

Summary
Fas apoptosis inhibitory molecule (FAIM) is a soluble cytosolic protein inhibitor of programmed cell death and is found in organisms throughout the animal kingdom. A short isoform (FAIM-S) is expressed in all tissue types, while an alternatively spliced long isoform (FAIM-L) is specifically expressed in the brain. Here FAIM-S is shown to consist of two independently folding domains in contact with one another. The NMR solution structure of the C-terminal domain of murine FAIM is solved in isolation and revealed to be a novel protein fold, a noninterleaved seven-stranded beta sandwich. The structure and sequence reveal several residues that are likely to be involved in functionally significant interactions with the N-terminal domain or other binding partners. Chemical shift perturbation is used to elucidate contacts made between the N- and C-terminal domains.
Keywords: FAIM, apoptosis, Fas, NMR structure, beta sandwich
Introduction
Programmed cell death, or apoptosis, plays a vital role in numerous physiological processes. During embryonic development, it is essential for the formation of anatomical structures such as the fingers and some tubular organs. It also has numerous regulatory roles in adult life, notably in regulation of the immune system and deletion of autoreactive lymphocytes. Dysregulation of apoptosis is also thought to play a role in many types of neoplastic disease.
Many stimuli can trigger apoptosis; one such stimulus is engagement of members of the transmembrane tumor necrosis factor receptor (TNFR) superfamily with a cognate ligand. Fas/FasL is an example of this type of receptor/ligand pair: binding of FasL can promote the binding of the Fas-associated protein with death domain (FADD) to the intracellular portion of Fas via a “death domain” carried by each protein. FADD also contains a death effector domain (DED) that binds to procaspase-8 and promotes its proteolytic activation. This leads to activation of downstream “executioner caspases” such as caspase-3, -6, and -7 that carry out the apoptotic process and represent the final step common to all apoptotic pathways.
FAIM was first identified as an inducible inhibitor of apoptosis by Schneider et al. (1999)[1] using differential mRNA display between primary murine B lymphocytes stimulated with antibodies directed against the surface immunoglobulin receptor (anti-IgM) in the presence of a soluble dimeric CD40 ligand (CD40L) construct and B lymphocytes stimulated with CD40L alone. Reverse transcription and PCR resulted in construction of a probe that was used to screen a murine cDNA library, from which was isolated a full length clone of a short isoform of FAIM that became known as FAIM-S. In the same work, the antiapoptotic activity of FAIM was confirmed by overexpressing FAIM-S in a murine B lymphoma cell line (BAL-17) and subjecting transfected and control cells to Fas-mediated apoptotic stimuli including Fas ligand-bearing AE7 T cells and Jo-2 anti-Fas antibody; overexpression of FAIM gave partial protection against cell lysis (51Cr release) and the DNA degradation characteristic of apoptosis.
FAIM is a 20 kDa, soluble, evolutionarily conserved protein known to be expressed at the mRNA level in a wide variety of mouse tissues. In murine brain (and to a lesser extent in the testis and embryonic tissue), alternative splicing is known to yield an mRNA whose protein product, FAIM-L, is longer than FAIM-S by 22 residues at the N-terminus [2]. The FAIM gene localizes to chromosome 9 (9f1) in mice and chromosome 3 (3q21-25) in humans [2]; these authors note that abnormalities of this region have been associated with a number of types of human cancer, hinting at a possible role for FAIM in oncogenesis.
Sole et al. (2004)[3] reported that overexpression of FAIM-S, but not FAIM-L, in conjunction with nerve growth factor (NGF), promoted the outgrowth of neurites in cultured neurons from the superior cervical ganglion (SCG) of the mouse and in the neuron-like PC12 cell line. Conversely, knockdown of endogenous FAIM with siRNA decreased neurite outgrowth. More recently, the same group [4] also found that FAIM-L, but not FAIM-S, is induced in cultured neurons in response to NGF signaling and that overexpression of FAIM-L antagonizes apoptotic signaling from FasL and TNFα. Thus, curiously, it would appear that FAIM-S is an inducible inhibitor of Fas-mediated apoptosis in cultured murine lymphocytes, while that function is performed exclusively by FAIM-L in the neuron model despite the presence of FAIM-S.
The molecular mechanisms of FAIM are not understood in detail, but studies have shed some light on the subject. With regard to apoptosis inhibition in lymphocytes, an experiment by Schneider et al. (1999)[1] showed that the anti-apoptotic activity of FAIM occurs upstream of poly (ADP-ribose) polymerase (PARP) cleavage by demonstrating a decrease in PARP cleavage in FAIM-transfected cells treated with the Jo-2 anti-Fas antibody. The neurite outgrowth activity of FAIM has also been investigated in molecular detail: it has been found that NF-κB is activated by FAIM-S in the presence of NGF and that transfection of a stabilized form of IκB, an inhibitor of NF-κB, into cultured neurons abolished the ability of FAIM-S to promote neurite outgrowth [3]. The same study showed immunoprecipitation evidence of an interaction between FAIM-S and both the TrkA and p75NTR neurotrophin receptors when NGF was present; FAIM-L was not studied in this context. The later study [4], using similar techniques, found an interaction between FAIM-L and the Fas receptor that was blocked by overexpression of the known Fas binding partner FADD. These researchers also showed that the antiapoptotic activity of FAIM-L in neurons is effective upstream of both caspase-8 and caspase-3, since activity of these caspases in the presence of apoptotic stimuli is blocked by FAIM-L.
Presence of FAIM nucleotide sequences in several vertebrate and invertebrate animal species has been confirmed by Southern blotting[1] and by analysis of publicly available nucleotide databases [1] [3]. Wiens et al. (2004)[5] even report a homolog of FAIM 41% identical to human FAIM in Suberites domuncula, a poriferan lacking lymphocytes and neurons but possessing a receptor, DD2, that contains Fas-like death domains [6]. Although the faim gene product is highly conserved among species, there is no known sequence homology to other antiapoptotic proteins [1] or indeed to any protein of known structure. It has been noted that these facts point toward a vital physiological function for FAIM.
Although progress has been made toward understanding the activities of FAIM at the cellular level, little is known about its biochemistry or the molecular mechanisms by which it acts. Because FAIM has no significant primary sequence homology to any protein of known structure or function, homology modeling currently can provide no insight. Experimental structure determination will therefore be necessary to interpret mechanistic data and may give clues to the precise function or functions of FAIM as well.
In this paper, we (1) describe a construct of FAIM and a set of buffer conditions conducive to NMR spectroscopy, in the process gaining insight into its domain organization; (2) report and analyze the NMR solution structure of the FAIM C-terminal domain (FAIM-CTD), a novel protein fold, noting residues likely to be involved in functionally significant interactions; and (3) identify certain sites of interaction between the N- and C-terminal domains.
Results
FAIM is composed of two independently folding domains
Wild-type FAIM-S exhibits fair quality NMR spectra and has limited solubility. Previous work for a similar situation [7] suggested that introducing rationally chosen point mutations could improve the physical and/or spectral properties of a protein. Point mutations to improve solubility were selected from among the residues seen at the homologous position in the orthologs identified by Schneider et al. (1999)[1], namely the Caenorhabditis elegans, mouse, and human forms. Substitutions were chosen subjectively based on criteria including the lack of conservation among the orthologs, the qualitative decrease in hydrophobicity or the reduction in pI achieved by the mutation, and the degree to which the substitution could be considered conservative. The murine FAIM-S triple mutant F55Y/N92D/G148D was identified as a candidate for further study because it was well-behaved at reasonable NMR concentrations and temperatures in the low-salt 1010TBS buffer and its 15N-HSQC spectrum was similar to that of the wild-type (data not shown). Backbone assignment of the triple mutant FAIM-S revealed that the majority of the peaks visible in the 15N-HSQC spectrum were from the C-terminal half of the protein: residues 91-179 were assigned with 85% completeness (76/89), while residues 1 to 90 were assigned with only 36% completeness (32/90). This result suggested that the N-terminal half of the protein might be responsible for the poor overall spectral quality and perhaps the poor physical behavior of the wild type as well. This idea was supported by limited proteolysis experiments, in which the triple mutant FAIM-S could be cleaved, resulting in one or several smaller, more stable fragments (see Figure 1a).
Figure 1. Biochemical, physical, and spectral properties of FAIM constructs.

(a) Limited proteolysis of FAIM constructs. Truncation mutants FAIM-NTD (CΔ111) and FAIM-CTD reproduce the same stable, low molecular weight band as FAIM-S itself when subjected to limited tryptic proteolysis, indicating that both constructs are well-folded and that truncation has not removed residues necessary for the stability of either domain. 0.5% bed volume of immobilized TPCK-treated trypsin (15-25 U/mL; Sigma) was added to protein in TBS and shaken gently at 4 °C. The reactions were centrifuged and aliquots were removed from the supernatant at the indicated time points.
(b) The G148D mutation only minimally perturbs the structure of FAIM-CTD. 15N-HSQC spectra of native G148 (blue) and mutant G148D (red) NΔ90 FAIM-CTD constructs are shown. The most strongly perturbed residues, near the site of the mutation, are labeled.
(c) FAIM-CTD is monomeric by ultracentrifugation at NMR concentration. Equilibrium ultracentrifugation of FAIM-CTD at 1.7 mM was performed for 12 hours at 22,500 rpm (34,000 × g) in freshly prepared, degassed 1010TBS identical to that used for NMR spectroscopy. The slope of the line corresponds to 11 kDa, assuming a partial specific volume of 0.73. The residuals are shown at the top of the figure.
(d) The N-terminal domain of FAIM-S has well-dispersed backbone amide chemical shifts and is largely structured. 15N-HSQC spectra at 500 MHz of the CΔ97/F55Y FAIM-NTD construct at 15 °C (blue), 25 °C (purple), and 35 °C (red) are overlaid. Contour levels are the same for each spectrum. Only about 60-70% of non-proline residues are visible, and a significant portion of these exhibit temperature-dependent signal broadening or loss (see insets). There is no trend toward improved spectra at any temperature: some peaks exhibit broadening at higher temperature while others are broadened at lower temperature.
(e) Alignment of selected FAIM sequences from publicly available databases. Secondary structural elements, where known, are shown above the sequence. Identical residues are boxed and colored according to the degree of substitution at each position: red, strictly conserved; orange, highly similar; yellow, less similar; white, not conserved. Numbering is according to murine FAIM-S.
(f) Mutation of cysteine residues reveals that they exist within a hydrophobic environment but are dispensable to folding of FAIM-S. SDS-PAGE of wild-type and cysteine-free full-length FAIM-S at various stages during purification shows that alanine substitutions are well-tolerated while serine substitutions are not. All constructs have a C-terminal cloning artifact. Results were similar when single cysteine substitutions were expressed (data not shown).
Purification of a construct (NΔ90/G148D) incorporating residues 91-179 of murine FAIM-S and the G148D mutation revealed that the peaks of this construct are well-dispersed in the 15N-HSQC spectrum. Thus, this construct is well-folded and is an independent domain, termed FAIM-CTD. FAIM-CTD had no requirement for detergent and could readily be concentrated to more than 1 mM. Limited proteolysis experiments confirmed that it was similar in size to one of the more trypsin-resistant products of triple mutant FAIM-S cleavage (see Figure 1a). It was extremely stable in low salt buffer as long as reducing conditions were maintained: samples showed no precipitation or changes in the 15N-HSQC spectrum when analyzed for several weeks at 25 °C and stored for more than one year at 4 °C. Comparison of the 15N-HSQC spectra of FAIM-CTD and triple mutant FAIM-S did show some significant differences, as discussed below, suggesting that removal of the N-terminal region could have caused structural perturbation within FAIM-CTD. Structural perturbation associated with the G148D mutation was minimal and was generally limited to the region near the site of mutation (see Figure 1b). FAIM-CTD appeared to be monomeric by ultracentrifugation. At 22,500 rpm (34,000 × g) the ultracentrifugation data were well fit by a straight line (see Figure 1c). The temperature, buffer conditions, and protein concentrations used for ultracentrifugation were very similar to those subsequently used for NMR spectroscopy, except for the unavoidable radial gradient in protein concentration, so the absence of detectable dimerization of FAIM-CTD during ultracentrifugation strongly suggests its absence during NMR spectroscopy.
To ensure that the FAIM-CTD construct contained the complete domain, another construct composed of residues 82-179 (NΔ81) was synthesized, expressed, and purified. Its 15N-HSQC spectrum was very similar to that of the shorter 91-179 (NΔ90) FAIM-CTD construct (data not shown), demonstrating that residues 82-90 do not interact strongly with FAIM-CTD and implying that the NΔ90 construct encompasses all the residues involved in folding of FAIM-CTD.
A FAIM-NTD construct (CΔ97/F55Y), incorporating residues 1-96 of murine FAIM-S and the F55Y mutation, was also expressed and evaluated. Although its 15N-HSQC spectrum shows heterogeneous peak intensity, it was stable at NMR concentrations (0.1-0.2 mM) for hours in 1010TBS with 3% Tween-20 at 35 °C. 15N-HSQC spectra of this construct at 0.1-0.2 mM, acquired at 500 MHz in 1010TBS and 3% Tween-20 (without DTT) at three different temperatures, are shown in Figure 1d. The visible residues are well-dispersed, indicating that large regions within this construct are also well-folded. However, the number of visible peaks is about 1/3 less than expected. The intensity and line shape of some of these peaks are highly temperature-dependent, suggesting the presence of chemical exchange or temperature-dependent partial unfolding.
A very short truncation of murine FAIM-S, incorporating residues 1-62 (CΔ63), was also made by introducing a stop codon into the FAIM-S gene at residue 63. This protein expressed at very low levels under the usual expression conditions and could not be otherwise evaluated. A longer C-terminal protein consisting of residues 52-179 was also synthesized and expressed, but expression yield was very poor, quantitative aggregation was seen by FPLC, and NMR spectroscopy of the aggregated fraction failed to detect a protein signal. These results indicate that truncation within the N-terminal domain yields unstable protein constructs.
FAIM tolerates apolar but not polar substitution of cysteine residues
To assess the structural significance, if any, of the two cysteine residues (Cys122 and Cys149) in FAIM-CTD, sequence alignments and mutation studies were undertaken. Both cysteine residues in murine FAIM-S are conserved among vertebrate orthologs but not among more distant relatives [3](see Figure 1e). Cysteine to serine mutations were thought to be conservative and most likely to preserve native structure in a surface-exposed position, while mutations to alanine would be less hydrophilic and also reasonably conservative. Substitution of Cys122 with alanine is observed in a known homolog, indicating that the protein was likely to tolerate this mutation. In one experiment, both cysteines were mutated to serine (C122S/C149S). Another construct was likewise made in which both residues were mutated to alanine (C122A/C149A). Finally, since there was no known homolog in which Cys149 was replaced by alanine, Cys149 was also mutated to phenylalanine to yield a third cysteine-free construct, C122A/C149F.
Expression levels of various cysteine mutants were compared; results are shown in Figure 1f. The majority of the C122S/C149S double mutant protein was found in the cell pellet after lysis. Expression of the cysteine to alanine double mutant C122A/C149A, however, was similar to expression of the wild type. Its behavior was also similar to that of the wild type during purification. The 15N-HSQC spectrum at 400 MHz showed some differences from that of wild-type FAIM-S. Many of these differences were from residues near the sites of mutation. Remarkably, the C149F mutation was also well-tolerated (data not shown). Taken together, these results imply that both cysteine residues reside within a hydrophobic environment that cannot tolerate disruption by polar substitutions. However, the sulfhydryl moieties of the cysteine sidechains themselves are not vital to the stability of the protein.
FAIM-CTD exhibits a novel beta sandwich fold
Due to the low molecular weight of the FAIM-CTD construct (10 kDa), TROSY experiments were not used and deuteration was unnecessary during NMR spectroscopy. Triple-resonance NMR spectra were of generally high quality, permitting backbone assignment of 100% of the non-proline amide protons between residues 96 and 179 inclusive. Because the backbone assignment was done without the benefit of a CO pair, and was later checked against the CO pair and found to agree, it is very likely to be error-free. Sidechain atoms were assigned using standard techniques. Stereospecific methyl assignments were obtained for 10 of 15 valine and leucine residues. NOESY peaks were integrated with Sparky, and CYANA was used to calculate 100 structures of which the lowest 20% in energy are presented. The structure was then validated by measuring N-HN residual dipolar couplings (RDCs) and calculating Qfree. Final NOE statistics and structural parameters are tabulated in Table 1.
Table 1. Figures of merit for the FAIM-CTD structure.
Dihedral angles and NOE restraints were analyzed with PROCHECK (Laskowski et al., 1996)[26]. Qfree was computed using PALES as described in the text. Other statistics were calculated using CYANA.
| Restrictive NOE distance restraints (unique) | 934 |
| Short range | 399 |
| Medium range | 114 |
| Long range | 421 |
| TALOS dihedral angle restraints | 106 |
| Sidechain dihedral angle restraints | 37 |
| Hydrogen bond distance restraints | 16 |
| Dihedral angle statistics | |
| Residues in most favored regions | 78.9% |
| Residues in additionally allowed regions | 20.9% |
| Residues in generously allowed regions | 0.1% |
| Residues in disallowed regions | 0.2% |
| Average backbone RMSD to mean structure (residues T96 - T178) | 0.51 Å |
| Average heavy atom RMSD to mean structure (residues T96-T178) | 1.02 Å |
| Maximum NOE violation | 0.37 Å |
| Cornilescu Qfree from RDC validation (lowest-energy structure, secondary structural elements and turns only) | 0.505 |
A ribbon diagram showing the tertiary structure is shown in Figure 2a. The quality of the structure is illustrated in Figure 2b with a superposition of the peptide backbones of all accepted structures. FAIM-CTD is a “beta sandwich” reminiscent of the classical immunoglobulin fold; however, it is somewhat smaller, has a simpler topology, and is stable in the absence of a disulfide bond between the two sheets. The two beta sheets are canted with respect to one another by an angle of approximately 30°.
Figure 2. FAIM-CTD is a non-interleaved beta sandwich.
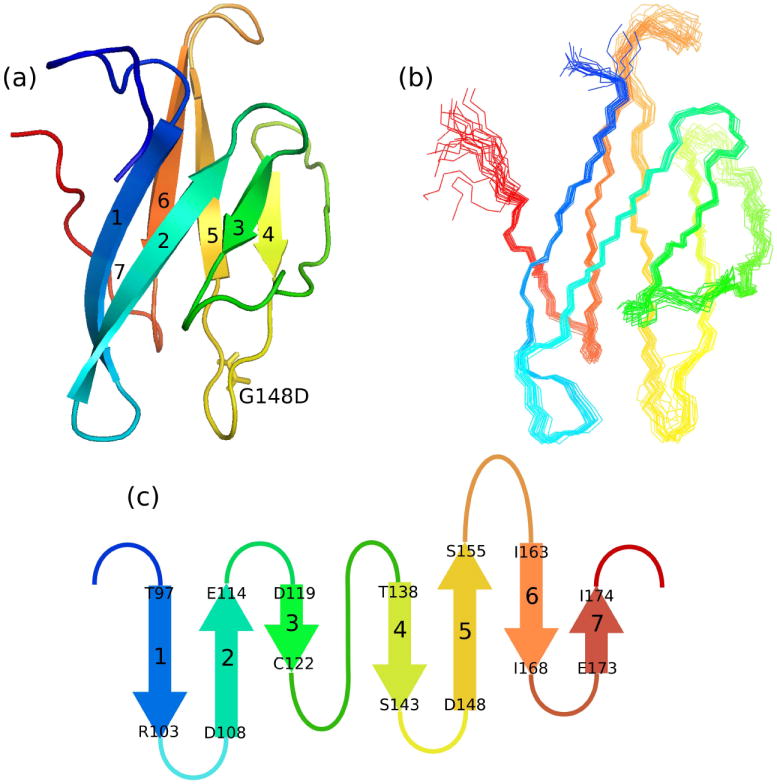
(a) Ribbon diagram of FAIM-CTD, with N-terminus colored red. The sidechain of the non-native D148 is also shown.
(b) Bundle of 20 structures of FAIM-CTD, colored as in (a). Nine residues preceding K95 are omitted from this figure.
(c) Topology diagram of FAIM-CTD.
All molecular models were rendered with PyMOL.
The topology of FAIM-CTD is shown schematically in Figure 2c. The topology is simple: there is no interleaving of β-strands and all strands are antiparallel. Successive strands are connected by four-residue turns in all but two cases. Between strands 5 and 6 is a longer loop comprising exclusively hydrophilic residues. Residues in the distal region of this loop, from K158 to K160, as well as the residues preceding T97, are not constrained by any experimental data. Only weak cross-strand NOE crosspeaks are seen between the backbone amide protons of residues Ser156 and Gly162, implying that the beta structure becomes more disordered toward the end of the hydrophilic loop under the conditions used in this study. The extended loop connecting the two sheets between strands 3 and 4 is well ordered but does not adopt a regular secondary structure, and its primary sequence, GQKMETAGEFVDDG, does not have the characteristic pattern of alternating hydrophobic and hydrophilic residues that would be expected of beta structure.
A search of the DALI protein structure database [8] in May 2008 yielded 664 matches with Z-scores ranging from 2.0 to 4.5. However, the vast majority of these had structural homology only with the C-terminal four-stranded β-sheet of FAIM-CTD and were therefore discarded. All other matches were examined manually and determined to be false positives, indicating that FAIM-CTD represents a novel fold.
The N-terminal face of FAIM-CTD interacts with FAIM-NTD
There are a number of changes seen in the 15N-HSQC spectrum when comparing the peak positions of residues in free FAIM-CTD and the corresponding residues in the CTD of the (full-length) triple mutant FAIM-S, as seen in Figure 3a. A plot of these chemical shift differences is shown in Figure 3b. There is a trend toward larger differences at the N-terminal end of FAIM-CTD. This is particularly true if one interprets the inability to assign a number of residues in the range V112-Q125 in the full-length FAIM-S construct as additional evidence of perturbation. The chemical shifts of the full-length triple mutant FAIM-S are unaffected by dilution from 150 μM to 50 μM and, at lower concentration, the 20 kDa protein is found to exhibit an apparent molecular weight of approximately 28 kDa on a size exclusion column (data not shown). Taken together, these data imply that FAIM-S is largely monomeric and the NTD and CTD of FAIM-S interact in cis.
Figure 3. FAIM-CTD is perturbed by removal of the N-terminal domain.
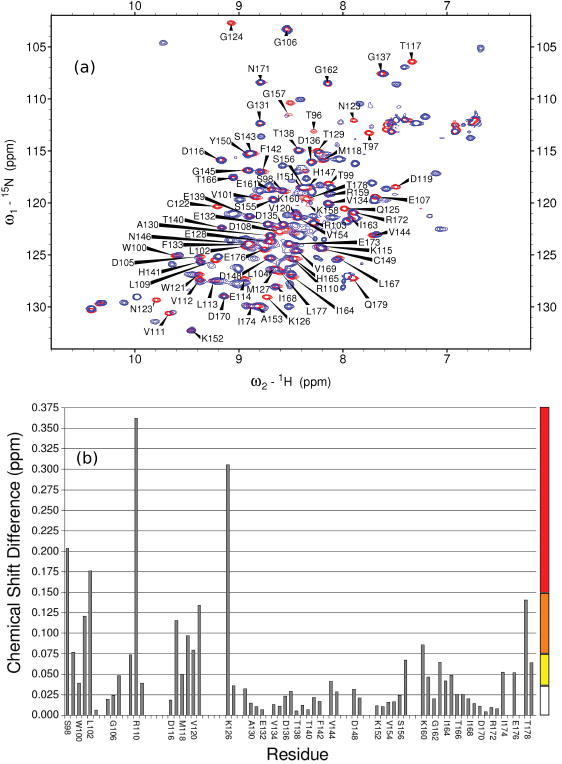
(a) Overlaid 15N-HSQC spectra of isolated FAIM-CTD (red) and full-length triple mutant FAIM-S (blue). Assignments are of FAIM-CTD. Buffer pH is 6.9-7.1. The buffer of full-length FAIM-S has 3% Tween-20. Titration experiments (not shown) indicated that chemical shift perturbation of FAIM-CTD by 3% Tween-20 was minimal and does not explain the observed differences.
(b) Chemical shift differences in the 15N-HSQC spectra in (a) are calculated according to the formula , plotted as a function of residue, and used to color Figure 4b according to the scale at right. No value is shown for residues that could not be assigned in one or both spectra.
It is remarkable that the most perturbed residue, R110, is one of the few residues that is strictly conserved among all known orthologs of FAIM (see Figure 4). One could therefore hypothesize that this residue plays a key role in mediating interactions between FAIM-NTD and FAIM-CTD. Notably, Glu38 in FAIM-NTD is also strictly conserved and could interact electrostatically with Arg110. Other candidates for this role include position 52, which contains a Glu or Asp residue in known homologs, and positions 3, 20, 22, 43, 68, and 76, which have a strong bias toward negatively charged residues as well.
Figure 4. Structure and sequence analysis highlight residues of FAIM-CTD involved in interactions with FAIM-NTD and other entities.
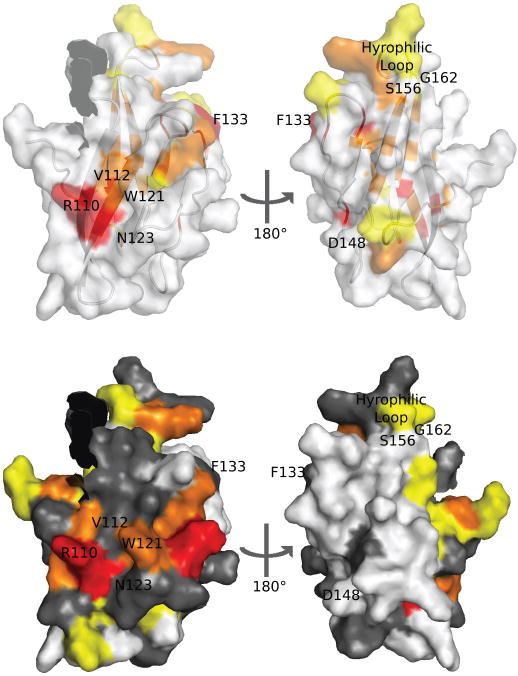
Surface-exposed residues discussed in the text are labeled in this figure. The position of the non-native G148D mutation is shown. Nine residues preceding K95 are omitted.
(a) Superimposed surface and ribbon models of FAIM-CTD with conserved residues colored as in Figure 2d.
(b) Surface model of FAIM-CTD with residues colored according to chemical shift perturbation (see Figure 3b). Residues for which chemical shifts are not known in one or both constructs are shown in gray.
To identify additional surface-exposed sites of high conservation, the FAIM-CTD structure and FAIM-S multiple sequence alignment were submitted to the online ConSurf server [9]. In addition to the residues already discussed, ConSurf identified Val112, Asn123, Gly137, and Gly157 as solvent-exposed and highly conserved. Because the glycine residues have no sidechains, they are not highly visible when plotted on the surface of FAIM-CTD. Val112 and Asn123, however, are adjacent to the strictly conserved Arg110 on the N-terminal surface of FAIM-CTD (data not shown). Thus, ConSurf discerns a larger region on this face of the protein that is likely to be involved in interactions with FAIM-NTD or other binding partners.
Discussion
The well-dispersed 15N-HSQC spectra of FAIM-CTD and FAIM-NTD show that each can fold independently of the other and clearly constitute distinct domains, although the structure of each domain may be affected by the loss of inter-domain interactions. At 90 residues or less, both domains are relatively small in size.
FAIM-CTD shares some topological features with the “up-and-down barrel” proteins in the fatty acid binding protein (FABP) and P2 families [10]. By definition, these proteins are composed of antiparallel β-strands with no interleaving. They differ from FAIM-CTD by their larger molecular weight and the angle at which the strands on opposite sides of the protein appear to cross (approximately 90° in FABP) when viewed in a two-dimensional projection in the plane of the strands. Unlike FABP, FAIM-CTD appears to be too small to accommodate a comparable fatty acid within a cavity and has no known role in lipid binding. The size and architecture of FAIM-CTD are also quite similar to that of the Type III fibronectin domains, but the β-strands of the latter have an interleaved topology. Structurally important residues such as Trp100 and Leu167 in FAIM-CTD (see below) are not conserved in Type III fibronectin domains.
Both cysteine residues appear to be embedded in the hydrophobic core in the FAIM-CTD structure (see Figure 5a), although Cys122 is near the surface. This observation is consistent with the findings that FAIM-S becomes insoluble during expression if either residue is substituted by serine, but that alanine substitutions are well tolerated (see Figure 1f). Similarly, while neither residue is strictly conserved, substitutions to polar residues are not found in known sequences. Despite their relative positions, several lines of argument support the absence of an intramolecular disulfide bond between these residues. FAIM is physiologically cytosolic [4] and would not likely have a disulfide bond in vivo. Secondly, NMR samples of murine FAIM were kept under reducing buffer conditions and were, in fact, not stable under oxidizing conditions. Thirdly, the Cβ chemical shifts of both Cys122 and Cys149 (28.7 ppm and 30.0 ppm) are consistent with the absence of a disulfide bond. Finally, these cysteine residues are not conserved in the more distant homologs of FAIM [3] and thus cannot be of vital structural importance.
Figure 5. Structural features of FAIM-CTD.
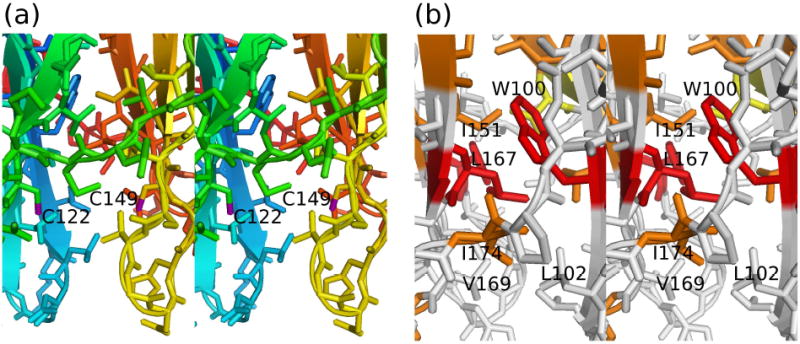
(a) Stick model in stereo of FAIM-CTD, colored as in Figure 2a, showing cysteine residues 122 and 149. The distance between the two sulfur atoms (purple) in this figure is 8.2 Å.
(b) Stick model in stereo of FAIM-CTD with residues colored according to residue conservation as in Figure 1e, illustrating the network of hydrophobic contacts involving Trp100, Leu167, Leu102, Val169, and Ile174 that hold together the two beta sheets.
There exists a network of hydrophobic contacts among Leu167, Trp100, Ile174, Val169, and Leu102 that appears to be responsible for holding the N- and C-terminal edges of the protein in contact and preventing clamshell-like movements of the two beta sheets (see Figure 5b). In particular, the sidechain of Leu167 packs closely against the indole sidechain of Trp100, consistent with the observed upfield shift of the sidechain protons of Leu167. These two residues are strictly conserved and may therefore be essential to maintaining the stability of this fold. Ile174 is highly conserved as well, and Val169 and Leu102, like much of the hydrophobic core of FAIM-CTD, suffer only relatively conservative substitutions. The FAIM-CTD structure calculation is also affected by unambiguous NOE restraints between Glu176 and Trp100, although the physical basis for the close contact between these two residues is less clear. One possible explanation for the high degree of conservation at this site is that the lack of interleaving of β-strands in FAIM-CTD increases the mechanical demands on the interface between the two β-sheets, thereby applying strong selective pressure to maintain an optimized set of hydrophobic contacts throughout evolutionary history.
Conversely, one can predict that conserved, non-structural features likely reflect involvement in interactions with FAIM-NTD or other binding partners. These features may be seen in Figure 4. Notably, the hydrophilic character of the loop between Ser156 and Gly162 is maintained among all the known homologs of FAIM, although no single residue is conserved absolutely. Arg110 is strictly conserved but serves no structural role in FAIM-CTD. Phe133 also serves no structural purpose and is in an energetically unfavorable solvent-exposed position, yet it is also strictly conserved. While conservation of the hydrophilic loop could simply reflect the deleterious effect on protein stability of introducing hydrophobic residues into a solvent-exposed position, conservation of the other two residues is difficult to explain in the absence of functional significance. Phe133 is therefore likely to be involved in interactions with a binding partner of FAIM-CTD. Arg110 is strongly perturbed in the presence of FAIM-NTD and is surrounded by similarly perturbed residues, as discussed above, and is thus likely to play a vital role in interacting with FAIM-NTD (perhaps via Glu38). It remains possible, however, that the essential function of Arg110 could involve interaction with a binding partner of FAIM-CTD; its interaction with FAIM-NTD could then be incidental.
N-terminal sequencing of the fragments of triple mutant FAIM-S yielded by limited tryptic proteolysis (data not shown) shows that trypsin was able to cleave at residues deep within the first beta sheet (Arg103 and Arg110), while products of cleavage at the unstructured Lys95 were not observed. In contrast, one would expect that the resistance to proteolysis of the folded segments would be much greater than that of the unstructured region. It seems possible that Lys95 could be protected within full-length FAIM or that partial unfolding events are common within FAIM-CTD.
The presence of inward-oriented cysteine and histidine residues raises the question of whether FAIM-CTD might bind metal ions. The solution structure of FAIM-CTD reveals that at least one of the conserved histidine residues (His141) is completely solvent-exposed and is in a poor position to chelate a metal ion to stabilize the fold. His147 is not well conserved across all phyla, indicating that it likely does not play a role in chelation of a structurally important metal ion either. His165, in addition to the lack of strict conservation, is also poorly situated to cooperate with other functional groups near the hydrophobic core of the protein. Neither cysteine residue is conserved in the insect gene products. Another residue that could potentially assist in chelation of a metal ion is Thr140, which is buried within the protein (though not deeply). Conservation of this residue is high but not absolute, with a serine and an alanine residue being the only known substitutions. Although backbone carbonyl groups have been known to interact directly with cations, as seen, for example, in the EF hand motif of calmodulin [11] and in the selectivity filter of the KcsA potassium channel [12], the backbone of FAIM-CTD is primarily involved in a hydrogen bonding network and does not project these groups into the core of the protein with suitable geometry. Thus, the absence of well positioned, conserved residues able to act as electron donors implies that FAIM-CTD does not bind metal ions at a central location within the protein.
With the exception of the Trp121 indole sidechain proton restraints, no NOE-derived distance restraints were discarded during calculation of the structures presented here. The Trp121 restraints were treated as an exception to the rule only because it was felt that violation of these restraints could readily be rationalized on the grounds that the large sidechain may be mobile, possibly flipping, and thereby sampling different environments on the timescale of the NOESY mixing delay.
The lack of structural homology to other proteins means that the FAIM-CTD structure cannot yet play a direct role in efforts to determine the molecular mechanisms of FAIM activity. Interpretation of the structure from a functional point of view must await the identification of, for example, a protein binding partner or substrate. However, based on the chemical shift differences between FAIM-CTD and full-length FAIM-S, FAIM-CTD clearly interacts with FAIM-NTD. The extent of the structural perturbations induced by this interaction is unknown at this time, but it is now apparent that the inter-domain contact surface on FAIM-CTD is on the N-terminal sheet of the protein. This information may be important to properly orient the two domains when modeling the structure of full-length FAIM-S, interpreting functional data, and designing mutation studies. However, preliminary experiments (T.R., unpublished results) indicate that the activity of FAIM-CTD in B cells is as high as, if not higher than, the activity of FAIM-S. This suggests that interaction with FAIM-NTD is not required for activity in this system, and underscores the importance of FAIM-CTD in mediating FAIM activity. It is possible that inter-domain interaction serves a regulatory purpose or plays a role in activity of FAIM in another context; this is likely true in cultured neurons, for example, in which FAIM-S and FAIM-L have different functions but share a common FAIM-CTD.
Because all FAIM-NTD constructs known to date have far fewer peaks than residues in the 15N-HSQC spectrum, it is not clear whether it is feasible to solve the solution structure of FAIM-NTD. Limited proteolysis results (see Figure 1a) indicate that the trypsin resistance of FAIM-NTD is comparable to or greater than that of FAIM-CTD despite the presence of at least ten arginine and lysine residues, implying that there are few unstructured regions within FAIM-NTD. Taken together, these results suggest that there are structured regions of considerable size within FAIM-NTD that are inaccessible to study by NMR under any currently known conditions.
Distinct functions can often be attributed to individual domains of proteins. It is therefore interesting that a protein-protein (blastp) BLAST search (against the non-redundant database, September 2008) revealed no homology of either domain with any sequences other than the orthologous animal gene products, suggesting that FAIM-CTD only occurs in nature when covalently attached to FAIM-NTD and vice versa. Although the structural basis for FAIM function is not yet fully understood, one could speculate that the small size of the domain could limit the diversity of functions that could have evolved to make use of this particular fold, thereby preventing FAIM-CTD from acquiring functions that would be useful in other contexts. The same could be true of FAIM-NTD, if it too were found to lack structural homology to other known proteins. However, full-length FAIM-S has a molecular weight of more than 20 kDa and also has no known paralogs, so it would seem that a more parsimonious explanation for the absence of paralogs of either domain would involve some property of full-length FAIM-S that is lost if the protein is truncated, perhaps a property that has not yet been identified. It is also possible that structurally homologous molecules do exist but simply lack sufficient primary sequence homology to be detected by BLAST. Although pairwise identity is high among known orthologs, there are few regions of strict conservation and few structurally irreplaceable residues in FAIM-CTD, so the pairwise primary sequence identity of a paralog could be low.
Materials and Methods
Primers and PCR Protocols
Point mutations were introduced using QuikChange PCR (Stratagene). Typically 100 ng or more of template DNA were used. FAIM-NTD and similar constructs were made by introducing a stop codon in the murine FAIM-S sequence with QuikChange PCR. For the FAIM-CTD constructs, primers containing restriction sites and annealing to the desired start and end sites were designed. The gene fragment was amplified by PCR, digested with BamHI and EcoRI, then ligated into a pGEX-6P-3 vector. Before use, all constructs were sequenced by the Dana-Farber/Harvard Cancer Center DNA Resource Core.
Protein expression
Murine FAIM constructs were cloned into pGEX-6P-3 GST fusion expression vectors and transfected into BL21 cells (Novagen, Stratagene). These were grown in M9 medium to an optical density of 0.6 and induced with 0.1 mM isopropyl β-D-1-thiogalactopyranoside (IPTG) for four hours at 37 °C or overnight at 20 °C. Both temperature profiles yielded acceptable expression levels. For expression in LB, cultures were grown to a final OD of about 3.0. To confirm protein identity, a sample of [15N]-FAIM-CTD in 1010TBS was analyzed by MALDI-TOF mass spectrometry by the Biopolymers Laboratory at Harvard Medical School. As an exception, the 1.8 mM [13C]-FAIM-CTD sample used for the 13C-NOESY-HSQC spectrum was purified from 1 L of BL21 cell culture and 2 L of XL-1 Blue (Stratagene) cell culture. Protein yield per liter of culture from the latter was 71% of the former, and induction at an optical density of 0.7 to 0.8 with 0.1 M IPTG for 52 hours at 20 °C was required.
Deuterated protein expression
A representative protocol is as follows. For triple mutant [2Hα, 13C, 15N]-FAIM-S and [2Hα, 15N]-FAIM-S, aliquots of a starter culture grown in H2O LB were transferred to 90% D2O LB to adapt the cultures to growth in D2O. The culture was then added to 1 L D2O M9 with protonated glucose for 24 to 28 hours of growth at 37 °C; at this point the optical density was 0.3 to 0.4, and the culture was induced to a final optical density of 1.5 with 0.1 mM IPTG for 16 to 19 hours at 20 °C. The notation 2Hα indicates that sidechains beyond the Hα position were not fully deuterated because protonated glucose was used during expression.
Residue-specific labeling
For residue-specific labeling with 15N, 500 mL of a modified M9 medium was used for protein expression. The medium was supplemented with 150 mg/L of the 15N-labeled amino acid and 200 mg/L of all 19 other amino acids, as well as 500 mg/L adenine, 650 mg/L guanine, 200 mg/L thiamine, 500 mg/L uracil, and 200 mg/L cytosine. The medium was then filtered to remove undissolved solids. Induction was for 4 hours at 37 °C. Scrambling appeared to be minimal, since the 15N-HSQC spectra of labeled FAIM-S showed no more strong peaks than expected based on the primary sequence (12 of 17 valine and 11 of 11 isoleucine).
Reagents
Tris-buffered saline (TBS: 150 mM NaCl, 50 mM Tris HCl pH 7.5) was used for routine handling of FAIM constructs. An optimized low-salt buffer (1010TBS: 10 mM NaCl, 10 mM Tris HCl pH 7.5) was also used. 0.1% Tween-20 (Calbiochem) was added when handling FAIM-NTD and full-length FAIM-S constructs. 5 mM dithiothreitol (DTT) was also added to all TBS buffers except where noted. In preliminary experimental work, Tris HCl pH 7.0 was used instead of Tris HCl pH 7.5, and 0.1% 2-ME was substituted for DTT. Deuterated 1010TBS, using Tris-d11 (D, 98%; Cambridge Isotope Labs) and DTT-d10 (D, 98%; CIL), was prepared in small batches prior to use. pH was adjusted with DCl and NaOD; nominal pH values given for D2O buffers are pH* [13].
Protein purification
All steps were carried out on ice or at 4 °C unless otherwise noted. A representative protocol is as follows. Fresh or frozen cell pellets were resuspended in 30 to 100 mL ice-cold lysis buffer per liter of cell culture. Lysis was accomplished using either sonication on ice or disruption in an Avestin Emulsiflex C5 homogenizer in TBS with 0.1% Tween-20, 200 μM AEBSF, and trace amounts of DNase I; 1 mg/mL lysozyme was also added during early investigations. Lysate was centrifuged at 10,000 rpm for 25 minutes. The supernatant was filtered at 0.22 μm or 0.45 μm, then incubated with glutathione-conjugated agarose beads (Glutathione Sepharose 4 Fast Flow; Amersham; 6 mL bed volume per liter of culture). Beads were washed with 150 mL TBS or 1010TBS (without Tween-20, regardless of construct). GST-fused rhinoviral 3C protease was then added to the column to remove the GST fusion protein. 0.1% Tween-20 was also added before cleaving full-length or N-terminal domain FAIM-S constructs. Cleaved protein was eluted with 1010TBS or TBS. Eluted protein was typically greater than 90% pure by SDS-PAGE, with the major visible contaminants (if any) being uncleaved GST fusion protein and free GST. Eluted protein was purified by gel filtration in TBS or 1010TBS. Full-length FAIM-S constructs typically eluted at 66-73 mL, FAIM-CTD at 73-77 mL, and FAIM-NTD at 84-94 mL.
Limited proteolysis
For N-terminal sequencing, limited proteolysis was carried out at 0 °C using 350 μg/mL wild-type FAIM-S and 4 μg/mL trypsin (Sigma) in 110 mM ammonium bicarbonate in a volume of 80 μL. 40 μL aliquots were removed at 10 and 90 minutes. The reaction was stopped by addition of SDS loading buffer and heating to 70 °C for 5 minutes. SDS-PAGE and electroblotting were performed essentially according to Matsudaira et al. (1987)[14]. N-terminal sequencing using Edman chemistry was done by the Molecular Biology Core Facilities at the Dana Farber Cancer Institute.
NMR spectroscopy
The pH of the buffer used for NMR spectroscopy (1010TBS with 5 mM DTT) was 7.3 at room temperature. 0.1% Tween-20 was added for full-length FAIM-S and FAIM-NTD constructs. Because detergent was retained during concentration of the protein sample, the concentration of Tween-20 was approximately 3-5% for the most concentrated samples during NMR spectroscopy. For experiments in deuterated 1010TBS, however, the buffer was prepared in small batches, and the buffer pH was titrated to 7.0. In titration experiments (not shown) it was found that, in the 15N-HSQC spectrum of FAIM-CTD, only the peak corresponding to V144 had a highly pH-dependent chemical shift between pH 6.8 and 8.0. NMR samples were filtered and the sample tube was flushed with argon. All spectra were acquired at 25 °C unless stated otherwise. Linear prediction was used to double the number of points in one indirect dimension when processing most three-dimensional experiments. Spectra were processed with NMRPipe [15]. Assignment was done manually using Sparky version 3.110 [16].
Backbone assignment of triple mutant FAIM-S
Standard non-TROSY triple resonance experiments (HNCO, HN(CA)CO, HNCA, HN(CO)CA, HNCACB, and HN(CO)CACB) were acquired on a 250 μM [2Hα, 13C, 15N] sample, using a Varian Inova 500 spectrometer except where noted. A 15N-NOESY-HSQC spectrum with 120 ms mixing time, acquired at 600 MHz on a Bruker Avance 600, was also used to assist in backbone assignment. For this experiment, a 0.7 mM triple mutant [2H, 15N]-FAIM-S sample was used. Residue-specific labeling with isoleucine and valine was done to help confirm the assignment and match sequences of successive unidentified residues onto the known primary sequence. In this case, 15N-HSQC spectra were acquired on a Varian UnityPlus 400 MHz spectrometer.
Backbone assignment of FAIM-CTD
Standard non-TROSY triple resonance experiments excluding HN(CA)CO (15N-HSQC, HNCA, HNCO, HN(CO)CA, HNCACB, CBCA(CO)NH, 15N-NOESY-HSQC) were acquired on a Varian Inova 500 MHz spectrometer. Knowledge of the backbone assignment of the C-terminal domain of the triple mutant FAIM-S was incorporated into the assignment procedure. An HN(CA)CO experiment was then acquired after completion of the FAIM-CTD backbone assignment and used to confirm the assignment.
Sidechain assignment of FAIM-CTD
Most aliphatic sidechain protons were assigned based on 13C-HSQC/D2O, C(CO)NH, H(CCO)NH, HCCH-COSY/D2O, HCCH-TOCSY/D2O, and 2-dimensional NOESY/D2O spectra. A 13C-NOESY-HSQC/D2O spectrum acquired at 900 MHz spectrometer was also used. Stereospecific assignments were obtained for 10 of the 15 valine and leucine residues according to Neri et al. (1989)[17]. Aromatic protons were assigned using the 15N-NOESY-HSQC, 2-dimensional NOESY/D2O, aromatic 13C-HSQC/D2O, and CCH-COSY [18] spectra. Histidine imidazole sidechain protons were assigned using the 15N-NOESY-HSQC spectrum and a modified version of the procedure used by Pelton et al. (1993) employing an 15N-HSQC spectrum in lieu of an 15N-HMQC spectrum. The Hε protons of M127 were assigned, after calculation of preliminary structures, on the basis of numerous crosspeaks in the 13C-NOESY-HSQC/D2O spectrum.
NOE spectroscopy of FAIM-CTD
A 15N-NOESY-HSQC spectrum was acquired in H2O at 500 MHz with a 100 ms mixing time on a 0.9 mM [15N]-FAIM-CTD sample over a period of 3.5 days. A 13C-NOESY-HSQC/D2O spectrum was acquired at 900 MHz with a 120 ms mixing time on a 1.8 mM [13C]-FAIM-CTD sample in 1010TBS/D2O in a 5-day acquisition. To minimize the effects of spin diffusion, a 2-dimensional NOESY/D2O spectrum at 900 MHz on a 1.4 mM unlabeled FAIM-CTD sample in D2O 1010TBS pH 7.0 was also acquired with 70 ms mixing time; any distance restraint arising from a peak in the 13C-NOESY-HSQC/D2O spectrum for which a corresponding peak was not seen in the 2-dimensional NOESY/D2O spectrum was removed.
Other restraints
TALOS [19] was used to obtain backbone torsion angle constraints. These reduced the backbone RMSD by approximately 0.5 Å when added to the force field of the simulated annealing calculations. Proton chemical shifts were referenced to an external reference sample of DSS dissolved in 1010TBS; nitrogen and carbon chemical shifts were referenced using virtual referencing [20]. Sidechain rotamer constraints were added based on work presented in Matuso et al. (1997)[21] and on data compiled by Dunbrack and Karplus (1993)[22], but individual constraints were removed if they were consistently violated. Hydrogen bonding restraints were introduced between adjacent β-strands when there was clear spectroscopic evidence that this structure existed. The secondary structural elements formed in the absence of hydrogen bonding restraints; the restraints were used only to decrease the RMSD of the structure bundle.
Structure calculation
CYANA version 2.1 was used to calculate structures. For final structure bundles, 100 structures were calculated using 100,000 steps of simulated annealing from a random starting structure. The lowest 20% in energy were accepted. In early investigations it was found that residue Trp121 was involved in large numbers of constraint violations. The NOE assignments were double-checked and found to be correct. As Trp121 is solvent-exposed in the calculated structures (see Discussion) and appears to make a NOE crosspeak with water (not shown), its large sidechain could be highly mobile, resulting in inaccurate distance restraints. Therefore, all NOE restraints involving a sidechain proton of Trp121 were removed.
Validation with residual dipolar couplings
6 mg/mL of Pf1 Magnetic Resonance Co-solvent (ASLA Biotech, Latvia) was added to one 250 μg/mL sample of [15N, 13C]-FAIM-CTD in 1010TBS. Buffer was added to an identical sample to ensure that the buffer conditions were identical in the isotropic sample. The splitting of the HDO peak in the aligned sample was 7 Hz. The one bond scalar coupling constant 1JNH was measured by two redundant means. One method used the J-modulated HSQC pulse sequence described by Tjandra et al. (1996)[23]. The second method was based on the chemical shift difference between TROSY and non-TROSY HNCO spectra. Values from each method were compared to ensure consistency and estimate error. The alignment tensor, predicted RDCs, and Qfree factor [24] were calculated using PALES [25]. RDCs for residues not known to be involved in the formation of secondary structural elements were excluded; this modestly improved the overall correlation between observed and predicted RDCs.
Accession numbers
Atomic coordinates for FAIM-CTD have been deposited in the Protein Data Bank with accession number 2KD2. Chemical shift assignments have been deposited in the Biological Magnetic Resonance Data Bank with accession number 16103.
Supplementary Material
Acknowledgments
The authors would like to acknowledge Dmitri Ivanov and Alexey Lugovskoy for preliminary work related to the expression and purification of FAIM. This research was supported by a fellowship from The Whitaker Foundation and by NIH grants GM47467, EB002026, and GM066360.
Abbreviations
- FAIM
Fas Apoptosis Inhibitory Molecule
- FAIM-CTD
C-terminal domain of FAIM
- FAIM-NTD
N-terminal domain of FAIM
- TNFR
tumor necrosis factor receptor
- FADD
Fas-associated protein with death domain
- DED
death effector domain
- PARP
poly (ADP-ribose) polymerase, FABP, fatty acid binding protein
- HSQC
heteronuclear single quantum coherence
- NOESY
nuclear Overhauser effect spectroscopy
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Schneider TJ, Fischer GM, Donohoe TJ, Colarusso TP, Rothstein TL. A Novel Gene Coding for a Fas Apoptosis Inhibitory Molecule (FAIM) Isolated from Inducibly Fas-resistant B lymphocytes. J Exp Med. 1999;189(6):949–955. doi: 10.1084/jem.189.6.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhong X, Schneider TJ, Cabral DS, Donohoe TJ, Rothstein TL. An alternatively spliced long form of Fas apoptosis inhibitory molecule (FAIM) with tissue-specific expression in the brain. Mol Immunol. 2001;38:62–76. doi: 10.1016/s0161-5890(01)00035-9. [DOI] [PubMed] [Google Scholar]
- 3.Sole C, Dulcet X, Segura M, Gutierrez H, Diaz-Meco MT, Gozzelino R, Sanchis D, Bayascas JR, Gallego C, Moscat J, Davies AM, Comella JX. The death receptor agonist FAIM promotes neurite outgrowth by a mechanism that depends on ERK and NF-κB signaling. J Cell Biol. 2004;167(3):479–492. doi: 10.1083/jcb.200403093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Segura MF, Sole C, Pascual M, Mubarak RS, Perez-Garcia MJ, Gozzelino R, Iglesias V, Badiola N, Bayascas JR, Llecha N, Rodriguez-Alvarez J, Soriano E, Yuste VJ, Comella JX. The long form of Fas apoptosis inhibitory molecule is expressed specifically in neurons and protects them against death receptor-triggered apoptosis. J Neurosci. 2007;28(42):11228–11242. doi: 10.1523/JNEUROSCI.3462-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Wiens M, Perović-Ottstadt S, Müller IM, Müller WEG. Allograft rejection in the mixed cell reaction system of the desmosponge Suberites domuncula is controled by differential expression of apoptotic genes. Immunogenetics. 2004;56(8):597–610. doi: 10.1007/s00251-004-0718-6. [DOI] [PubMed] [Google Scholar]
- 6.Wiens M, Müller WEG. Cell death in Porifera: molecular players in the game of cell death in living fossils. Can J Zool. 2006;84:307–321. [Google Scholar]
- 7.Ito T, Wagner G. Using codon optimization, chaperone co-expression, and rational mutagenesis for production and NMR assignments of human eIF2α. J Biol NMR. 2004;28:357–367. doi: 10.1023/B:JNMR.0000015405.62261.cb. [DOI] [PubMed] [Google Scholar]
- 8.Holm L, Sanders C. Dali: a network tool for protein structure comparison. Trends Biochem Sci. 1995;20:478–480. doi: 10.1016/s0968-0004(00)89105-7. [DOI] [PubMed] [Google Scholar]
- 9.Armon A, Grauer D, Ben-Tal N. ConSurf: An Algorithmic Tool for the Identification of Functional Regions in Proteins by Surface Mapping of Phylogenetic Information. J Mol Biol. 2001;307:447–463. doi: 10.1006/jmbi.2000.4474. [DOI] [PubMed] [Google Scholar]
- 10.Jones TA, Bergfors T, Sedzik J, Unge T. The three-dimensional structure of P2 myelin protein. EMBO J. 1988;7(6):1597–1604. doi: 10.1002/j.1460-2075.1988.tb02985.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Babu YS, Bugg CE, Cook WJ. Structure of Calmodulin Refined at 2.2 Angstrom Resolution. J Mol Biol. 1988;204:191–204. doi: 10.1016/0022-2836(88)90608-0. [DOI] [PubMed] [Google Scholar]
- 12.Doyle DA, Cabral JM, Pfuetzner RA, Kuo A, Gulbis J, Cohen SL, Chait BT, MacKinnon R. The Structure of the Potassium Channel: Molecular Basis of K+ Conduction and Selectivity. Science. 1998;280(5360):69–77. doi: 10.1126/science.280.5360.69. [DOI] [PubMed] [Google Scholar]
- 13.Krężel A, Bal W. A formula for correlating pKa values determined in D2O and H2O. J Inorg Chem. 2003;98:161–166. doi: 10.1016/j.jinorgbio.2003.10.001. [DOI] [PubMed] [Google Scholar]
- 14.Matsudaira P. Sequence from Picomolar Quantities of Protein Electroblotted onto Polyvinylidene Fluoride Membrane. J Biol Chem. 1987;262(21):10035–10038. [PubMed] [Google Scholar]
- 15.Delaglio F, Grzesiek S, Vuister GW, Zhu G, Pfeifer J, Bax A. NMRPipe: a multidimensional spectral processing system based on UNIX pipes. J Biomol NMR. 1995;6:277–293. doi: 10.1007/BF00197809. [DOI] [PubMed] [Google Scholar]
- 16.Goddard TG, Kneller DG. SPARKY 3. University of California; San Francisco: undated. [Google Scholar]
- 17.Neri D, Szyperski T, Otting G, Senna H, Wuthrich K. Stereospecific Nuclear Magnetic Resonance Assignments of the Methyl Groups of Valine and Leucine in the DNA-Binding Domain of the 434 Repressor by Biosynthetically Directed Fractional 13C Labeling. Biochemistry. 1989;28:7510–7516. doi: 10.1021/bi00445a003. [DOI] [PubMed] [Google Scholar]
- 18.Pelton JG, Torchia DA, Meadow ND, Roseman S. Tautomeric states of the active-site histidines of phosphorylated and unphosphorylated IIIGlc, a signal-transducing protein from Escherichia coli, using two-dimensional heteronuclear NMR techniques. Prot Sci. 1993;2:543–558. doi: 10.1002/pro.5560020406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cornilescu G, Delaglio F, Bax A. Protein backbone angle restraints from searching a database for chemical shift and sequence homology. J Biomol NMR. 1999;13:289–302. doi: 10.1023/a:1008392405740. [DOI] [PubMed] [Google Scholar]
- 20.Wishart DS, Bigam CG, Yao J, Abildgaard F, Dyson HJ, Oldfield E, Markley JL, Sykes Brian D. 1H, 13C, and 15N chemical shift referencing in biomolecular NMR. J Biomol NMR. 1995;6:135–140. doi: 10.1007/BF00211777. [DOI] [PubMed] [Google Scholar]
- 21.Matsuo H, Li H, McGuire A, Fletcher CM, Gingras A-C, Sonenberg N, Wagner G. Structure of translation factor eIF4E bound to m7GDP and interaction with 4E-binding protein. Nat Struct Biol. 1997;4(9):717–724. doi: 10.1038/nsb0997-717. [DOI] [PubMed] [Google Scholar]
- 22.Dunbrack RL, Karplus M. Backbone-dependent Rotamer Library for Proteins: Application to Side-Chain Prediction. J Mol Biol. 1993;230:543–574. doi: 10.1006/jmbi.1993.1170. [DOI] [PubMed] [Google Scholar]
- 23.Tjandra N, Grzesiek S, Bax A. Magnetic Field Dependence of Nitrogen-Proton J Splittings in 15N-Enriched Human Ubiquitin Resulting from Relaxation Interference and Residual Dipolar Coupling. J Am Chem Soc. 1996;118:6264–6272. [Google Scholar]
- 24.Cornilescu G, Marquadt JL, Ottinger M, Bax A. Validation of Protein Structure from Anisotropic Carbonyl Chemical Shifts in a Dilute Liquid Crystalline Phase. J Am Chem Soc. 1998;120:6836–6837. [Google Scholar]
- 25.Zwecksetter M, Bax A. Prediction of Sterically Induced Alignment in a Dilute Liquid Crystalline Phase: Aid to Protein Structure Determination by NMR. J Am Chem Soc. 2000;122:3791–3792. [Google Scholar]
- 26.Laskowski RA, Rullmann JAC, MacArthur MW, Kaptein R, Thornton JM. AQUA and PROCHECK-NMR: Programs for checking the quality of protein structures solved by NMR. J Biol NMR. 1996;8:477–486. doi: 10.1007/BF00228148. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.