

Abstract
Huntington's disease (HD) is caused by a polyglutamine repeat in the protein huntingtin (Htt) with mutant Htt (mHtt) expressed throughout the body and similarly in all brain regions. Yet, HD neuropathology is largely restricted to the corpus striatum. We report that the small guanine nucleotide–binding protein Rhes, which is localized very selectively to the striatum, binds physiologically to mHtt. Using cultured cells, we found Rhes induces sumoylation of mHtt, which leads to cytotoxicity. Thus, Rhes-mHtt interactions can account for the localized neuropathology of HD.
Huntington's disease (HD), a genetically dominant neurodegenerative disorder, reflects expansion of a polyglutamine repeat in the protein huntingtin (Htt) (1). Mutant Htt (mHtt) occurs uniformly throughout the brain and peripheral tissues. Yet, HD is brain-specific with profound abnormal movements related to selective, gross degeneration of the corpus striatum and lesser damage to the cerebral cortex eliciting dementia (2, 3). Molecular mechanisms causing mHtt cytotoxicity are unclear. mHtt forms protein aggregates, which may be neuroprotective with soluble mHtt linked to cytotoxicity (4–7). mHtt is sumoylated, which increases the soluble form of mHtt and elicits cytotoxicity and neurotoxicity in a Drosophila model of HD (8).
Rhes (Ras homolog enriched in striatum) is a small guanine nucleotide–binding protein (G protein) very selectively localized to the striatum (9). To determine whether Rhes binds to Htt, we overexpressed Rhes in HEK293 cells where it bound to both wild-type (wt) Htt and mHtt (Fig. 1A) (10). In conditionally immortalized Htt knock-in striatal neuronal cells (11), which lack endogenous Rhes (fig. S1C), overexpressed Rhes bound robustly to endogenous mHtt (Fig. 1B). In HD transgenic mice (12), endogenous striatal mHtt coprecipitated with Rhes (Fig. 1C). In the presence of purified Rhes and Htt, Rhes bound much more to mHtt than wtHtt protein (fig. S1A). Rhes did not bind to ataxin (fig. S1B), a polyglutamine-repeat protein involved in another neurodegenerative disorder, spinocerebellar ataxia.
Fig. 1.
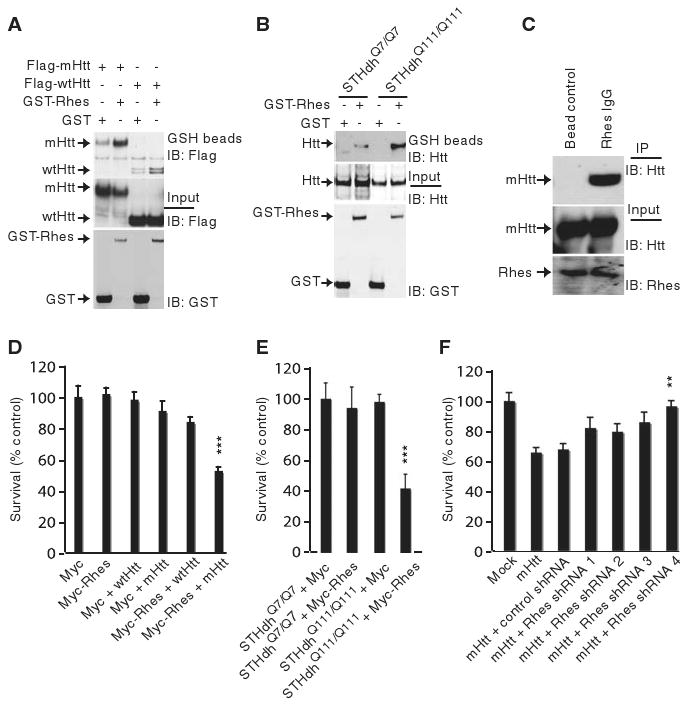
Rhes binds Htt and affects cell survival. (A) Rhes interacts with N-terminal Htt. HEK293 cells were transfected with glutathione S-transferase (GST) or GST-Rhes together with Flag-tagged Htt or the N-terminal fragment containing 171 amino acids and 18 glutamines (wtHtt) or 82 glutamines (mHtt). After 48 hours, cell lysates were glutathione (GSH) precipitated and immunoblotted (IB) for Flag. (B) Rhes interacts with full-length Htt. Striatal cells expressing wild-type Htt (STHdhQ7/Q7) or mutant Htt (STHdhQ111/Q111) were transfected with GST or GST-Rhes. After 48 hours, cell lysates were GSH-precipitated and immunoblotted for Htt. Htt and GST inputs are shown. (C) Rhes interacts with mHtt in striatum. Striatum of transgenic mice expressing mHtt was lysed and immunoprecipitated with Rhes antibody or immunoglobulin IgG alone (bead control). Immunoprecipitates were probed with an N-terminal–specific Htt antibody (N-Htt). (D) Rhes reduces cell survival. HEK293 cells were transfected with Myc/Myc-Rhes and wtHtt–mHtt constructs. ***P < 0.005 versus mHtt alone. (E) Wild-type (STHdhQ7/Q7) or mutant (STHdhQ111/Q111) striatal cells were transfected with Myc/Myc-Rhes. ***P < 0.005 versus Myc. (F) Depletion of Rhes prevents PC12 cell death. Control short hairpin–mediated (shRNA) or Rhes shRNA 1 to 4 were cotransfected with mHtt. Only shRNA4 was significantly cytoprotective (**P < 0.01 versus control shRNA). After 48 hours, cell survival was measured by MTT.
To ascertain whether Rhes influences mHtt cytotoxicity, we used several cell lines. In HEK293 cells, overexpression of mHtt or Rhes alone did not decrease cell survival. However, overexpression of Rhes together with mHtt reduced cell survival by 50%, whereas survival was normal in cells containing wtHtt and Rhes (Fig. 1D). We confirmed that survival of a striatal cell line with mHtt is the same as that in cells with wtHtt (13) (Fig. 1E). Overexpression of Rhes in mHtt knock-in striatal cells (STHdhQ111/Q111) (14) reduced cell survival by 60%, whereas over-expression of Rhes in wtHtt knock-in striatal cells (STHdhQ7/Q7) had no effect (Fig. 1E). Rhes's influences on striatal cell survival were concentration-dependent (fig. S2A). Cleaved caspase-3, an index of apoptosis, was selectively augmented in STHdhQ111/Q111 cells overexpressing Rhes (fig. S2B). We examined the role of endogenous Rhes in cytotoxicity in PC12 cells, which contain endogenous Rhes (fig. S1C). The reduction in cell survival associated with overexpression of mHtt was reversed by depleting Rhes with RNA interference (fig. S1D and Fig. 1F).
How might Rhes facilitate mHtt neurotoxicity? When expressed in cells, mHtt, but not wtHtt, formed robust aggregates (fig. S1E). mHtt is sumoylated, that is, the small ubiquitin-like modifier (SUMO) is covalently attached to the protein, which decreases mHtt aggregation and elicits neurotoxicity (8). We examined the influence of Rhes on mHtt aggregation. Rhes overexpression markedly reduced aggregation and increased levels of soluble mHtt (Fig. 2, A and B). We confirmed the sumoylation of mHtt, which was markedly augmented in cells overexpressing Rhes (Fig. 2C). By contrast, Rhes failed to increase wtHtt sumoylation (fig. S3A). Because mHtt is both sumoylated and ubiquitinated at the same lysine (8), we examined the effect of Rhes on mHtt ubiquitination. Rhes elicited a pronounced decrease in mHtt ubiquitination (fig. S3B). To ascertain whether sumoylation at specific lysines of mHtt determines disaggregation of the protein, we evaluated mHtt with lysine-to-arginine mutations at positions 6, 9, 15, and 91 (Fig. 2D). The combined mutations abolished mHtt sumoylation, as well as disaggregation, and reversed the cytotoxicity elicited by Rhes overexpression (Fig. 2D). Sumoylation of mHtt in cells involved multiple lysines, specifically K9, K15, and K91 (fig. S4A). When Arg re-placed Lys at residues 15 and 91 (K15R and K91R), these mutations of mHtt markedly diminished Rhes-elicited disaggregation of mHtt without influencing Rhes-mHtt binding (fig. S4, A and B).
Fig. 2.
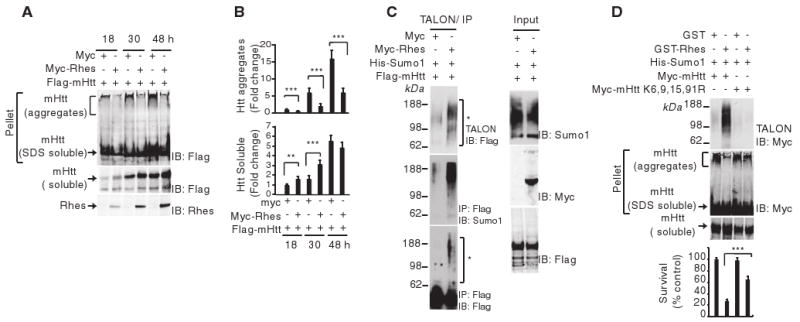
Rhes inhibits mHtt aggregate formation. (A) HEK293 cells were transfected with Myc or Myc-Rhes and Flag-mHtt. At the indicated time points, cells were lysed, the pellet fraction (containing mHtt aggregates plus SDS-soluble mHtt), and the soluble fractions (containing only soluble Htt) were immunoblotted (IB) for Flag or Rhes. (B) Quantification of aggregated and soluble Htt. Fold change compared with results at 18 hours. (C) Rhes increases mHtt sumoylation in cells. HEK293 cells were transfected with Myc or Myc-Rhes, His-SUMO1 and Flag-mHtt. After 36 hours, cell lysates were immunoprecipitated (IP) with antibody against Flag-IgG beads and immunoblotted for Flag. SUMO-tagged proteins were enriched with TALON metal-affinity resin and immunoblotted for Flag or SUMO1. Input lysates were immunoblotted for SUMO1, Myc, or Flag. (D) Effect of mHtt-K6,9,15,91R mutation on sumoylation and cell survival. HEK293 cells were transfected with GST or GST-Rhes, His-SUMO1, and Myc-tagged mHtt or Myc–mHtt-K6,9,15,91R. After 48 hours, the SUMO-tagged protein was enriched with TALON metal-affinity resin and immunoblotted for Myc. The pellet fraction of the cell lysate was subjected to aggregate detection assay. Cell survival was measured by MTT.
We next examined the effect of SUMO1 depletion in HEK293 cells. Depletion by RNA interference of SUMO1 (fig. S5A) increased aggregation of mHtt and abolished the cytotoxicity associated with overexpression of Rhes and mHtt (fig. S5B). Conversely, overexpression of SUMO1 caused disaggregation of mHtt and increased cell death (fig. S5C). We wondered whether SUMO2/3 might sumoylate mHtt in cells lacking SUMO1 (fig. S6A). We failed to detect any sumoylation of mHtt by SUMO2 in the SUMO1-deleted cells, consistent with the observation that SUMO2/3 is exclusively nuclear, whereas mHtt exhibited a granular distribution in the cytoplasm (fig. S6B).
We explored whether Rhes directly regulates sumoylation of mHtt. Rhes bound Ubc9, the cellular E2 ligase (Fig. 3A), and enhanced sumoylation of recombinant mHtt in a time- and concentration-dependent manner (Fig. 3B). Rhes did not influence wtHtt sumoylation (fig. S7A). In these experiments in vitro, sumoylated mHtt was a single discrete band. In contrast, with intact cells, sumoylated mHtt appeared as a smear, probably because of sumoylation at multiple lysine residues and/or formation of polySUMO chains. Sumoylation was abolished in mHtt-K6,9,15,91R mutants (Fig. 3B). We explored the substrate specificity of Rhes (Fig. 3, C and D). Rhes augmented sumoylation of Ran guanosine triphos-phatase (GTPase)–activating protein (RanGAP1) and SP100, but failed to elicit sumoylation of E2-25K, ubiquitin-conjugating E2 ligase (Fig. 3C). Rhes-dependent sumoylation of RanGAP1 was evident as early as 1 min (fig. S7B). Higher concentrations of Rhes were required to sumoylate inhibitor of nuclear factor kB (IkB) (Fig. 3D). Both recombinant Rhes and overexpressed Rhes in cultured cells were sumoylated (fig. S8, A and B), similar to E3 ligases (15, 16).
Fig. 3.

Rhes enhances sumoylation. (A) Rhes interacts with Ubc9. HEK293 cells were transfected with GST or GST-Rhes and Myc-Ubc9. After 48 hours, cells were lysed, precipitated with GSH beads and probed for Myc. Purified GST or GST-Rhes was incubated with purified Ubc9 and precipitated with GSH beads. The precipitates and inputs were immunoblotted for Ubc9. (B) Rhes sumoylates mHtt. Four μl of in vitro translated mHtt (control) and its K6,9,15,91R mutant were subjected to sumoylation [1× RB buffer, 250 ng E1, 125 ng E2, 1.5 μg SUMO1, 5 mM adenosine triphosphate (ATP), and 2 mM dithiothreitol (DTT)] in the presence of 500 ng Rhes (+) or bovine serum albumin (BSA) (−). Htt was detected by N-Htt antibody. Rhes sumoylates Htt in a time- (Rhes, 500 ng) and concentration-dependent manner. (C and D) Sumoylation of multiple substrates. Indicated substrates (500 ng) were subjected to sumoylation assay as in (B). Rhes sumoylates substrates in a (C) time- (Rhes, 200 ng) and (D) concentration-dependent manner.
We conducted a series of experiments to examine mechanisms whereby Rhes alters mHtt aggregation and cell death. Biological activity of small G proteins requires attachment to the cell membrane via fatty acid addition to conserved cysteines on CXXX domains (17). Rhes is farnesylated at cysteine 263 (18). Farnesylation mediates the localization of proteins to the plasma membrane, as well as to intracellular membranes (19). Mutation of Rhes-C263 abolished its sumoylation of mHtt, the disaggregation of mHtt, and cytotoxicity (Fig. 4A). Thus, these actions of Rhes require cysteine 263. The GTPase function of Rhes is mediated by serine 33. Rhes-S33N retained its ability to sumoylate and disaggregate mHtt and to elicit cytotoxicity and thus dissociated GTPase activity from sumoylation and/or cytotoxicity (Fig. 4, A and B). We explored the localization of the farnesylation-deficient mutant Rhes-C263S (Fig. 4C). Whereas wild-type Rhes, Ubc9, and mHtt occurred in granular structures restricted to the cytosol, Rhes-C263S was translocated to the nucleus with negligible staining in the cytoplasm. By contrast, GTPase mutant Rhes-S33N, which retains sumoylation activity, displayed the same intracellular localization as wild-type Rhes. Biochemical studies revealed a substantial reduction of Rhes-C263S binding to Ubc9 and mHtt (Fig. 4D). Finally, Rhes-induced mHtt sumoylation occurred both in the soluble and membrane fractions (fig. S9).
Fig. 4.
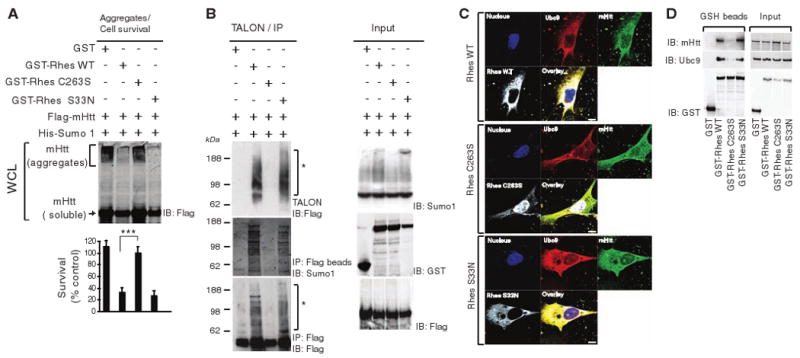
Rhes function requires cysteine 263. (A) Disaggregation and cell death. GST or GST-tagged Rhes (WT, C263S, or S33N) were transfected along with Flag-mHtt; Htt aggregation and cell survival were assessed at 48 hours. ***P < 0.001 versus WT. (B) Sumoylation. HEK293 cells were transfected with GST, GST-tagged Rhes (WT, C263S, or S33N), His-SUMO1, and mHtt. After 36 hours, cell lysates were either immunoprecipitated (IP) with Flag-IgG beads or enriched with TALON metal-affinity resin. The precipitate and input were immunoblotted (IB) for Flag, SUMO1, or GST. (C) Rhes/Ubc9/mHtt colocalization. STHdhQ111/Q111 cells were transfected with GST-Rhes WT, C263S, or S33N mutants. After 48 hours, cells were processed for nuclear staining and immunostaining with antibodies against Rhes, Ubc9, and Htt. (D) Rhes/Ubc9/mHtt interaction. STHdhQ111/Q111 cells were transfected with GST or GST-Rhes with WT, S33N, or C263S. After 48 hours, cells were lysed and precipitated with GSH beads. The precipitates and inputs were probed for mHtt, Ubc9, or GST.
In summary, Rhes binds to mHtt and elicits its sumoylation, which is associated with mHtt disaggregation and cell death. In some animal models overexpression of full-length mHtt augments aggregates in the striatum (20, 21), although, in other models, the overexpression leads to fewer or no aggregates (22, 23). In human HD patients and several animal models, aggregates are not correlated with cell death (5, 20, 24–26). Sumoylated mHtt represses nuclear transcription (8). We observed caspase-3 activation in Rhes-mHtt cells, and mHtt is known to induce cytochrome c release (27).
Rhes elicits sumoylation of mHtt via a mechanism independent of its GTPase activity but which does require cysteine at CXXX domains presumably for farnesylation and membrane attachment. Sumoylation of mHtt, RanGAP1, and SP100 occurs in the absence of Rhes but is markedly augmented by Rhes. The three well-studied SUMO E3 ligases, the PIASy family, Pc2, and RanBP2, do not share obvious sequence homology with each other (28) or with Rhes, the only G protein with demonstrated E3 ligase activity.
Dexras1, a close homolog of Rhes, displays the highest levels in the brain, but with no marked regional differences (29). Dexras1 mediates linking of nitric oxide (NO) signaling by CAPON, a scaffolding protein, which links Dexras1 to neuronal NO synthase (29). NO serves as a guanine nucleotide–exchange factor to activate Dexras1. Dexras1 also mediates neurotoxic iron influx following glutamate–N-methyl-D-aspartate neurotransmission (30).
Our discovery that the striatal-selective protein Rhes partners with mHtt to elicit cytotoxicity can account for the striatal pathophysiology of HD. Although Rhes is uniquely enriched in the striatum, it displays detectable cerebral cortical levels with negligible values in the cerebellum (9, 31). Cortical damage presumably elicits dementia; however, the cerebellum is relatively impervious to neurotoxic damage. Because HD can be diagnosed many years before the onset of symptoms, prophylactic therapy could, in principle, prevent or delay the onset of symptoms. Drugs that block the binding of Rhes and mHtt may thus have therapeutic potential.
Supplementary Material
Acknowledgments
We thank I. Rao, A. Hayashi, K. Ishizuka and M. Chakraborty for technical support. We thank N. Shahani and R. Mealer for critically reading the manuscript. We thank E. Fossale and M. Macdonald for generously providing striatal cell lines. We thank M. Matunis for providing SUMO-related constructs, H. Zoghbi for ataxin constructs, S. Li and X. J.Li for purified Htt, J. Nathans for use of his cell culture facility and Alessandro Usiello for providing antibody to Rhes. Supported by USPHS grant MH18501 and Research Scientist Award DA00074 (SHS).
References and Notes
- 1.Gusella JF, Macdonald ME. Trends Biochem Sci. 2006;31:533. doi: 10.1016/j.tibs.2006.06.009. [DOI] [PubMed] [Google Scholar]
- 2.Cowan CM, Raymond LA. Curr Top Dev Biol. 2006;75:25. doi: 10.1016/S0070-2153(06)75002-5. [DOI] [PubMed] [Google Scholar]
- 3.Sieradzan KA, Mann DM. Neuropathol Appl Neurobiol. 2001;27:1. doi: 10.1046/j.0305-1846.2001.00299.x. [DOI] [PubMed] [Google Scholar]
- 4.Saudou F, Finkbeiner S, Devys D, Greenberg ME. Cell. 1998;95:55. doi: 10.1016/s0092-8674(00)81782-1. [DOI] [PubMed] [Google Scholar]
- 5.Kuemmerle S, et al. Ann Neurol. 1999;46:842. [PubMed] [Google Scholar]
- 6.Arrasate M, Mitra S, Schweitzer ES, Segal MR, Finkbeiner S. Nature. 2004;431:805. doi: 10.1038/nature02998. [DOI] [PubMed] [Google Scholar]
- 7.Gong B, Lim MC, Wanderer J, Wyttenbach A, Morton AJ. Brain Res Bull. 2008;75:146. doi: 10.1016/j.brainresbull.2007.08.005. [DOI] [PubMed] [Google Scholar]
- 8.Steffan JS, et al. Science. 2004;304:100. doi: 10.1126/science.1092194. [DOI] [PubMed] [Google Scholar]
- 9.Falk JD, et al. J Neurosci Res. 1999;57:782. [PubMed] [Google Scholar]
- 10.Materials and methods are available as supporting material on Science Online.
- 11.Trettel F, et al. Hum Mol Genet. 2000;9:2799. doi: 10.1093/hmg/9.19.2799. [DOI] [PubMed] [Google Scholar]
- 12.Schilling G, et al. Hum Mol Genet. 1999;8:397. doi: 10.1093/hmg/8.3.397. [DOI] [PubMed] [Google Scholar]
- 13.Ruan Q, Lesort M, MacDonald ME, Johnson GV. Hum Mol Genet. 2004;13:669. doi: 10.1093/hmg/ddh082. [DOI] [PubMed] [Google Scholar]
- 14.Single-letter abbreviations for the amino acid residues are as follows: A, Ala; C, Cys; D, Asp; E, Glu; F, Phe; G, Gly; H, His; I, Ile; K, Lys; L, Leu; M, Met; N, Asn; P, Pro; Q, Gln; R, Arg; S, Ser; T, Thr; V, Val; W, Trp; Y, Tyr; and X, any amino acid.
- 15.Pichler A, Gast A, Seeler JS, Dejean A, Melchior F. Cell. 2002;108:109. doi: 10.1016/s0092-8674(01)00633-x. [DOI] [PubMed] [Google Scholar]
- 16.Kagey MH, Melhuish TA, Wotton D. Cell. 2003;113:127. doi: 10.1016/s0092-8674(03)00159-4. [DOI] [PubMed] [Google Scholar]
- 17.Magee T, Seabra MC. Curr Opin Cell Biol. 2005;17:190. doi: 10.1016/j.ceb.2005.02.003. [DOI] [PubMed] [Google Scholar]
- 18.Vargiu P, et al. Oncogene. 2004;23:559. doi: 10.1038/sj.onc.1207161. [DOI] [PubMed] [Google Scholar]
- 19.Wright LP, Philips MR. J Lipid Res. 2006;47:883. doi: 10.1194/jlr.R600004-JLR200. [DOI] [PubMed] [Google Scholar]
- 20.Wheeler VC, et al. Hum Mol Genet. 2000;9:503. doi: 10.1093/hmg/9.4.503. [DOI] [PubMed] [Google Scholar]
- 21.Lin CH, et al. Hum Mol Genet. 2001;10:137. doi: 10.1093/hmg/10.2.137. [DOI] [PubMed] [Google Scholar]
- 22.Reddy PH, et al. Nat Genet. 1998;20:198. doi: 10.1038/2510. [DOI] [PubMed] [Google Scholar]
- 23.Levine MS, et al. J Neurosci Res. 1999;58:515. [PubMed] [Google Scholar]
- 24.Hodgson JG, et al. Neuron. 1999;23:181. doi: 10.1016/s0896-6273(00)80764-3. [DOI] [PubMed] [Google Scholar]
- 25.Shelbourne PF. Hum Mol Genet. 1999;8:763. doi: 10.1093/hmg/8.5.763. [DOI] [PubMed] [Google Scholar]
- 26.Gutekunst CA. J Neurosci. 1999;19:2522. doi: 10.1523/JNEUROSCI.19-07-02522.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Choo YS, Johnson GV, MacDonald M, Detloff PJ, Lesort M. Hum Mol Genet. 2004;13:1407. doi: 10.1093/hmg/ddh162. [DOI] [PubMed] [Google Scholar]
- 28.Geiss-Friedlander R, Mechior F. Nat Rev Mol Cell Biol. 2007;8:947. doi: 10.1038/nrm2293. [DOI] [PubMed] [Google Scholar]
- 29.Fang M, et al. Neuron. 2000;28:183. doi: 10.1016/s0896-6273(00)00095-7. [DOI] [PubMed] [Google Scholar]
- 30.Cheah JH, et al. Neuron. 2006;51:431. doi: 10.1016/j.neuron.2006.07.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Usui H, et al. J Neurosci. 1994;14:4915. doi: 10.1523/JNEUROSCI.14-08-04915.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.