--442-VWFpp. (A) AtT-20 cells were transfected with wild-type VWFpp (1) or Δ437--442-VWF (2, 3). Transfected cells were stained for VWFpp and adrenocorticotrophic hormone (ACTH) (1, 2), or VWFpp and GRP78 (3). Intracellular localization of proteins was examined by confocal microscopy. The merged image of the two stains is shown in the last column, with colocalization shown in yellow. Δ437--442-VWFpp does not traffick to ACTH-containing granules. (B) AtT-20 cells expressing wild-type VWFpp or Δ437--442-VWFpp were incubated with either OptiMEM or 5 mmol L−1 8-Br-cAMP. The amount of released VWFpp was determined by enzyme-linked immunosorbent assay. Wild-type VWFpp was released in response to agonist, whereas Δ437–442-VWFpp was not.
The effect of Δ437–442-VWF on secretion of coexpressed wild-type VWF
To determine the effect on VWF from the ‘normal’ allele, coexpression experiments were performed. The amount of transfected wild-type VWF was constant (0.5 μg per well), and 0–0.5 μg per well of Δ437–442-VWF was added. As the amount of Δ437–442-VWF was increased, the total amount of secreted VWF decreased, indicating that the variant VWF affects the secretion of VWF expressed from the normal allele (Fig. 4).
Fig. 4.
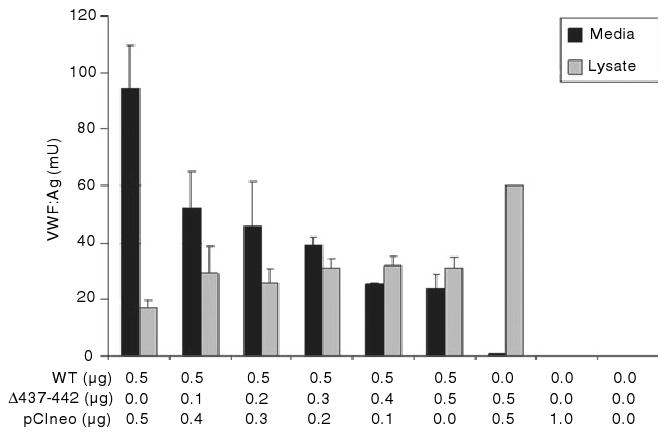
The effect of Δ437--442-von Willebrand factor (VWF) on secretion of coexpressed wild-type (WT) VWF. HEK293T cells were transfected with varying amounts of Δ437--442-VWF and wild-type VWF. The amounts of transfected wild-type VWF, Δ437--442-VWF and pCIneo are listed on the x-axis. The concentration of VWF in the conditioned media and cell lysate was assessed by VWF enzyme-linked immunosorbent assay. The data are derived from three separate transfections, with each vector expressed in duplicate. Results shown are mean VWF level, with error bars indicating standard deviation. As the amount of transfected Δ437–442-VWF is increased, total VWF secretion decreases.
Multimerization of coexpressed Δ437–442-VWF and wild-type VWF
To further analyze the heterozygous interaction, GFP or cmyc tags were added to the C-terminus of VWF (Δ437–442-VWF-GFP, WT-VWF-GFP, WT-VWF-cmyc). These constructs were expressed individually and together in HEK293T cells, and multimers were analyzed by western blotting with an anti-GFP or anti-cmyc antibody (Fig. 5A,B). Both WT-VWF-c-myc and WT-VWF-GFP multimerized normally when expressed alone or together (Fig. 5A,B). Δ437–442-VWF-GFP did not form multimers, similar to Δ437–442-VWF lacking a GFP tag. When Δ437–442-VWF-GFP was coexpressed with WT-VWF-c-myc and blotted for GFP, Δ437–442-VWF-GFP was incorporated into mid-molecular mass multimers, whereas WT-VWF-c-myc appeared to have a normal multimer distribution (Fig. 5A,B). Wild-type- VWF appears to function in trans to partially restore multimerization of Δ437–442-VWF.
Fig. 5.
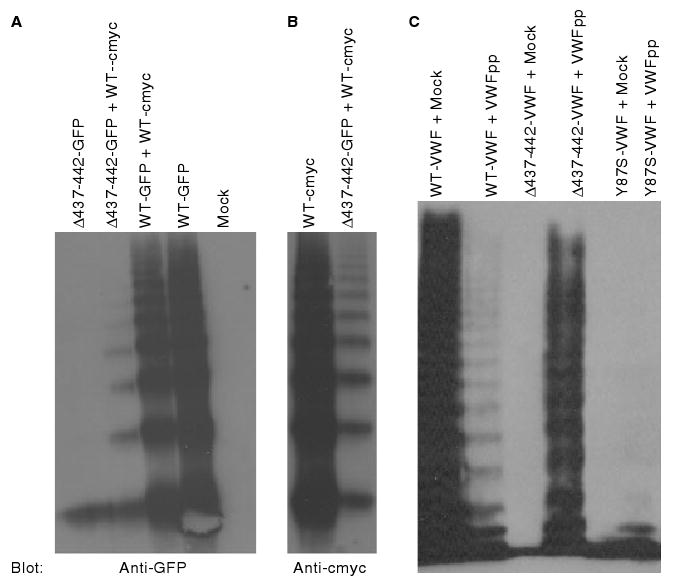
Multimeric composition of coexpressed Δ437--442-von Willebrand factor (VWF) and wild-type (WT) VWF. (A, B) HEK293T cells were transfected with green fluorescent protein (GFP)-tagged Δ437--442-VWF (Δ437--442-GFP) or wild-type VWF (WT-GFP), and cmyc-tagged wild-type VWF (WT-cmyc). The multimer composition of VWF proteins was assessed by sodium dodecylsulfate--agarose electrophoresis and western blotting with (A) anti-GFP antibody or (B) anti-c-myc antibody. Δ437--442-GFP forms only dimer (A, lane 1), but is incorporated into mid-molecular mass multimers when coexpressed with wild-type VWF (A, lane 2). Controls included coexpressed WT-GFP and WT-cmyc (A, lane 3), WT-GFP alone (A, lane 4), WT-cmyc (B, lane 1), and mock (A, lane 5). (C) The ability of VWF propeptide (VWFpp) to function in trans to facilitate multimerization was assessed by coexpression of wild-type VWFpp with full-length Δ437–442-VWF (lane 4), wild-type VWF (lane 2), and Y87S-VWF (lane 6). As controls, wild-type VWF (lane 1), Δ437–442-VWF (lane 3) and Y87S-VWF (lane 5) were also coexpressed with pCIneo (mock). Coexpression with wild-type VWFpp partially rescues multimerization of Δ437–442-VWF as a result of the trans function of VWFpp.
Previous studies have established the role of VWFpp in VWF multimerization [6,8,13,14]. Our data suggest that VWFpp from the wild-type allele can function in trans to facilitate multimerization of VWF from the Δ437–442-VWF allele. To address this, we coexpressed wild-type VWFpp with Δ437–442-VWF or Y87S-VWF (Fig. 5C) [6]. Coexpression of wild-type VWFpp with wild-type VWF appeared to result in a decrease in high molecular mass multimers, suggesting that free VWFpp may interfere with the normal VWFpp function of pro-VWF. When Y87S-VWF was coexpressed with VWFpp, VWFpp did not multimerize the variant Y87S-VWF. However, coexpression of Δ437–442-VWF with VWFpp resulted in essentially normal VWF multimerization, indicating that VWFpp is capable of functioning in trans to overcome the defective multimerization of a variant allele.
Intracellular localization of coexpressed Δ437–442-VWF and wild-type VWF
The effect of coexpressed Δ437–442-VWF/wild-type VWF on intracellular localization was examined. Coexpressed WT-VWF-GFP and WT-VWF-cmyc proteins were stored, colocalized in granules (Fig. 6C). When Δ437–442-VWF-GFP was coexpressed with WT-VWF-cmyc (Fig. 6A,B), the majority of cells expressing both proteins showed diffuse staining consistent with ER localization. In some cells, we observed a mixture of colocalized granular and diffuse staining patterns. We could not identify any transfected cells that showed a distinctly granular appearance. Together, these data suggest that Δ437–442-VWF may have a negative effect on the granular trafficking of VWF from the ‘normal’ allele.
Fig. 6.
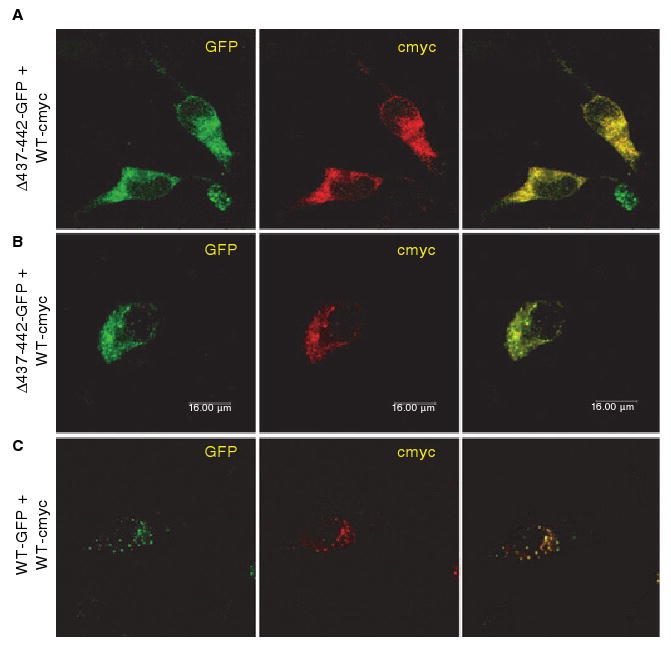
The effect of Δ437--442-von Willebrand factor (VWF) on intracellular localization of coexpressed wild-type (WT) VWF. (A, B) AtT-20 cells were transfected with green fluorescent protein (GFP)-tagged Δ437--442-VWF (Δ437--442-GFP) and cmyc-tagged wild-type VWF (WT-cmyc). (C) GFP-tagged wild-type VWF (WT-GFP) was coexpressed with cmyc-tagged wild-type VWF (WT-cyc). Transfected cells were stained using an anti-cmyc antibody. Δ437--442-GFP and WT-GFP were detected by GFP fluorescence. Intracellular localization of VWF was determined by confocal microscopy. The merged image is shown in the last column, and colocalization is shown in yellow. Coexpressed Δ437--442-VWF and cmyc-tagged wild-type VWF did not appear to be routed to storage granules.
Discussion
We have identified a mutation in the VWF gene of a VWD patient, an 18-bp deletion that predicts the deletion of amino acids 437–442 in the D2 domain of VWF. From the data presented in this article, we can conclude that: (i) this mutation causes a complete loss of VWF multimerization, decreased secretion, and loss of platelet-binding and collagen-binding functions; (ii) the deletion in VWFpp abolishes regulated VWFpp storage and, secondarily, VWF storage; (iii) Δ437–442-VWF negatively influences the secretion and storage of coexpressed wild-type VWF; and (iv) although VWFpp may normally function in a homotypic manner upon its own mature VWF, VWFpp may have the ability to function in trans on VWF expressed from the alternative allele.
On the basis of the current VWD classification, our patient could be classified as having type 2A VWD [1]. The increased VWF dimer band, small decrease in high molecular mass multimers and mutation in the VWF propeptide (Fig. 1A) are consistent with the historical subclassification of type IIC characterized by recessive inheritance, phenotypic heterogeneity, increased concentration of VWF protomer band, lack of high molecular mass multimers, and decrease in number of multimer satellite bands [15–18]. However, the apparent dominant inheritance does not fit the classic IIC subset, so the patient may best be described under the more general classification of type 2A VWD. A number of mutations in VWFpp have been identified in type 2A patients, emphasizing the importance of VWFpp in VWF multimer assembly [16–19].
The Δ437–442 VWF mutation results in complete loss of VWF multimerization and impaired function (Fig. 1D, Table 1). It is not unexpected that a six amino acid deletion in VWFpp abolishes multimerization [5,8,20]. Unexpectedly, the variant had decreased botrocetin-induced platelet binding and differed from dimeric Y87S-VWF in this respect, suggesting that Δ437–442-VWF has defects in folding/secondary structure that prohibit platelet binding. The increased intracellular retention provides further evidence of aberrant structure. Misfolded proteins are selected in the ER and targeted for degradation, although cells may also retain misfolded proteins in the ER [21]. The six amino acid deletion eliminates one cysteine residue (Cys441). Deletion of a cysteine in VWFpp is likely to disrupt normal disulfide bonding and impair proper protein folding, impacting on further processing and secretion.
The patient in this study was heterozygous for the Δ437–442 mutation in VWF. At a 1 : 1 ratio of Δ437–442-VWF to wild-type VWF (recapitulating heterozygosity), secretion was reduced (Fig. 4). In contrast, the patient had a plasma VWF:Ag within the normal range. This discrepancy could be due to a number of factors, including increasing VWF level with increasing age or the effect of other modifier genes [22]. Many patients with low VWF levels have no detectable mutation in the VWF gene, indicating that other genes may modify plasma VWF levels [23,24]. This points to the possibility of differential expression of VWF modifiers that affect VWF secretion in HEK293T cells vs. endothelial cells. This variation of transfected mammalian cells in completely recapitulating the biological phenotype has been previously reported [25–27]. Also, the wild-type allele may be preferentially translated or transcribed, or have greater stability, than the variant allele. Plasma VWF levels are also influenced by other modifiers, including ABO blood group, ADAMTS13, and other unidentified modifiers that influence VWF degradation and clearance.
The role of VWFpp in the trafficking of VWF to the regulated secretory pathway has been well established [4,5,13]. The VWF variants Y87S, R273W, C788R, C1157F, C1225G and C1234W have multimer abnormalities, and yet are stored in granules [6,7,26]. We have demonstrated a unique loss of regulated VWF storage with the Δ437–442 deletion (Fig. 2), resulting from defective VWFpp trafficking (Fig. 3). Coexpression with wild-type VWF (Fig. 6) did not appear to correct the storage defect, although a subset of cells did appear to contain VWF granules. On the basis of our AtT-20 expression data, we would predict that the patient had a reduction in endothelial cell Weibel–Palade bodies and a diminished response to desmopressin. Previously, using a site-directed mutagenesis approach, we identified amino acids in VWFpp and the D3 domain of VWF that are critical for regulated storage. Recent studies have demonstrated the importance of VWFpp and the D'′–D3 domains in Weibel–Palade body formation [9,28,29]. Many mutations have been identified in these domains in VWD patients (ISTH SSC VWF database, http://www.vwf.group.shef.ac.uk/). Whether loss of Weibel–Palade body formation is a common phenomenon in other VWD types remains to be determined.
The role of VWFpp in VWF multimerization is firmly established [5,14]. Purvis et al. observed the formation of an intrachain, transient intracellular disulfide bond between VWFpp and mature VWF, suggesting that VWFpp functions in a homotypic manner, facilitating the multimerization of its own mature VWF [20]. An interesting observation from our study (Fig. 5) is the partial rescue of multimerization of variant Δ437–442-VWF when coexpressed with wild-type VWF. The incorporation of Δ437–442-VWF-GFP into high molecular mass multimers suggests the possibility that VWFpp from the normal allele is functioning in trans to partially correct the dysfunctional mutant VWFpp. The rescue of multimerization was much greater with VWFpp coexpression than with full-length wild-type VWF, suggesting that only a fraction of VWFpp expressed from the wild-type allele is available to function in trans. However, coexpression of wild-type VWFpp could not restore multimerization of the dimeric Y87S-VWF (Fig. 5). These observations are consistent with patient plasma VWF multimers; plasma Y87S-VWF was primarily dimeric with very faint staining of a normal multimer pattern, whereas plasma Δ437–442-VWF demonstrated an over-representation of dimeric VWF among a relatively normal intensity and distribution of multimers (Fig. 1A). The difference in reactivity could stem from a potentially reactive free cysteine created in Δ437–442-VWF. Unexpectedly, expression of VWFpp with full-length wild-type VWF resulted in a decrease in the highest molecular mass multimers (Fig. 5). Overexpression of VWFpp may interfere with the normal VWF multimerization process. In total, these results suggest that VWFpp may normally function in a homotypic manner, acting upon its own mature VWF subunit, but may retain the ability to function in trans upon VWF expressed from the alternative allele.
In conclusion, our results demonstrate the detrimental effect of the Δ437–442 mutation on VWF multimerization, secretion, function, and regulated storage. By examining the interaction of the Δ437–442-VWF allele with VWF expressed from the ‘normal’ allele, we show that whereas VWFpp may typically facilitate multimerization of its own mature VWF subunit, VWFpp has the ability to function in trans on VWF expressed from the variant allele.
Supplementary Material
Acknowledgments
This work was supported by National Institutes of Health grants HL-33721, HL-081588, HL-44612 and American Heart Association SDG 0435466N.
Footnotes
Addendum: A. M. Allmann and M. A. Jozwiak collected and analyzed data; J. C. Gill was involved in study design and data collection; R. R. Montgomery was involved in study design and data analysis, S. L. Haberichter was involved in study design, collected and analyzed data, and wrote the manuscript; all authors were responsible for review and approval of the final manuscript.
Disclosure of Conflict of Interests: The authors state that they have no conflict of interest.
Supporting Information: Additional Supporting Information may be found in the online version of this article:
S1. Construction of expression plasmids.
S2. pCIneo-VWF-(Δ437–442).
S3. pCIneo-VWFpp-(Δ437–442).
S4. pEGFP-VWF-WT.
S5. pCIneo-VWF-(Δ437–442)-GFP.
Please note: Wiley-Blackwell are not responsible for the content or functionality of any supporting materials supplied by the authors. Any queries (other than missing material) should be directed to the corresponding author for the article.
References
- 1.Sadler JE, Budde U, Eikenboom JC, Favaloro EJ, Hill FG, Holmberg L, Ingerslev J, Lee CA, Lillicrap D, Mannucci PM, Mazurier C, Meyer D, Nichols WL, Nishino M, Peake IR, Rodeghiero F, Schneppenheim R, Ruggeri ZM, Srivastava A, Montgomery RR, et al. Update on the pathophysiology and classification of von Willebrand disease: a report of the Subcommittee on von Willebrand Factor. J Thromb Haemost. 2006;4:2103–14. doi: 10.1111/j.1538-7836.2006.02146.x. [DOI] [PubMed] [Google Scholar]
- 2.Verweij CL, Diergaarde PJ, Hart M, Pannekoek H. Full-length von Willebrand factor (vWF) cDNA encodes a highly repetitive protein considerably larger than the mature vWF subunit. EMBO J. 1986;5:1839–47. doi: 10.1002/j.1460-2075.1986.tb04435.x. [DOI] [PMC free article] [PubMed] [Google Scholar]; EMBO J. 1986;5:3074. erratum. [Google Scholar]
- 3.Wagner DD. Cell biology of von Willebrand factor. Annu Rev Cell Biol. 1990;6:217–46. doi: 10.1146/annurev.cb.06.110190.001245. [DOI] [PubMed] [Google Scholar]
- 4.Wagner DD, Saffaripour S, Bonfanti R, Sadler JE, Cramer EM, Chapman B, Mayadas TN. Induction of specific storage organelles by von Willebrand factor propolypeptide. Cell. 1991;64:403–13. doi: 10.1016/0092-8674(91)90648-i. [DOI] [PubMed] [Google Scholar]
- 5.Haberichter SL, Fahs SA, Montgomery RR. Von Willebrand factor storage and multimerization: 2 independent intracellular processes. Blood. 2000;96:1808–15. [PubMed] [Google Scholar]
- 6.Rosenberg JB, Haberichter SL, Jozwiak MA, Vokac EA, Kroner PA, Fahs SA, Kawai Y, Montgomery RR. The role of the D1 domain of the von Willebrand factor propeptide in multimerization of VWF. Blood. 2002;100:1699–706. doi: 10.1182/blood-2002-03-0789. [DOI] [PubMed] [Google Scholar]
- 7.Michaux G, Hewlett LJ, Messenger SL, Goodeve AC, Peake IR, Daly ME, Cutler DF. Analysis of intracellular storage and regulated secretion of 3 von Willebrand disease-causing variants of von Willebrand factor. Blood. 2003;102:2452–8. doi: 10.1182/blood-2003-02-0599. [DOI] [PubMed] [Google Scholar]
- 8.Voorberg J, Fontijn R, van Mourik JA, Pannekoek H. Domains involved in multimer assembly of von willebrand factor (vWF): multimerization is independent of dimerization. EMBO J. 1990;9:797–803. doi: 10.1002/j.1460-2075.1990.tb08176.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Haberichter SL, Jacobi P, Montgomery RR. Critical independent regions in the VWF propeptide and mature VWF that enable normal VWF storage. Blood. 2003;101:1384–91. doi: 10.1182/blood-2002-07-2281. [DOI] [PubMed] [Google Scholar]
- 10.Andrews RK, Booth WJ, Gorman JJ, Castaldi PA, Berndt MC. Purification of botrocetin from Bothrops jararaca venom. Analysis of the botrocetin-mediated interaction between von Willebrand factor and the human platelet membrane glycoprotein Ib–IX complex. Biochemistry. 1989;28:8317–26. doi: 10.1021/bi00447a009. [DOI] [PubMed] [Google Scholar]
- 11.Haberichter SL, Balistreri M, Christopherson P, Morateck P, Gavazova S, Bellissimo DB, Manco-Johnson MJ, Gill JC, Montgomery RR. Assay of the von Willebrand factor (VWF) propeptide to identify patients with type 1 von Willebrand disease with decreased VWF survival. Blood. 2006;108:3344–51. doi: 10.1182/blood-2006-04-015065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Blagoveshchenskaya AD, Hannah MJ, Allen S, Cutler DF. Selective and signal-dependent recruitment of membrane proteins to secretory granules formed by heterologously expressed von Willebrand factor. Mol Biol Cell. 2002;13:1582–93. doi: 10.1091/mbc.01-09-0462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Journet AM, Saffaripour S, Wagner DD. Requirement for both D domains of the propolypeptide in von Willebrand factor multimerization and storage. Thromb Haemost. 1993;70:1053–7. [PubMed] [Google Scholar]
- 14.Wise RJ, Pittman DD, Handin RI, Kaufman RJ, Orkin SH. The propeptide of von Willebrand factor independently mediates the assembly of von Willebrand multimers. Cell. 1988;52:229–36. doi: 10.1016/0092-8674(88)90511-9. [DOI] [PubMed] [Google Scholar]
- 15.Ruggeri ZM, Nilsson IM, Lombardi R, Holmberg L, Zimmerman TS. Aberrant multimeric structure of von Willebrand factor in a new variant of von Willebrand's disease (type IIC) J Clin Invest. 1982;70:1124–7. doi: 10.1172/JCI110700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Gaucher C, Dieval J, Mazurier C. Characterization of von Willebrand factor gene defects in two unrelated patients with type IIC von Willebrand disease. Blood. 1994;84:1024–30. [PubMed] [Google Scholar]
- 17.Holmberg L, Karpman D, Isaksson C, Kristoffersson AC, Lethagen S, Schneppenheim R. Ins405AsnPro mutation in the von Willebrand factor propeptide in recessive type 2A (IIC) von Willebrand's disease. Thromb Haemost. 1998;79:718–22. [PubMed] [Google Scholar]
- 18.Schneppenheim R, Thomas KB, Krey S, Budde U, Jessat U, Sutor AH, Zieger B. Identification of a candidate missense mutation in a family with von Willebrand disease type IIC. Hum Genet. 1995;95:681–6. doi: 10.1007/BF00209487. [DOI] [PubMed] [Google Scholar]
- 19.Allen S, Abuzenadah AM, Hinks J, Blagg JL, Gursel T, Ingerslev J, Goodeve AC, Peake IR, Daly ME. A novel von Willebrand disease-causing mutation (Arg273Trp) in the von Willebrand factor propeptide that results in defective multimerization and secretion. Blood. 2000;96:560–8. [PubMed] [Google Scholar]
- 20.Purvis AR, Sadler JE. A covalent oxidoreductase intermediate in propeptide-dependent von Willebrand factor multimerization. J Biol Chem. 2004;279:49982–8. doi: 10.1074/jbc.M408727200. [DOI] [PubMed] [Google Scholar]
- 21.Hampton RY. ER-associated degradation in protein quality control and cellular regulation. Curr Opin Cell Biol. 2002;14:476–82. doi: 10.1016/s0955-0674(02)00358-7. [DOI] [PubMed] [Google Scholar]
- 22.Gill JC, Endres-Brooks J, Bauer PJ, Marks WJ, Jr, Montgomery RR. The effect of ABO blood group on the diagnosis of von Willebrand disease. Blood. 1987;69:1691–5. [PubMed] [Google Scholar]
- 23.Goodeve A, Eikenboom J, Castaman G, Rodeghiero F, Federici AB, Batlle J, Meyer D, Mazurier C, Goudemand J, Schneppenheim R, Budde U, Ingerslev J, Habart D, Vorlova Z, Holmberg L, Lethagen S, Pasi J, Hill F, Hashemi SM, Baronciani L, et al. Phenotype and genotype of a cohort of families historically diagnosed with type 1 von Willebrand disease in the European study, Molecular and Clinical Markers for the Diagnosis and Management of Type 1 von Willebrand Disease (MCMDM-1VWD) Blood. 2007;109:112–21. doi: 10.1182/blood-2006-05-020784. [DOI] [PubMed] [Google Scholar]
- 24.James PD, Notley C, Hegadorn C, Leggo J, Tuttle A, Tinlin S, Brown C, Andrews C, Labelle A, Chirinian Y, O'Brien L, Othman M, Rivard G, Rapson D, Hough C, Lillicrap D. The mutational spectrum of type 1 von Willebrand disease: results from a Canadian cohort study. Blood. 2007;109:145–54. doi: 10.1182/blood-2006-05-021105.. [DOI] [PubMed] [Google Scholar]
- 25.Eikenboom JC, Matsushita T, Reitsma PH, Tuley EA, Castaman G, Briet E, Sadler JE. Dominant type 1 von Willebrand disease caused by mutated cysteine residues in the D3 domain of von Willebrand factor. Blood. 1996;88:2433–41. [PubMed] [Google Scholar]
- 26.Hommais A, Stepanian A, Fressinaud E, Mazurier C, Meyer D, Girma JP, Ribba AS. Mutations C1157F and C1234W of von Willebrand factor cause intracellular retention with defective multimerization and secretion. J Thromb Haemost. 2006;4:148–57. doi: 10.1111/j.1538-7836.2005.01652.x. [DOI] [PubMed] [Google Scholar]
- 27.Hilbert L, Gaucher C, Mazurier C. Identification of two mutations (Arg611Cys and Arg611His) in the A1 loop of von Willebrand factor (vWF) responsible for type 2 von Willebrand disease with decreased platelet-dependent function of vWF. Blood. 1995;86:1010–18. [PubMed] [Google Scholar]
- 28.Huang RH, Wang Y, Roth R, Yu X, Purvis AR, Heuser JE, Egelman EH, Sadler JE. Assembly of Weibel–Palade body-like tubules from N-terminal domains of von Willebrand factor. Proc Natl Acad Sci USA. 2008;105:482–7. doi: 10.1073/pnas.0710079105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Michaux G, Abbitt KB, Collinson LM, Haberichter SL, Norman KE, Cutler DF. The physiological function of von Willebrand's factor depends on its tubular storage in endothelial Weibel–Palade bodies. Dev Cell. 2006;10:223–32. doi: 10.1016/j.devcel.2005.12.012. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.