

Abstract
Triplex-forming oligonucleotides (TFOs) are gene targeting tools that can bind in the major groove of duplex DNA in a sequence-specific manner. When bound to DNA, TFOs can inhibit gene expression, can position DNA-reactive agents to specific locations in the genome, or can induce targeted mutagenesis and recombination. There is evidence that third strand binding, alone or with an associated cross-link, is recognized and metabolized by DNA repair factors, particularly the nucleotide excision repair pathway. This review examines the evidence for DNA repair of triplex-associated lesions.
Keywords: DNA repair, gene targeting, gene modification, interstrand cross-link repair
Introduction
Triple helical structures, or triplexes, were first reported in 1957 by Felsenfeld and Rich, who inferred from RNA diffraction studies that poly(U) (polyuridylate) and poly(A) (polyadenylate) strands can associate in a stable complex in a 2:1 ratio [1]. The discovery thirty years later that a synthetic 15 base pair oligonucleotide can form a stable triple helix in a sequence-specific manner, launched the triplex field as tools for directed gene modification [2-7]. The inherent sequence specificity of TFO binding underscores the potential of these synthetic oligonucleotides to direct gene manipulation to desired locations in the genome. In the past 20 years, TFOs have been successfully used to inhibit transcription initiation and elongation, induce site-specific damage and cleavage, and target disease-related genes for site-directed mutagenesis or correction (Figure 1).
Figure 1.
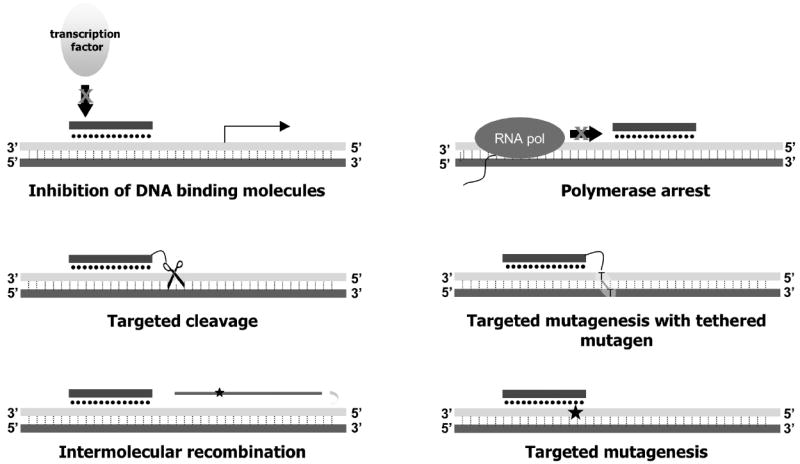
Triplex-forming oligonucleotides can mediate various biological activities upon binding to duplex DNA. Reprinted with permission from [58].
TFO sequence specificity comes from base-specific interactions between the bases of the third strand, and the purines of the target duplex DNA. TFOs bind to polypurine stretches of duplex DNA, because purines have an available hydrogen bond necessary for Hoogsteen bonding to the third strand. These Hoogsteen bonds are generally weaker than corresponding Watson-Crick base pairing but can be stabilized in the presence of divalent cations. TFOs of the pyrimidine motif, that is, consisting primarily of thymines or protonated cytosines, form the canonical base triads T:AT and C+:GC with duplex DNA (Figure 2). Pyrimidine-rich TFOs tend to bind duplex DNA in a parallel orientation with respect to the polypurine strand. In contrast, purine-rich TFOs bind to DNA via reverse Hoogsteen bonds in an anti-parallel orientation, to form the base triads G:GC, A:AT, and T:AT.
Figure 2.
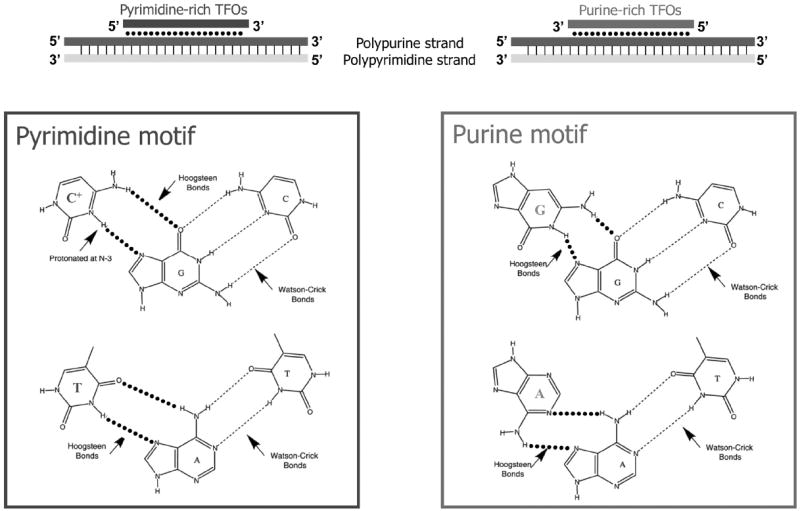
TFOs bind to polypurine sequences in DNA via Hoogsteen base-pairing. Pyrimidine and purine TFOs differ in third strand base composition and third strand orientation with respect to the polypurine strand of DNA. Reprinted with permission from [58].
TFOs can be composed of DNA, RNA, or synthetic base analogs. For example, various base analogs have been used to address the requirement for cytosine protonation on pyrimidine TFOs, which does not occur under physiological conditions. Base analogs such as 5’methylcytosine or pseudoisocytidine have higher pK values than does cytosine, and therefore have a hydrogen at physiological pH for bonding with the N7 of the target guanine (Figure 2). Recently, TFOs with locked nucleic acids (LNAs), RNA-like analogs in which the 2’O and 4’C are linked via a methylene bridge, have demonstrated impressive affinity to complementary DNA and RNA and increased triplex stability at physiological pH. When used in cells, LNA-TFOs have biological activity in the nanomolar range at chromosomal targets [8,9]. Similarly, TFOs with 2’ aminoethylribose modifications appear to have improved thermal stability and association kinetics, thought to be due to their RNA-like sugar conformation and reduced charge repulsion between the third strand and the target duplex [10]. Base analogs have also been used for guanines in purine-rich TFOs, particularly to reduce the formation of G-tetrads that are favored under physiological K+ conditions, and therefore may reduce effective TFO bioavailability for targeting [11].
The TFO backbone may also be modified to improve intracellular resistance to nucleases and improve binding to duplex DNA. Modifications include phosphoramidate, N,N-diethylethylenediamine (DEED), phosphorothioate, or a peptide-like backbone to form so-called peptide-nucleic acids (PNAs). PNAs, in particular, may be useful gene targeting agents, because they are nuclease and protease resistant, and form very stable complexes with DNA [12]. We and others have shown that two PNAs linked together to form a bis-PNA can efficiently form a stable PNA/DNA/PNA clamp at chromosomal targets, leading to site-specific gene modification [13].
TFOs bind in the major groove of DNA, and there is evidence that duplex DNA undergoes helical distortions upon TFO binding to accommodate the added electrostatic repulsions of the third strand [14]. It is thought that these distortions may trigger DNA repair mechanisms in the cell, and the subsequent metabolism of the triplex structure may lead to the observed mutagenic and recombinatory activity at the TFO binding site [15]. As well, there is some evidence to suggest the involvement of transcription in TFO-associated processing. There is some disagreement in the literature, however, about the extent of repair of TFO-directed lesions, with some groups suggesting efficient repair of complex TFO lesions [16] and others finding evidence for minimal repair [17]. This review will focus primarily on the evidence for the repair of TFO-directed lesions, and the effects of repair on TFO-mediated mutagenesis and recombination.
Targeted mutagenesis
A TFO, either alone or with a tethered mutagen, can induce mutagenesis on duplex DNA, primarily at the binding site itself but also in the surrounding sequence. Psoralen is one widely used mutagen that can intercalate into DNA to create interstrand cross-links (ICLs) when activated with UVA irradiation. When conjugated to a TFO, the psoralen-induced cross-link is thus “delivered” by the TFO to a particular 5’-TpA-3’ site in the genome, resulting in increased mutagenesis of the region surrounding the cross-link, both in vitro and in mammalian cells. TFO binding is necessary for mutagenesis, and in fact, TFO-induced frequencies of mutagenesis are directly correlated with the binding affinities of purine TFOs targeting the supF reporter gene. In one study, TFOs with KD’s of 10-9M were able to induce mutagenesis at frequencies 35-fold above TFOs with KD’s of 10-7M targeting the same region in plasmid DNA [18]. Sequence analysis of the mutants revealed that 71% were T-A to A-T transversions at the predicted psoralen-intercalation site; the rest of the mutations were point mutations at adjacent base pairs, or deletions spanning the TFO binding region. TFO binding affinity to its cognate site is not the only determinant of TFO biological activity, however, particularly for TFOs of the pyrimidine motif. As will be discussed below, base substitutions of pyrimidine TFOs such as 2’aminoethyl or the use of LNA moieties may increase binding affinity, but these TFOs also tend to adopt an RNA-like conformation that minimally distorts the helix. In these cases it appears that TFO binding affinity may be less important for TFO-induced mutagenesis than the ability of the bound TFO to provoke an altered helical structure [19].
Psoralen-conjugated TFOs can also be used to mutate chromosomal target sites in mammalian cells in culture. A psoralen-conjugated pyrimidine-rich TFO targeting a 17-mer or 23-mer sequence in the CHO hypoxanthine phosphoribosyl transferase (hprt) gene induced mutagenesis frequencies several hundredfold above spontaneous mutagenesis [20]. The majority of induced mutations (85%) were in the triplex target region, but the character of these chromosomal mutations differed from the plasmid mutagenesis observed by Wang et al. [18] described above: 77% of the hprt mutations had deletions of 4-50 bases and 4% had base substitution mutations at or adjacent to the site of the cross-link. The authors hypothesized from the mutation pattern that the major repair intermediate following the placement of a TFO-directed ICL was a double-strand break (DSB), leading to simple end joining at microhomologies.
It is important to note, however, that neither UVA irradiation nor psoralen conjugation are absolute requirements for TFO-induced mutagenesis; that is, TFOs by themselves can provoke mutagenesis in cells and in transgenic mice. This was demonstrated in plasmids transfected into mammalian cells; a 30-mer purine TFO was able to induce mutagenesis in the plasmid transfected into mammalian cells at a frequency of 0.27%, five fold above that of a scrambled TFO [15]. This induction was abrogated in mammalian cell lines deficient in the nucleotide excision repair (NER) factor Xeroderma pigmentosa group A (XPA).
TFOs also induced mutagenesis in somatic cells of mice, when delivered systemically and without any associated mutagen such as irradiation. The transgenic mice contained about 15 copies of a chromosomally integrated shuttle vector containing the TFO binding site. Intraperitoneal injection of the TFO for 5 days resulted in a 5-fold increase in mutation frequency, compared to mice treated with a control oligonucleotide that contains mismatches to the TFO binding site [21]. Mutagenesis was seen in various tissues, including small intestine, liver, kidney, lung and skin, but not in the brain, where oligonucleotide uptake is minimal due to the presence of the blood-brain barrier. Mutagenesis frequencies in mice treated with control TFO were similar to that of saline-treated mice, and non-targeted genes on the shuttle vector (that do not contain the TFO binding site) did not show any mutagenesis, highlighting TFO specificity for the target site and the low toxicity of TFOs even with systemic delivery.
Enhanced recombination
TFO binding can augment recombination frequencies of “donor” DNA fragments into plasmid or chromosomal targets, although the mechanisms by which TFOs are able to do this are not yet clearly defined. Because the presence of double-stranded breaks can greatly increase recombinatory activity in bacteria, yeast, and mammalian systems, many groups have explored the use of TFOs tethered to DNA cleaving agents, such as Fe-EDTA, or selected nucleases, to stimulate high levels of recombination in vitro and in mammalian cells [22-24]. Recently, the DNA cleaving agent, orthophenanthroline (OP), was conjugated to a TFO to target a specific chromosomal site in HeLa cells [9]. The authors demonstrated that transfection of the OP-TFO into HeLa cells induced DSBs that were associated with phosphorylated H2AX (a DSB marker) by immunofluorescence colocalization, and by chromatin immunoprecipitation. As expected with the induction of DSBs, OP-TFO treatment led to high levels of mutagenesis, at a frequency of 10% as estimated by direct sequencing of the targeted DNA region. Mutations were small deletions, consistent with non-homologous end-joining mechanism (NHEJ) of the DSB, or substitutions. This OP-TFO was then used in a chromosomal recombination assay, in which the TFO binding site with a mutated eGFP sequence was chromosomally integrated into HeLa cells. These cells were then treated with OP-TFO, with a plasmid containing the reverting, wild-type eGFP sequence, to promote recombination and restoration of GFP expression. The authors demonstrated a recombination frequency of 1.5% in treated HeLa cells, at similar levels induced by I-SceI with donor plasmid, and representing nearly a 35-fold increase over spontaneous recombination by the donor plasmid alone. Therefore, DNA cleaving agents can be used in conjunction with TFOs to promote mutagenesis and recombination at targeted sites within the genome, and with significant efficiency.
TFOs by themselves are recombinogenic, however. This is advantageous for in vivo gene manipulation because it does not require prior insertion of an I-SceI recognition site, for example, to stimulate recombination. TFO recombinogenicity was demonstrated in a plasmid construct in which two tandem supF reporter genes, each containing inactivating mutations in different positions, flank a TFO-binding site. A G-rich TFO bound to this site can stimulate intramolecular recombination at a frequency of 0.37% to generate a wild-type supF gene (fivefold above background); furthermore, TFO conjugation to psoralen augmented the frequency to 1.5% (25-fold above background) [25]. This induction was found to be reduced in cells deficient in the NER factors XPA, XPG and XPF. XPA cDNA complementation partially restores the induced recombination. Interestingly, the induced recombination was not found to be dependent on the mismatch repair (MMR) factors MSH2 and MLH1. Mutation events were also analyzed from this recombination assay. As deletion events constituted 15-25% of the observed mutations (with most of the deletion end points joined at sites of 3 to 4 bp of homology), the observations were consistent with an NHEJ model of repair [26].
The demonstration of TFO-stimulated recombination in an intramolecular setting, led us to design an intermolecular system in which a TFO is used to guide and position a short fragment of homologous DNA to a specific location in the genome. We have demonstrated in episomal and chromosomal settings that a TFO, either covalently linked to a donor DNA fragment or simply co-transfected with the donor DNA, can lead to recombination at the TFO binding site, thereby resulting in increased rates of site-directed information transfer from the donor DNA to plasmid or to the chromosome itself. TFOs with donor DNAs were more active than either component alone, and activity was dependent on TFO binding and the presence of XPA [27]. Similarly, TFOs of both the purine and pyrimidine motifs were able to induce recombination in the absence of tethered mutagen [19].
Interestingly, when we substituted pyrimidine TFOs with base analogs that in theory provoked the largest relative amount of duplex helical distortion with TFO binding, we found the highest recombinatory frequencies between the reporter construct and a co-introduced donor DNA [19]. An NMR-based structural analysis of helical distortion compared TFOs composed of 2’-deoxy (DNA), 2’OH (RNA), and 2’O-methyl (2’OMe) ribose moieties, and found that the relative degrees of distortion were DNA > RNA > 2’OMe [28]. Consistent with this, pyrimidine TFOs with RNA-like 2’-substituted sugars resulted in the least recombinatory activity, despite TFO KD’s in the range of 10-10M. This latter result suggests that TFO binding and affinity are not the only factors that influence TFO metabolism and downstream events, and that TFO composition (in addition to affecting TFO delivery and bioavailability) can have a profound effect on TFO activities. Moreover, we found that while the frequency of TFO-induced recombination was greatest at target sites close to (<50 bp) of the TFO binding site of episomal target in mammalian cells, recombination can also be seen at distances up to 800 bp from the TFO binding site [29]. This suggests that a change in local topology (due to third strand binding) can have relatively distant effects.
Repair of triplex structures
The initial evidence that triplex binding can lead to DNA repair came from repair synthesis assays, in which we observed 32P-dCTP incorporation into plasmid constructs pre-treated with TFO, and incubated with human whole cell extracts to test for in vitro repair. This TFO-induced repair synthesis required the presence of both the specific TFO and its cognate site. Only TFOs that induced high rates of mutagenesis were able to induce DNA repair synthesis, suggesting that TFO-induced mutagenesis and repair were correlated. Furthermore, these studies suggested a role for the NER pathway in triplex structure recognition. Whereas a purine TFO induced mutagenesis at frequencies of 0.27%, 13-fold above spontaneous mutagenesis, the same TFO did not augment mutagenesis when the assay was performed using cells deficient in XPA or the transcription-coupled repair factor Cockayne’s Syndrome B (CSB; Table 1) [15].
Table 1.
Summary of repair factors studied in TFO-associated processing. NER, nucleotide excision repair; GGR, global genome repair; TCR, transcription-coupled repair; TLS, translesion synthesis; MMR, mismatch repair; HR, homologous recombination; NHEJ, non-homologous end-joining; FA, Fanconi anemia repair; ICL, interstrand cross-link
| Repair factor | Function | TFO-associated repair activity |
|---|---|---|
| XPA | GGR and TCR damage recognition | XPA deficiency |
| Decreased TFO-induced plasmid mutagenesis [15] | ||
| Decreased TFO-induced plasmid recombination [26] | ||
| Decreased PNA-induced plasmid repair synthesis [31] | ||
| Decreased PNA-induced plasmid recombination with ssDNA donor [31] | ||
| No effect on intracellular cross-link efficiency with pso-TFOs targeting chromosomal DNA [51] | ||
| Decreased alkylation repair in plasmid bound to chlorambucil-conjugated TFOs [55] | ||
| XPA binds in complex with RPA to cross-linked triplex structures; may reduce nonspecific RPA binding [37] | ||
| Presence of third strand interferes with XPA binding to TFO-directed ICLs on plasmid [49] | ||
| RPA | GGR damage recognition | Binds specifically to cross-linked triplex substrates, in complex with XPA [37] |
|
XPC HR23B |
GGR damage recognition and molecular matchmaker | XPC deficiency |
| Decreased removal of TFO-induced gene repression in plasmid substrate [30] | ||
| Decreased pso-TFO-induced plasmid mutagenesis in mammalian cells [39] | ||
| Decreased de-repression of reporter gene expression by pso-TFO [39] | ||
| Decreased pso-TFO-induced plasmid repair synthesis [39] | ||
| XPC/hHR23B complex recognizes cross-linked triplex structures [38] | ||
| XPD (ERCC2) | 5’→3’ helicase; part of TFIIH complex | XPD deficiency |
| Increased NHEJ-like deletion mutations in CHO hprt gene with TFO-directed ICL [44] | ||
| Increased TFO-recombination of ssDNA into CHO cell hprt target [45] | ||
| XPG (ERCC5) | GGR and TCR 3’ incision | XPG deficiency |
| Decreased TFO-induced plasmid intramolecular recombination [26] | ||
|
XPF (ERCC4) ERCC1 |
GGR and TCR 5’ incision | XPF deficiency |
| Decreased TFO-induced plasmid intramolecular recombination [26] | ||
| Decreased de-repression of reporter gene inhibition and associated ICL removal [43] | ||
| Increased NHEJ-like deletion mutations in CHO hprt gene with TFO-directed ICL [44] | ||
| Decreased base-pair substitutions in CHO hprt gene with TFO-directed ICL [44] | ||
| ERCC1 deficiency | ||
| Increased pso-TFO-induced recombination of ssDNA into CHO cell hprt target [45] | ||
| CSA | TCR | CSA deficiency |
| Decreased removal of TFO-induced gene repression in HeLa cell extracts [30] | ||
| CSB | TCR | CSB deficiency |
| Increased NHEJ-like deletion mutations in CHO hprt gene with TFO-directed ICL [44] | ||
| XPV (POLH) | TLS | XPV mutant |
| Decreased 125I-TFO induced mutagenesis in plasmid construct [56] | ||
|
MutSbeta MSH2 MSH3 MSH6 |
MMR | MSH2 deficiency |
| No effect on TFO-induced plasmid intramolecular recombination [26] | ||
| Decreased plasmid repair synthesis due to presence of TFO-directed ICLs [52] | ||
| No effect on mutagenesis nor mutation spectra on plasmid with TFO-directed ICL [52] | ||
| No effect on pso-TFO-induced recombination of ssDNA into CHO cell hprt target [45] | ||
| MSH3, MSH6, or MutSbeta deficiency | ||
| No change in mutagenesis frequency nor mutation spectra in CHO hprt target with TFO-directed ICL [44] | ||
| No effect on pso-TFO-induced recombination of ssDNA into CHO cell hprt target [45] | ||
| Mlh1 | HR | Mlh1 deficiency |
| No effect on TFO-induced plasmid intramolecular recombination [26] | ||
| Rad51 | HR | Rad51 deficiency |
| No TFO-induced recombination of tethered donor DNA into shuttle vector [32] | ||
| Increased mutagenesis in plasmid with TFO-directed ICL repaired in yeast [36] | ||
| Rad51 overexpression | ||
| Increased TFO-induced recombination of tethered donor DNA into shuttle vector [32] | ||
|
Artemis Ku86 DNA-PKcs WRN |
NHEJ | Artemis, Ku86, or Werner helicase deficiency |
| No change in mutagenesis frequency nor mutation spectra in CHO hprt target with TFO-directed ICL [44] | ||
| RecQ-family DNA helicase | DNA-PKcs deficiency | |
| No effect on pso-TFO-induced recombination of ssDNA into CHO cell hprt target [45] | ||
|
FANC-B FANC-C |
FA | FA complementation group B or C deficiency |
| No change in mutagenesis nor mutation spectra in shuttle vector with TFO-directed ICL [16] | ||
Transcription-coupled repair (TCR) is not only involved in TFO-induced mutagenesis; it is also critical for removal of bound TFO that inhibits reporter gene expression [30]. When a TFO-bound plasmid is incubated in cell extracts lacking TFIIB, TFIIH, or RNA polymerase II, or otherwise incompetent in transcription (e.g. rNTP depleted), gene expression remains inhibited, implying lack of repair of the TFO-associated lesion in the promoter region of the reporter gene. Similarly, TCR factor CSA was found to be necessary for TFO-induced repair. In addition to TCR, the authors also found evidence for global genome repair (GGR) of TFO-associated lesions, that is, NER that takes place outside of actively transcribed regions. Specifically, XPC, thought to be the primary recognition factor of the GGR subpathway, was found to be necessary for TFO repair [30].
Repair synthesis induced by TFOs is also correlated with recombination. As described above, a purine-rich TFO, without an associated mutagen, can provoke recombination of a tethered donor DNA fragment in a shuttle vector assay, and this recombination was associated with repair synthesis [31]. A triplex-forming peptide nucleic acid provoked even higher levels of recombination (whether or not the donor DNA was covalently linked), and this augmented recombination frequency corresponded with an increase in repair synthesis. Both repair and recombination of the purine TFO and PNA were dependent on the presence of XPA in the HeLa cell-free extracts.
As discussed above, the NER factors XPA, XPF and XPG were involved in TFO-induced recombination. When a purine TFO was bound to a reporter construct, in which tandem repeats of the supF reporter gene flank a TFO binding site, and then transfected into monkey COS cells, the resulting recombination frequency of the plasmid was 0.37% [26]. When the same TFO bound to the reporter plasmid was transfected in human fibroblasts deficient in XPA, XPF or XPG, the recombination frequency drops to 0.03%, near background frequencies. Complementation with XPA cDNA partially restored TFO-induced recombination, at a frequency of 0.14%, suggesting that NER plays an important role in TFO-induced recombination, but is not the only mechanism by which these structures are metabolized. However, MLH1 and MSH2, candidate repair factors from other canonical repair pathways, did not show any effect in observed recombination frequency.
Not surprisingly, TFO-induced recombination requires hsRad51, the human recA homolog that mediates strand exchange. Overexpression of hsRad51 increased recombination of donor DNA, in this case tethered to a purine TFO, in an episomal vector. In contrast, hsRad51 immunodepletion of whole cell extracts abrogated TFO-induced recombinogenic activity [32]. It is unclear whether it is the presence of the single-stranded donor DNA that triggers the involvement of Rad51, as Rad51 can nucleate ssDNA in preparation for homology scanning and strand invasion, or if HR mechanisms are invoked by the binding of the third strand by itself. Indeed, HR is known to be involved in processing stalled replication forks, in addition to DSB repair. Consistent with evidence that triplex structures (endogenous or with TFOs) can induce transcription arrest [33-35], bound third strands may prevent DNA polymerase progression, thereby promoting local recombination.
Repair of pso-TFO lesions
As discussed above, TFOs are commonly conjugated to psoralen to augment TFO-mediated biological activity. Psoralen is a photoreactive DNA intercalator, which, following UVA irradiation, can form monoadducts (in which psoralen is covalently linked to one strand of the targeted DNA), or interstrand cross-links (both strands of the target DNA are covalently linked to the psoralen moiety). The use of pso-TFO targeting to episomal or chromosomal environments offers investigators a covalent, stable cross-link for the study of targeted repair, in contrast to third strand binding, which may be transient and difficult to detect in an assay. Processing of adducts formed by psoralen conjugated TFOs have been studied in E. coli, in yeast and in mammalian systems. There is evidence that multiple repair pathways, error-prone and error-free, participate in the repair of TFO-associated ICLs. Their relative contributions to the processing of TFO-associated lesions may shift depending on chromosomal context, the cell cycle stage, and transcription state. Accordingly, a mutation in the HR factor Rad51 in yeast leads to an increase in pso-TFO-induced mutagenesis [36], offering evidence that both error-free (HR, single-strand annealing) and error-prone (NER, NHEJ) pathways are involved intracellularly.
As with the repair of structures induced by TFOs by themselves, TFO-directed psoralen interstrand cross-links are processed by NER factors. In vitro recognition of triple helical structures by the NER recognition factors replication factor A (RPA) and XPA was explored with a series of binding assays, using pso-TFO binding to duplex DNA as substrates [37]. RPA was found to bind specifically to covalent psoralen-monoadducted or psoralen-cross-linked triplex structures with high affinity. In contrast, RPA did not bind to duplex DNA by itself, nor duplex DNA treated with free psoralen (that is, in the absence of the third strand). In the absence of RPA, XPA by itself does not appear to bind to either triplex or duplex DNA. However, because XPA is present in RPA-triplex complexes, it is thought that XPA’s role is to reduce nonspecific RPA binding to triplex, possibly as an RPA-XPA complex.
Other NER factors, the global genome repair factor XPC, and the human homolog of RAD23B (hHR23B), can also bind to triplex-directed ICLs formed in vitro [38]. Recognition of triplex structures was seen to be rapid, occurring within 10 seconds of incubation of the triplex structure to purified repair proteins. At low concentrations, RPA can compete with and displace XPC-hHR23B on cross-linked triplex; however, at high concentrations, RPA and XPC-hHR23B form a multimeric complex with the cross-linked triplex.
Consistent with in vitro data, XPC deficiency has been shown to lead to a decrease in frequencies of plasmid mutagenesis by pso-TFOs in mammalian cells [39]. Using a 10-mer TFO conjugated to psoralen, the authors showed that XPC deficiency in a primary fibroblast cell line was correlated with a decrease in ICL removal as demonstrated by persistent reporter gene repression, and decreased levels of repair synthesis at the cross-linking site. This work suggests that XPC is involved in error-prone repair of the TFO-directed ICLs.
Following recognition of a lesion, NER factors typically make a dual excision on the damaged DNA strand six nucleotides 3’ to the damage, and 22 nucleotides 5’ from the damage site to excise the cross-link [40,41]. For an interstrand cross-link lesion, this creates a gap that is still attached to the non-incised strand via the cross-link. In E. coli, this gap is repaired by recA-mediated homologous recombination or lesion bypass synthesis [42]. Therefore, the mammalian homologs of recA, the nucleases XPD and ERCC1/XPF, and the translesion bypass synthesis polymerase zeta, are candidate proteins in the repair of TFO-directed ICLs in mammalian systems. An early study showed that whereas wild-type cells were able to remove TFO-directed ICLs within 48 hours of plasmid transfection, similar ICLs were not removed when transfected into primary cells deficient in XPF [43].
Furthermore, a mutagenesis assay in CHO cells deficient in these NER excision factors revealed a change in the type of mutation induced by a TFO-directed ICL, relative to mutations resulting from repair in wild-type CHO cells [44]. XPF, ERCC1, or XPD deficiencies resulted in an increase in the frequency of deletion mutations, and sequencing of these deletions revealed that repair around the ICL lesion was consistent with NHEJ processing. This suggested to the authors that in wild-type cells, NER may repair TFO-directed ICLs, but in the absence of NER factors, ICLs persist and disrupt replication, which may in turn lead to strand discontinuities, double-stranded breaks, and other substrates of NHEJ repair. Transcription coupled repair may have also been involved in pso-TFO mutagenesis, since cells deficient in CSB also resulted in an increase in deletion mutants. Notably, the frequency of basepair substitutions decreased in mutants derived from cells lacking XPF or ERCC1 (relative to frequencies from wild-type cells), but there was no change in basepair substitution frequency in cells that lacked XPD. Deficiencies in Artemis or Ku86 of the NHEJ pathway did not result in changes in mutation spectra, nor did deficiencies in the MMR proteins MSH3 and MSH6 [44].
The authors then extended these results to assess the involvement of NER factors on TFO-induced recombination [45]. In this assay, pso-TFOs were introduced into CHO cells in conjunction with a short, single-stranded donor oligonucleotide for recombination. They demonstrated that the absence of XPD or ERCC1 (which acts in concert with XPF) leads to increased recombinogenic activity of pso-TFOs targeting the hamster hprt gene. The authors interpreted the data to mean that XPD and ERCC1/XPF deficiency results in unrepaired ICLs and double-stranded breaks, whose presence, in turn, promote recombinatory mechanisms for repair. The presence of a short, homologous donor oligonucleotide in this setting may also bias repair towards recombination. In contrast, these authors did not find any change in recombination frequencies using cells deficient in DNA-PKcs, MSH2, or MSH3, relative to wild-type cells.
Repair of the TFO-directed cross-link may be affected by the orientation of the asymmetric psoralen moiety with respect to the duplex DNA. Psoralen has a furane on one end, and a pyrone on the other, and free psoralen can intercalate into DNA in either direction, resulting in equal probabilities of the two cross-link orientations in the duplex DNA. When conjugated to a TFO, however, the furane side of the psoralen molecule reacts preferentially with the polypurine strand of the target duplex DNA, leading to asymmetric repair patterns. Barre [46] investigated this issue by inserting the polypurine TFO binding site in opposite orientations to force psoralen-ICL polarity: in one construct, the target polypurine sequence was on the coding strand of the DNA (“direct”), whereas in the “inverted” sequence, the TFO binding site was on the transcribed strand of DNA (that is, the template strand for RNA polymerase). Following introduction of a psoralen-conjugated TFO and UVA photoactivation, the cross-linked plasmids were allowed time to repair in yeast, recovered and sequenced to look at mutation patterns. The investigators found that regardless of the orientation of the triplex binding site, the mutations (primarily substitutions and insertions at the thymine involved in psoralen linking) were predominantly found on the coding strand of the recovered plasmid, implying that repair is coupled with transcription. However, the authors found that the frequencies of mutagenesis generated in the two constructs were different: higher frequencies were found when the TFO binding site (and furane-adduct) was on the transcribed strand of the DNA, almost double that of mutation frequencies using the “direct” construct. Moreover, the authors discovered that the repair machinery preferentially excised the furane-adducted strand (that is, the polypurine strand) relative to the pyrone-adducted strand. Excision of the furane-adduct, in turn, led to translesion bypass of and a subsequent base substitution on the pyrone-adducted strand (that is, the polypyrimidine strand). Taken together, the authors concluded that when both the TFO binding site and the furane ring of the psoralen cross-link were on the transcribed strand, there were relatively increased levels of mutagenesis, implying increased levels of repair, due to a concert of repair efforts from both translesion bypass repair and NER [46]. It is unclear, however, if the lower mutagenesis frequencies resulting from the “direct” sequence was due to decreased repair (which would result in decreased plasmid survival, which the authors did not observe), or increased error-free repair (the latter is not detected in their assay). In fact, this is a general concern of most mutagenesis assays, in that mutagenesis frequencies likely underestimate the extent of repair, given that they do not account for error-free repair.
To assess how the presence of a third strand would affect repair of an ICL in mammalian cells, Wang et al. compared mutagenesis and repair induced by a 10nt and 30nt TFO in a supF reporter mutagenesis assay [47]. The authors speculated that if NER were a predominant mode of ICL removal, the longer TFO may preclude access to the preferred excision sites flanking the ICL. Indeed, excision products were only found when the 10nt TFO was used to target an ICL on a plasmid, and none were visualized when a 30nt TFO was bound. This is consistent with in vitro data showing that although the E. coli Uvr(A)BC excinuclease can form a complex with triplex DNA covalently linked at a specific site, excinuclease activity is significantly reduced in the context of a bound third strand [48]. Furthermore, using a shuttle vector assay, Wang et al. determined that the 10nt TFO generated mostly A:T to T:A transversions, while the 30nt TFO generated a diverse set of mutations, including large deletions. The change in mutation spectra is thought to reflect a shift in the predominance of the NER pathway (leading to transversions) to a NHEJ repair pathway (leading to deletions).
Several other groups have also found that the presence of a bound TFO may interfere with multiple steps of the ICL repair process, in addition to the excision step as shown above. Using a cleavable form of a psoralen-conjugated TFO (by disulphide linkage, which is cleaved with reduction), Guillonneau et al. were able to induce a site-specific ICL on a plasmid, and study the effect of third strand binding on ICL repair by introducing the reducing agent TCEP, versus leaving the third strand intact [49]. They found that in the presence of an associated third strand, a site-specific ICL provokes reduced levels of repair synthesis, decreased incision products, decreased levels of XPA binding, and decreased religation, relative to ICLs in which the TFO had been cleaved off. Functionally, this interference of ICL repair by the third strand results in the persistence of pso-TFO-mediated inhibition of reporter gene expression [50].
In fact, Musso et al. found that reporter gene expression is repressed by 90% up to 72 hours following introduction of pso-TFOs, compared with plasmid with no ICLs. Gene repression correlated with decreased PCR efficiency through the ICL site, and protection from restriction enzyme cleavage [17], implying persistence of the ICL. Barre et al. estimated that only 10% of TFO-directed ICLs are repaired in yeast, and only about half of these by error-prone mechanisms [36]. As well, the chromosomal environment may change the degree of ICL repair, as shown in Oh and Hanawalt’s chromosomal targeting assay [51]. They used a 15-mer pso-TFO targeting the human collagenase gene in human fibrosarcoma cells, and detected TFO-directed ICLs in about 15-40% of recovered chromosomal DNA by primer extension assay and by modified single-stranded ligation PCR through the ICL site. The percentage of cells with ICLs did not change when they used the pso-TFOs to target the same gene in XPA-deficient fibroblasts, implying there is minimal ICL removal by an NER-dependent mechanism in these cells. ICL removal did not change when TPA was added to the cells to augment transcription, suggesting that repair at this particular site was not linked to transcription. Taken together, these data indicate that many TFO-directed lesions are resistant to removal, particularly in the context of the third strand. The persistence of TFO-associated lesions, combined with the augmentation of localized mutagenesis by TFOs, may be particularly attractive features of TFOs conjugated to DNA reactive molecules, for applications in long-term gene repression and gene inactivation.
Because ERCC1/XPF is also involved in other repair pathways, such as mismatch repair and translesion bypass synthesis, there is some interest in these other repair pathways for TFO-directed ICL processing. Cells deficient in MSH2 (HEC59) show increased sensitivity to plasmid ICLs created by free psoralen exposure, but HEC59 cell extracts show a decrease in repair synthesis ability following a TFO-directed ICL, relative to extracts from an isogenic control repair-proficient cell line [52]. This suggests that MSH2 is involved in TFO-directed ICL processing. However, repair of a cross-linked shuttle vector in HEC59 cells result in similar frequencies of mutagenesis as the isogenic (MSH2 proficient) control cells, and the resulting mutation spectra are similar from both cell lines (44% transversions in the targeted region, 40% single- or double-base mutations at adjacent sites, and 11% deletions). This observation, that there is MSH2-mediated repair synthesis in the absence of MSH2-mediated mutagenesis, led the authors to conclude that MMR is involved in error-free processing of TFO-directed psoralen ICLs [52].
Similarly, two cell lines with deficiencies in the Fanconi anemia (FA) pathway were studied with respect to their abilities to process a TFO-directed ICL on a similar shuttle vector assay as above [53]. The Fanconi anemia pathway is an HR-related pathway that specializes in cross-link repair. FA cells display a diagnostic sensitivity to DNA cross-linking agents, and so far, 13 complementation groups have been identified in the FA repair pathway [54]. When TFO-directed ICLs were introduced into FA complementation group B or C cell lines (HSC230 and GM04510, respectively), Bredberg et al. observed mutation frequencies in the shuttle vector that were similar to those from pso-ICL-treated wild-type cells, after accounting for the generally higher levels of “spontaneous” mutagenesis in FA cells. Moreover, the mutation spectra of plasmids obtained from FA cells was similar to that from wild-type cells. These results were surprising given that FA cells are generally more sensitive to ICLs and have a high frequency of deletions following 8-methoxypsoralen plus UVA radiation treatment. However, free psoralen induces many more ICLs to genomic DNA, whereas with pso-TFO treatment, only one ICL is targeted to DNA. The difference in ICL sensitivity and processing, therefore, may reflect decreased repair capacity of FA cells. At the time these studies were reported, however, the molecular repair deficiencies of these cells were not yet characterized, so it would be interesting to follow up these early studies with more direct genetic evidence. The contribution of the presence of the third strand to ICL repair in FA cells would be an interesting corollary to the study of general ICL repair.
Repair of other TFO-directed lesions
Site-specific DNA alkylation was studied with a chlorambucil-conjugated TFO targeting the HER2/neu promoter. Significant repair synthesis was observed in vitro using TFO-directed chl-guanine adducts as substrates, and transfection of chl-TFO treated plasmid into HeLa cells resulted in repair of the alkylation site within 24 hours of transfection. In contrast, when XP12BE cell extracts were used instead (which are NER deficient), alkylation repair was not observed [55].
As well, several groups have used radionuclide-conjugated TFOs to induce double-stranded breaks to a specific site in the genome. 125I decay, for example, in proximity of nuclear DNA imparts local radiotoxicity to the site, resulting in an estimated one DSB per decay. Consistent with work with nucleases showing that the presence of DSBs can sensitize sites to repair, radionuclide-conjugated TFOs induce 7-fold higher levels of mutagenesis than pso-TFOs in a similar plasmid assay; mutagenesis is correlated with the number of DSBs present. In cells with defective (but not deficient) XPV, which codes for polymerase eta, this mutagenesis frequency is reduced 3-fold, indicating that translesion bypass synthesis is involved in radionuclide-induced DSB repair [56]. However, TFO-directed, radionuclide-induced DSB lesions are not identical to relatively “clean” DSBs by restriction enzyme cleavage, as shown by Odersky et al. [57]. This group found that while NHEJ may mediate DSB repair in plasmid DNA following 125I-TFO treatment, the repair efficiency is dramatically decreased relative to DSBs made by various restriction enzymes. Furthermore, the presence of the third strand by itself may affect repair, as it does in TFO-directed psoralen-induced cross-links as discussed above.
Summary & Conclusions
TFOs have enormous potential as gene targeting agents, but also are useful reagents in the study of site-specific DNA lesions. The repair of TFO-associated lesions is complex, and involves multiple repair pathways that may act in concert or in competition with one another. While nucleotide excision repair and the NER subpathway, transcription coupled repair, appear to play the predominant role in processing triplex structures, there is evidence that recombinatory, mismatch repair, and end-joining mechanisms are also involved. In addition, the presence of the third strand appears to not only change the kinetics of repair, perhaps by interfering with repair factor binding and duplex DNA accessibility, but also may recruit different repair factors in response to TFO-induced local topological changes in the DNA structure. In the field of targeted gene therapy, a better understanding of TFO metabolism will aid in augmenting their biological activity, for example by prolonging third strand binding for targeted gene repression, or by promoting TFO recombinogenicity of a donor DNA for directed information transfer. TFOs can also aid in the study of general DNA repair, by localizing lesions to particular sites for the study of local repair factor recruitment and lesion processing.
Acknowledgments
The authors acknowledge members of the Glazer Lab for helpful discussions. This work is supported by NIH grants to PMG (R01CA64186 and R01HL082655), and an MSTP Training Grant to JYC (5T32GM07205).
Abbreviations
- TFO
Triplex-forming oligonucleotide
- pso
psoralen
- ICL
interstrand cross-link
- DSB
double-strand break
- NER
nucleotide excision repair
- TCR
transcription-coupled repair
- HR
homologous recombination
- NHEJ
non-homologous end-joining
- MMR
mismatch repair
- FA
Fanconi anemia
References
- 1.Felsenfeld G, Davies DR, Rich A. Formation of a three-stranded polynucleotide molecule. J Am Chem Soc. 1957;79:2023–2024. [Google Scholar]
- 2.Moser HE, Dervan PB. Sequence-specific cleavage of double helical DNA by triple helix formation. Science. 1987;238(4827):645–650. doi: 10.1126/science.3118463. [DOI] [PubMed] [Google Scholar]
- 3.Praseuth D, Perrouault L, Le Doan T, Chassignol M, Thuong N, Helene C. Sequence-specific binding and photocrosslinking of alpha and beta oligodeoxynucleotides to the major groove of DNA via triple-helix formation. Proc Natl Acad Sci U S A. 1988;85(5):1349–1353. doi: 10.1073/pnas.85.5.1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Letai AG, Palladino MA, Fromm E, Rizzo V, Fresco JR. Specificity in formation of triple-stranded nucleic acid helical complexes: studies with agarose-linked polyribonucleotide affinity columns. Biochemistry. 1988;27(26):9108–9112. doi: 10.1021/bi00426a007. [DOI] [PubMed] [Google Scholar]
- 5.Cooney M, Czernuszewicz G, Postel EH, Flint SJ, Hogan ME. Site-specific oligonucleotide binding represses transcription of the human c-myc gene in vitro. Science. 1988;241(4864):456–459. doi: 10.1126/science.3293213. [DOI] [PubMed] [Google Scholar]
- 6.Perrouault L, Asseline U, Rivalle C, et al. Sequence-specific artificial photo-induced endonucleases based on triple helix-forming oligonucleotides. Nature. 1990;344(6264):358–360. doi: 10.1038/344358a0. [DOI] [PubMed] [Google Scholar]
- 7.Hanvey JC, Shimizu M, Wells RD. Site-specific inhibition of EcoRI restriction/modification enzymes by a DNA triple helix. Nucleic Acids Res. 1990;18(1):157–161. doi: 10.1093/nar/18.1.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Simon P, Cannata F, Concordet JP, Giovannangeli C. Targeting DNA with triplex-forming oligonucleotides to modify gene sequence. Biochimie. 2008 doi: 10.1016/j.biochi.2008.04.004. [DOI] [PubMed] [Google Scholar]
- 9.Cannata F, Brunet E, Perrouault L, et al. Triplex-forming oligonucleotide-orthophenanthroline conjugates for efficient targeted genome modification. Proc Natl Acad Sci U S A. 2008;105(28):9576–9581. doi: 10.1073/pnas.0710433105. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 10.Alam MR, Majumdar A, Thazhathveetil AK, et al. Extensive sugar modification improves triple helix forming oligonucleotide activity in vitro but reduces activity in vivo. Biochemistry. 2007;46(35):10222–10233. doi: 10.1021/bi7003153. [DOI] [PubMed] [Google Scholar]
- 11.Cheng AJ, Wang JC, Van Dyke MW. Self-association of G-rich oligodeoxyribonucleotides under conditions promoting purine-motif triplex formation. Antisense Nucleic Acid Drug Dev. 1998;8(3):215–225. doi: 10.1089/oli.1.1998.8.215. [DOI] [PubMed] [Google Scholar]
- 12.Nielsen PE. PNA Technology. Mol Biotechnol. 2004;26(3):233–248. doi: 10.1385/MB:26:3:233. [DOI] [PubMed] [Google Scholar]
- 13.Chin JY, Kuan JY, Lonkar PS, et al. Correction of a splice-site mutation in the beta-globin gene stimulated by triplex-forming peptide nucleic acids. Proc Natl Acad Sci U S A. 2008 doi: 10.1073/pnas.0711793105. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hartman DA, Kuo SR, Broker TR, Chow LT, Wells RD. Intermolecular triplex formation distorts the DNA duplex in the regulatory region of human papillomavirus type-11. J Biol Chem. 1992;267(8):5488–5494. [PubMed] [Google Scholar]
- 15.Wang G, Seidman MM, Glazer PM. Mutagenesis in mammalian cells induced by triple helix formation and transcription-coupled repair. Science. 1996;271(5250):802–805. doi: 10.1126/science.271.5250.802. [DOI] [PubMed] [Google Scholar]
- 16.Sandor Z, Bredberg A. Repair of triple helix directed psoralen adducts in human cells. Nucleic Acids Res. 1994;22(11):2051–2056. doi: 10.1093/nar/22.11.2051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Musso M, Wang JC, Van Dyke MW. In vivo persistence of DNA triple helices containing psoralen-conjugated oligodeoxyribonucleotides. Nucleic Acids Res. 1996;24(24):4924–4932. doi: 10.1093/nar/24.24.4924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang G, Levy DD, Seidman MM, Glazer PM. Targeted mutagenesis in mammalian cells mediated by intracellular triple helix formation. Mol Cell Biol. 1995;15(3):1759–1768. doi: 10.1128/mcb.15.3.1759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kalish JM, Seidman MM, Weeks DL, Glazer PM. Triplex-induced recombination and repair in the pyrimidine motif. Nucleic Acids Res. 2005;33(11):3492–3502. doi: 10.1093/nar/gki659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Majumdar A, Khorlin A, Dyatkina N, et al. Targeted gene knockout mediated by triple helix forming oligonucleotides. Nat Genet. 1998;20(2):212–214. doi: 10.1038/2530. [DOI] [PubMed] [Google Scholar]
- 21.Vasquez KM, Narayanan L, Glazer PM. Specific mutations induced by triplex-forming oligonucleotides in mice. Science. 2000;290(5491):530–533. doi: 10.1126/science.290.5491.530. [DOI] [PubMed] [Google Scholar]
- 22.Segal DJ, Carroll D. Endonuclease-induced, targeted homologous extrachromosomal recombination in Xenopus oocytes. Proc Natl Acad Sci U S A. 1995;92(3):806–810. doi: 10.1073/pnas.92.3.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Strobel SA, Dervan PB. Site-specific cleavage of a yeast chromosome by oligonucleotide-directed triple-helix formation. Science. 1990;249(4964):73–75. doi: 10.1126/science.2195655. [DOI] [PubMed] [Google Scholar]
- 24.Strobel SA, Dervan PB. Single-site enzymatic cleavage of yeast genomic DNA mediated by triple helix formation. Nature. 1991;350(6314):172–174. doi: 10.1038/350172a0. [DOI] [PubMed] [Google Scholar]
- 25.Faruqi AF, Seidman MM, Segal DJ, Carroll D, Glazer PM. Recombination induced by triple-helix-targeted DNA damage in mammalian cells. Mol Cell Biol. 1996;16(12):6820–6828. doi: 10.1128/mcb.16.12.6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Faruqi AF, Datta HJ, Carroll D, Seidman MM, Glazer PM. Triple-helix formation induces recombination in mammalian cells via a nucleotide excision repair-dependent pathway. Mol Cell Biol. 2000;20(3):990–1000. doi: 10.1128/mcb.20.3.990-1000.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chan PP, Lin M, Faruqi AF, Powell J, Seidman MM, Glazer PM. Targeted correction of an episomal gene in mammalian cells by a short DNA fragment tethered to a triplex-forming oligonucleotide. J Biol Chem. 1999;274(17):11541–11548. doi: 10.1074/jbc.274.17.11541. [DOI] [PubMed] [Google Scholar]
- 28.Asensio JL, Carr R, Brown T, Lane AN. Conformation and thermodynamic properties of parallel intramolecular triple helices containing a DNA, RNA, or 2’OMeDNA third strand. J Am Chem Soc. 1999;121(48):11063–11070. [Google Scholar]
- 29.Knauert MP, Lloyd JA, Rogers FA, et al. Distance and affinity dependence of triplex-induced recombination. Biochemistry. 2005;44(10):3856–3864. doi: 10.1021/bi0481040. [DOI] [PubMed] [Google Scholar]
- 30.Wang G, Chen Z, Zhang S, Wilson GL, Jing K. Detection and determination of oligonucleotide triplex formation-mediated transcription-coupled DNA repair in HeLa nuclear extracts. Nucleic Acids Res. 2001;29(8):1801–1807. doi: 10.1093/nar/29.8.1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rogers FA, Vasquez KM, Egholm M, Glazer PM. Site-directed recombination via bifunctional PNA-DNA conjugates. Proc Natl Acad Sci U S A. 2002;99(26):16695–16700. doi: 10.1073/pnas.262556899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Datta HJ, Chan PP, Vasquez KM, Gupta RC, Glazer PM. Triplex-induced recombination in human cell-free extracts. Dependence on XPA and HsRad51 J Biol Chem. 2001;276(21):18018–18023. doi: 10.1074/jbc.M011646200. [DOI] [PubMed] [Google Scholar]
- 33.Belotserkovskii BP, De Silva E, Tornaletti S, Wang G, Vasquez KM, Hanawalt PC. A triplex-forming sequence from the human c-MYC promoter interferes with DNA transcription. J Biol Chem. 2007;282(44):32433–32441. doi: 10.1074/jbc.M704618200. [DOI] [PubMed] [Google Scholar]
- 34.Giovannangeli C, Perrouault L, Escude C, Gryaznov S, Helene C. Efficient inhibition of transcription elongation in vitro by oligonucleotide phosphoramidates targeted to proviral HIV DNA. J Mol Biol. 1996;261(3):386–398. doi: 10.1006/jmbi.1996.0471. [DOI] [PubMed] [Google Scholar]
- 35.Samadashwily GM, Mirkin SM. Trapping DNA polymerases using triplex-forming oligodeoxyribonucleotides. Gene. 1994;149(1):127–136. doi: 10.1016/0378-1119(94)90421-9. [DOI] [PubMed] [Google Scholar]
- 36.Barre FX, Giovannangeli C, Helene C, Harel-Bellan A. Covalent crosslinks introduced via a triple helix-forming oligonucleotide coupled to psoralen are inefficiently repaired. Nucleic Acids Res. 1999;27(3):743–749. doi: 10.1093/nar/27.3.743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vasquez KM, Christensen J, Li L, Finch RA, Glazer PM. Human XPA and RPA DNA repair proteins participate in specific recognition of triplex-induced helical distortions. Proc Natl Acad Sci U S A. 2002;99(9):5848–5853. doi: 10.1073/pnas.082193799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Thoma BS, Wakasugi M, Christensen J, Reddy MC, Vasquez KM. Human XPC-hHR23B interacts with XPA-RPA in the recognition of triplex-directed psoralen DNA interstrand crosslinks. Nucleic Acids Res. 2005;33(9):2993–3001. doi: 10.1093/nar/gki610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chen Z, Xu XS, Yang J, Wang G. Defining the function of XPC protein in psoralen and cisplatin-mediated DNA repair and mutagenesis. Carcinogenesis. 2003;24(6):1111–1121. doi: 10.1093/carcin/bgg051. [DOI] [PubMed] [Google Scholar]
- 40.Batty DP, Wood RD. Damage recognition in nucleotide excision repair of DNA. Gene. 2000;241(2):193–204. doi: 10.1016/s0378-1119(99)00489-8. [DOI] [PubMed] [Google Scholar]
- 41.Reardon JT, Sancar A. Nucleotide excision repair. Prog Nucleic Acid Res Mol Biol. 2005;79:183–235. doi: 10.1016/S0079-6603(04)79004-2. [DOI] [PubMed] [Google Scholar]
- 42.Cole RS. Repair of DNA containing interstrand crosslinks in Escherichia coli: sequential excision and recombination. Proc Natl Acad Sci U S A. 1973;70(4):1064–1068. doi: 10.1073/pnas.70.4.1064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Degols G, Clarenc JP, Lebleu B, Leonetti JP. Reversible inhibition of gene expression by a psoralen functionalized triple helix forming oligonucleotide in intact cells. J Biol Chem. 1994;269(24):16933–16937. [PubMed] [Google Scholar]
- 44.Richards S, Liu ST, Majumdar A, et al. Triplex targeted genomic crosslinks enter separable deletion and base substitution pathways. Nucleic Acids Res. 2005;33(17):5382–5393. doi: 10.1093/nar/gki851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Majumdar A, Muniandy PA, Liu J, et al. Targeted gene knock in and sequence modulation mediated by a psoralen-linked triplex-forming oligonucleotide. J Biol Chem. 2008;283(17):11244–11252. doi: 10.1074/jbc.M800607200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Barre FX, Asseline U, Harel-Bellan A. Asymmetric recognition of psoralen interstrand crosslinks by the nucleotide excision repair and the error-prone repair pathways. J Mol Biol. 1999;286(5):1379–1387. doi: 10.1006/jmbi.1999.2550. [DOI] [PubMed] [Google Scholar]
- 47.Wang G, Glazer PM. Altered repair of targeted psoralen photoadducts in the context of an oligonucleotide-mediated triple helix. J Biol Chem. 1995;270(38):22595–22601. doi: 10.1074/jbc.270.38.22595. [DOI] [PubMed] [Google Scholar]
- 48.Duval-Valentin G, Takasugi M, Helene C, Sage E. Triple helix-directed psoralen crosslinks are recognized by Uvr(A)BC excinuclease. J Mol Biol. 1998;278(4):815–825. doi: 10.1006/jmbi.1998.1728. [DOI] [PubMed] [Google Scholar]
- 49.Guillonneau F, Guieysse AL, Nocentini S, Giovannangeli C, Praseuth D. Psoralen interstrand cross-link repair is specifically altered by an adjacent triple-stranded structure. Nucleic Acids Res. 2004;32(3):1143–1153. doi: 10.1093/nar/gkh267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Guieysse AL, Praseuth D, Giovannangeli C, Asseline U, Helene C. Psoralen adducts induced by triplex-forming oligonucleotides are refractory to repair in HeLa cells. J Mol Biol. 2000;296(2):373–383. doi: 10.1006/jmbi.1999.3466. [DOI] [PubMed] [Google Scholar]
- 51.Oh DH, Hanawalt PC. Triple helix-forming oligonucleotides target psoralen adducts to specific chromosomal sequences in human cells. Nucleic Acids Res. 1999;27(24):4734–4742. doi: 10.1093/nar/27.24.4734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wu Q, Christensen LA, Legerski RJ, Vasquez KM. Mismatch repair participates in error-free processing of DNA interstrand crosslinks in human cells. EMBO Rep. 2005;6(6):551–557. doi: 10.1038/sj.embor.7400418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Bredberg A, Sandor Z, Brant M. Mutational response of Fanconi anaemia cells to shuttle vector site-specific psoralen cross-links. Carcinogenesis. 1995;16(3):555–561. doi: 10.1093/carcin/16.3.555. [DOI] [PubMed] [Google Scholar]
- 54.Li X, Heyer WD. Homologous recombination in DNA repair and DNA damage tolerance. Cell Res. 2008;18(1):99–113. doi: 10.1038/cr.2008.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ziemba A, Derosier LC, Methvin R, et al. Repair of triplex-directed DNA alkylation by nucleotide excision repair. Nucleic Acids Res. 2001;29(21):4257–4263. doi: 10.1093/nar/29.21.4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Mezhevaya K, Winters TA, Neumann RD. Gene targeted DNA double-strand break induction by (125)I-labeled triplex-forming oligonucleotides is highly mutagenic following repair in human cells. Nucleic Acids Res. 1999;27(21):4282–4290. doi: 10.1093/nar/27.21.4282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Odersky A, Panyutin IV, Panyutin IG, et al. Repair of sequence-specific 125I-induced double-strand breaks by nonhomologous DNA end joining in mammalian cell-free extracts. J Biol Chem. 2002;277(14):11756–11764. doi: 10.1074/jbc.M111304200. [DOI] [PubMed] [Google Scholar]
- 58.Chin JY, Schleifman EB, Glazer PM. Repair and recombination induced by triple helix DNA. Front Biosci. 2007;12:4288–4297. doi: 10.2741/2388. [DOI] [PubMed] [Google Scholar]