

Abstract
It has been a major challenge to develop effective therapeutics for stroke, a leading cause of death and serious debilitation. Intensive research in the past fifteen years have implicated many regulators and the related mechanisms by which neuronal cell death is regulated. It is now clear that even a brief ischemic stroke may trigger complex cellular events that lead to both apoptotic and necrotic neuronal cell death in a progressive manner. Although efforts at developing specific chemical inhibitors for validated targets have been successful for in vitro enzymatic assays, the development of some of such inhibitors into human therapy has been often hindered by their in vivo bioavailability profile. Considerations for the ability to chemically target a cellular mechanism in manner compatible with disease targets in vivo might be emphasized early in the development process by putting a priority on identifying key targets that can be effectively targeted chemically. Thorough interrogation of cellular pathways by saturation chemical genetics may provide a novel strategy to identify multiple key molecular entities that can be targeted chemically in order to select a target suitable for the treatment of intended human diseases such as stroke.
Keywords: apoptosis, necroptosis, necrosis, stroke, neurodegeneration
Neurons, the crucial cell type that controls our ability to think and to move, are one of the most precious cell types in our body because of their limited ability to regenerate once mature. Devastating loss of neurons occur in stroke, either due to cerebral ischemia or hemorrhage, which triggers a complex series of biochemical events that leads to total breakdown of cellular integrity and eventually cell death. Although stroke is one of the leading causes of mortality and disability worldwide, spectacular failures in recent clinical trials and drug development programs for neuroprotective agents have led to the withdrawal of funding aimed at developing new drugs for stroke which still has no direct therapy available other than reopening an occluded artery with thrombolytic drugs. There is no ongoing clinical trial for stroke with a neuroprotective drug. At the moment, the current budget crisis at the NIH has impaired the efforts for developing new neurological drugs. Thus, acute neurological injury represents a huge unmet medical need that receives little attention.
A stroke lesion is characterized by a core of necrotic cell death formed rapidly after injury and may represent tissues that are irreversibly lost, and the penumbra, which surrounds the core and is defined as a moderately hypoperfused meta-stable region that retains structural integrity but has lost or impaired function. The penumbra is the battle ground for stroke therapy as it has been recognized as an area at risk and can be salvaged with appropriate intervention. The only available therapeutic strategy for ischemic stroke is to reopen an occluded artery by thrombolytic therapy to restore perfusion to the ischemic area, a procedure which in itself can sometimes induce secondary damage. Furthermore, the necessity of differentiating patients with ischemic stroke suitable for thrombolytic therapy from those with hemorrhagic stroke patients causes significant delay in hospital that contributes to the loss of a crucial window of opportunity when the penumbra might still be saved. There is an urgent need to develop safe and effective neuroprotective drugs that can protect cells in the penumbra under either ischemic or hemorrhagic conditions so that the drug might be given immediately after onset and prior to hospital arrival.
Multiple mechanisms have been identified that contribute to the loss of neurons in the penumbra. Here I will review some of the progress made in the past decade or so that contributes to our understanding of neuronal cell death induced by acute neurological injury, and discuss the merits of targeting different mechanisms for reducing neuronal loss after stroke.
Apoptosis
Apoptosis is a cellular suicide mechanism regulated by an evolutionarily conserved genetic program. The proteins in Bcl-2 family and caspase family play critical roles in the activation, signal transduction and execution of apoptosis (1). Transgenic mice overexpressing Bcl-2 are protected from ischemic injury as Bcl-2 protects against mitochondrial damage, an important signal amplification step during apoptosis. In vivo delivery of a Bcl-xL fusion protein, which contains the protein transduction domain (PTD) derived from the human immunodeficiency TAT protein fused with Bcl-xL, decreased cerebral infarction induced by focal ischemia (2). Inhibition of Bcl-2 by small molecule mimetic of the BH3 domain, a key protein-protein interacting domain involved in Bcl-2 function, leads to the activation of apoptosis and is currently under clinical trials for cancers (3). No attempt, however, had been developed to directly activate Bcl-2 as a neuroprotective strategy as overexpression of Bcl-2 is known to contribute to multiple forms of cancers, in particular B-cell malignancies including leukemia and lymphomas. However, since anti-apoptotic activity of Bcl-2 is negatively regulated by pro-apoptotic BH3-only members of Bcl-2 family, the loss of Bid, a BH3-only protein, protects against ischemic brain injury by reducing mitochondrial damage induced by transient occlusion of the middle cerebral artery (4, 5). An attempt has been made to use a NMR-based approach to develop small molecules of Bid by targeting a deep hydrophobic crevice on the surface of Bid (6). Further studies in this direction might be interesting.
Caspases
Caspases are a family of cysteine proteases that function as key mediators of apoptosis (1). Mammalian caspase family has 11 members. The first evidence indicating the involvement of caspases in mediating acute neurological injury was demonstrated by the ability of transgenic mice expressing a dominant negative mutant of caspase-1 (C285G) to resist ischemic brain injury induced by middle cerebral artery occlusion (7, 8). Intracerebral ventricular injection of peptide inhibitors of caspases (z-VAD.FMK) decreased caspase cleavage products and tissue immunoreactive IL-1β levels in ischemic mouse brain, reduced tissue damage and significantly improved behavioral deficits. Furthermore, intracerebral injection of a caspase inhibitor, z-VAD.FMK, protected against α-amino-3-hydroxy-5-methyl-4-isoxazole propionate-induced and to a lesser extent N-methyl-D-aspartate-induced excitotoxic brain damage. These experiments provide the first evidence for the roles of caspases in mediating ischemic and excitotoxic brain injury and ignite the interest in the research field and pharmaceutical industry to target caspases as a therapeutic strategy for stroke.
Although the functional roles of a number of caspases, including caspase-1 and caspase-3, in mediating ischemic neuronal cell death were confirmed in caspase-1-/- and caspase-3-/- mice (9, 10), the exact mechanisms by which the loss of different caspases protect against acute neurological injury still need investigation.
First of all, although as a downstream caspase, caspase-3 most likely mediates neuronal cell death intracellularly, the function of caspase-1 in neuronal cell death might be mediated in part through processing and secretion of mature IL-1β and IL-1α extracellularly. Hayashi et al. (11) examined the distribution of caspase-1 in the hippocampus of mongolian gerbils. Immunoreactivity of caspase-1 was found predominantly in microglia, astrocytes, and endothelial cells of capillaries as well as some non-pyramidal neurons. The major presence of caspase-1 in non-neuronal cells of the brain suggests neuronal injury induced by the activation of caspase-1 might be cell non-autonomous. Consistent with this possibility, exogenous administration or overexpression of IL-1RA, which is an endogenous antagonizer of IL-1β, is neuroprotective in diverse rodent models of cerebral ischaemia (12, 13), excitotoxicity (14) and trauma (15). Furthermore, double mutant mice of IL-1α and IL-1β are highly resistant to ischaemic brain damage (16). IL1Ra has been tested in Phase II clinical trials for stroke (17) and is a promising lead for stroke therapy. On the other hand, this observation also raised the question of contribution of glia in ischemic brain injury. Glia have been increasingly recognized to play an important role in neuronal function (18); however, very little is currently known about cell death mechanisms of glia.
Consistent with in vivo data, hypoxia-induced neuronal cell death in vitro can also be inhibited by IL-1Ra (19). IL-1Ra also inhibits apoptosis induced by trophic factor deprivation in primary neurons, as well as by TNFα in fibroblasts. However, in vitro evidence suggests that the extracellular mature IL-1β is most likely one of the factors promoting cell death under certain conditions as IL-1β alone is not sufficient to induce cell death. For example, the addition of mature IL-1β to G1/S phase arrested HeLa cells induces apoptosis, but not to exponentially growing HeLa cells. In another example, COS cells are normally resistant to apoptosis induced by overexpression of caspase-1 but can be sensitized by the extracellular IL-1β. Thus, IL-1β can be considered as a tonic factor mediating the sensitivity of cells to apoptosis. Recently, caspase-1 has been found to contribute to the secretion of a large number of leaderless proteins including IL-1α and FGF-2 (20). Although the role of caspase-1 in mediating secretion of these factors is yet to be confirmed in the central nervous system, this study projects a much more complex role of stress activated caspase-1 in mediating inflammation, cytoprotection, cell survival, and regenerative processes than previously proposed. Although such extracellular output is most extensively studied for caspase-1, other upstream caspases may have similar properties that remain to be explored in future.
The cell death process triggered by ischemic injury is an evolving process that is a function of both time and severity of injury. Consistent with this proposal, the activation of caspases after ischemic injury has been shown to occur in region specific and time specific manner. Benchoua et al. (21) showed that in the early stages of cerebral infarction, neurons of the necrotic core display a number of morphological, physiological, and biochemical features of early apoptosis, which include cytoplasmic and nuclear condensations and the involvement of caspase-8 and the caspase-1 pathways. In contrast, pathways that are activated during the secondary expansion of the lesion in the penumbral area mainly involve mitochondrial pathway mediated by caspase-9. The authors propose that apoptosis is the first commitment to death after acute cerebral ischemia and that the final morphological features observed results from aborted apoptotic process because of severe energy depletion in the core. In contrast, energy-dependent caspase activation cascades are observed in the penumbra in which apoptosis can fully develop because of residual blood supply.
Consistent with above observation, the protection offered by inhibition of caspases might be dependent upon the ability of injured brain tissues to undergo apoptosis. Li et al. (22) found that although blocking the activation of caspases by caspase inhibitors z-VAD.FMK and z-DEVD.FMK can reduce ischemic neuronal injury after transient cerebral ischemic insult, they are not effective in blocking injury following complete cessation of blood flow such as during global ischemia. The authors conclude that inhibition of caspases might be beneficial for moderately severe focal but not global cerebral ischemia. On the other hand, inability of caspase inhibitors to reduce infarct size after permanent ischemia suggests that residual blood flow (albeit low) and reperfusion might be required for the activation of caspases. Consistent with this possibility, Manabat et al. (23) showed that although a marked increase in caspase-3 activity was observed within 24 hours of reperfusion after transient MCA occlusion, caspase-3 activity remained significantly lower within 24 hours of permanent MCA occlusion. Therefore, persistent perfusion deficits result in less caspase-3 activation and appear to favor caspase-independent injury.
Although the contribution of apoptosis to neuronal cell death induced by stroke may depend upon the severity of condition and may be spatially heterogeneous, it is unequivocal that apoptosis is activated after ischemic brain injury and inhibition of apoptosis provides an important therapeutic strategy for stroke. In this regard, it is regrettable that after considerable efforts in major pharmaceutical companies to develop small molecule inhibitors of caspases for inhibiting apoptosis induced under pathological conditions, none has reached clinics. At least one of the major obstacles here is the bioavailability, i.e. the inability of chemical inhibitors of caspases, which are cysteine proteases, to reach the site of injury because they are efficiently retained by liver and degraded in situ. Thus, we have a clear example where the limitations in chemistry might have prohibited the translation of a great biological discovery into treatments for human diseases. The failure in developing chemical inhibitors of caspases suggests that we need to look for important apoptosis targets that can be chemically targeted in a manner that is compatible for the treatment of human diseases.
JNK
c-Jun N-terminal kinase (JNK) is an important stress inducible kinase that has been shown to be activated after focal or global ischemia (24), in both permanent and transient models (25). Kuan et al. found that the expression of c-Jun, a downstream target of JNK, correlated with pyknotic nuclei, suggesting the induction of JNK signaling in dying neurons. Systemic delivery of SP600125, a reversible ATP competitive JNK inhibitor (26), diminished JNK activity and infarct volume after transient focal ischemia in mice (27) and global ischemia/reperfusion in the vulnerable hippocampal CA1 subregion in rats (28).
JNK family has three isoforms, JNK1, JNK2, and JNK3. Although JNK1 is the major isoform responsible for the high level of basal JNK activity in the brain, JNK3 is the major family member that is responsible for the stress-induced JNK activity because a deficiency in Jnk3, but not Jnk1, protects mice from brain injury after cerebral ischemia-hypoxia (29). MKK4, an upstream kinase that activates JNK, was highly phosphorylated after cerebral ischemia–hypoxia in both wild-type and Jnk3 deficient mice. In contrast, the phosphorylation level of c-Jun after ischemia–hypoxia was also markedly reduced in Jnk3 deficient mice, consistent with a major block in stress induced JNK activity.
JNK has been shown to mediate the induction and/or activation of a number of pro-apoptotic proteins including Bim (30, 31) and Fas (32). Consistent with an important role of JNK3 in apoptosis induced by ischemic injury, the induction of Bim, a BH3-only proapoptotic member of Bcl-2 family, and Fas were both inhibited in Jnk3 deficient mice. Inhibition of Bim and Fas expression can also be accomplished by a chemical inhibitor of JNK, SP600125 (27). These results confirm the role of JNK as a critical cell death mediator in ischemic brain injury. Development of specific inhibitors for JNK3 should provide a promising lead for inhibiting acute neurological injury such as stroke.
Necrosis
Although apoptosis has been recognized as the predominant form of cell death in developing immature neurons, the role of apoptosis in mature neurons might be less significant. Immature neurons, which are the type that can be easily cultured and therefore extensively studied, readily undergo apoptosis under growth factor deprivation condition or stimulated with pro-apoptotic stimuli, mature neurons are relatively apoptotic resistant, which raises the question as to the ability of mature neurons to undergo apoptosis after insult of adult brains. Finally, it was discovered that although there was a reduction of apoptotic neurons in the developing nervous system of apoptotic deficient mice, elimination of supernumerary neurons, which normally occur through apoptosis, still happened but may be mediated through a different mechanism as dead neurons exhibit non-apoptotic morphology. Thus, neurons might have a back-up cell death mechanism(s) that can be activated in the absence of full apoptotic machinery.
A number of morphological studies addressed the role of apoptosis vs. necrosis after ischemic brain injury. Wei et al. (33) used a combination of TTC (2,3,5-triphenyltetrazolium chloride) staining, hematoxylin-eosin (H&E) staining, the terminal deoxyribonucleotidyl transferase-mediated dUTP nick end labeling (TUNEL), and caspase-3 staining as well as ultrastructural studies using electron microscopy to study the morphology of cell death in ischemic cortex and non-ischemic thalamus after focal ischemia in the cerebral cortex in rats. It was discovered that the morphology of TUNEL positive cells in the ischemic cortex were best described as necrotic, whereas the TUNEL positive cells in the thalamus and the cortex penumbra region show both apoptotic and necrotic morphology. The cell death in the thalamus and the cortex penumbra was at least partially mediated by apoptotic mechanism as it can be attenuated by delayed administration of caspase inhibitor benzyloxycarbonyl-Val-Ala-Asp fluoromethylketone (Z-VAD-FMK). Thus, different types of neurons might have different propensity to undergo apoptosis or necrosis.
The concomitant activation of apoptosis and necrosis after ischemic injury is corroborated by a similar study using markers including lysosomal leakage and TUNEL positivity in mice (34). Consistent with evidence from in vitro studies as described above, the tendency to undergo necrotic cell death may increase as neurons mature. Liu et al. (35) compared the tendency of newborn vs. older brains to undergo apoptosis after ischemic insult. In newborn pups, most neurons after ischemic insult are positive for activated caspase-3. The number of activated caspase-3 positive neurons decline significantly with a concomitant increase in numbers of necrotic neurons in older rats after a comparable insult. These results strongly suggest that predominant mechanisms of neuronal cell death shifts from apoptosis to necrosis as neurons mature.
The involvement of apoptosis or necrosis may depend on the severity of injury as well as the presence of reperfusion. As discussed above, Manabat et al. found significant caspase activation only in the penumbra after reperfusion but not in the core (23) and inhibition of caspase protects against focal but not global ischemia (22).
Although necrotic cell death had been traditionally believed to be caused by overwhelming stress and therefore not regulated by cellular mechanisms, emerging evidence suggests the existence of alternative cell death mechanisms characterized by necrotic morphology. Below, I will discuss a number of necrotic cell death mechanisms that have been proposed to be involved in ischemic brain injury.
Poly(ADP-ribose)-polymerase (PARP)
Nitric oxide is a messenger molecule that has functions as a neurotransmitter, in addition to its role in mediating macrophage killing of tumor cells and bacteria, and blood vessel relaxation (36). When produced in large quantities in response to actions of the excitatory neurotransmitter glutamate acting at N-methyl-D-aspartate (NMDA) receptors, NO mediates neuronal killing which contributes to neuronal cell death induced by stroke. Multiple mechanisms have been proposed to mediate the neurotoxicity of NO. In particular, Ca2+ influx through activated NMDA receptors activates calcium-dependent cytoplasmic proteins to mediate nNOS and NO production which in turn increases the accumulation of reactive oxygen species and formation of peroxynitrite (37). Peroxynitrite causes DNA damage which activates the DNA repair mechanism including PARP-1, a member of poly(ADP-ribose)polymerase family (PARPs).
PARPs are a family of proteins that mediate the formation of poly(ADP-ribose) chain on selected proteins. Using NAD+ as a substrate, PARPs catalyze the covalent attachment of ADP-ribose units on the glutamic or aspartic acid residues of target proteins, to generate long linear and branched poly(ADP-ribose)(PAR) chains. The human genome encodes 18 different PARP genes. Poly-ADP-ribosylation is now known to play a role in a wide range of biological structures and processes, including DNA repair and maintenance of genomic stability, transcriptional regulation, cell division and cell death by modifying a diverse array of proteins. Like many other posttranslational protein modification mechanisms involved in signaling, poly-ADP-ribosylation is transient and reversible in nature, due to the rapid action of poly(ADP-ribose) glycohydrolase (PARG) which catalyzes the hydrolysis of PAR into free ADP-ribose (38). The role of PARP-1 activation following DNA damage is well established to play an important role in mediating DNA repair. Caspase-mediated cleavage of PARP-1 has been recognized as a hallmark of apoptosis (39). The cleavage separates the DNA binding domain and catalytic domain of PARP-1. Thus, inhibition of caspase activation alone might prevent the inactivation of PARP-1 by caspase cleavage and led to excessive activation of PARP-1 (40). Since poly-ADP-ribosylation consumes NAD+, a high energy product as well as key coenzyme required in the mitochondria for maintaining proton gradient in the electron transport chain to generate ATP, over-activation of PARP-1 has been proposed to lead to necrosis by excessive energy loss.
Neurons contain high levels of PARP-1 activity following trauma, ischemic injury, or oxidative stress (41). Studies utilizing PARP-1-/- mice demonstrated the importance of the role of PARP-1 in the neuronal injury following stroke. In experimental models of ischemia in vitro, PARP-1-/- primary neuronal cultures are resistant to the toxicity elicited by combined oxygen-glucose deprivation (OGD), NMDA treatment, or treatment with NO donors (42). Reduced infarct volume is observed following middle cerebral artery occlusion in PARP-1 null mice. PARP-1 inhibitors also provide neuroprotection against excitotoxicity (36).
The exact mechanism by which PARP-1 deficiency protects against acute neurological injury is still under investigation. One study reported that in PARP-1-/- ischemic brain, the markers of apoptosis, such as oligonucleosomal DNA damage, total DNA fragmentation, and the density of terminal deoxynucleotidyl transferase dUTP nick-end-labelled (TUNEL +) cells, were not different from that of control, although there was a decrease in infarct size (43). Although the resistance of PARP-1-/- mice to ischemic brain injury has been generally attributed to a protection against NAD depletion and energy failure, since poly-ADP-ribosylation is a signaling mechanism, it is at least equally likely that the resistance of PARP-1-/- mice is due to inhibition of specific event(s) in mediating damage signaling. Since the markers for apoptosis were not altered in PARP-1-/- mice after ischemic brain injury, it is reasonable to propose that the loss of PARP-1 protects mostly against necrotic cell death. In this regard, the activation of PARP-1 has been shown to mediate mitochondrial damage and the release of AIF from the mitochondria to induce necrotic cell death (41).
Interestingly, the protection of ischemic damage offered by PARP deficiency shows gender specificity. In contrast to the male PARP-1-/- mice, which are more resistant to ischemic injury, the female PARP-1-/- mice have exacerbated injury compared to that of wt control after ischemic insult (44). Although it has not been examined if the activity of PARP-1 might exhibit gender specific regulation, a sexually dimorphic role of PARP-1 in ischemic neuronal death is consistent with the signaling hypothesis, rather than in regulation of energy levels per se. In this regard, it is interesting to note that PARP-1 has recently been proposed to be a part of histone code. Poly-ADP-ribosylation mediated by PARP-1 and PARP-2 has been recognized as a part of epigenetic regulation of chromatin structures that have the potential to orchestrate various chromatin-based biological tasks including transcription, DNA repair and differentiation. PARP-1 was found to be a regulated promoter-specific exchange factor required for the activation of specific gene programs. The catalytic function of PARP-1 was shown to facilitate the release of histone H1 from a subset of PARP-1-targeted PolII-transcribed promoters (45), and the consequent recruitment of the chromatin architectural protein HMGB1 on the estrogen-stimulated pS2 promoter (45). A role of PARP-1 in mediating the expression of the estrogen-regulated genes such as TFF1 gene might explain the sexual dimorphic response to inhibition of PARP-1(46). A sexual dimorphic role of PARP activation in ischemic brain injury may make it difficult to target PARP as a therapeutic target for stroke.
Calpains
The intracellular Ca2+ concentration is tightly regulated under physiological condition. Ischemic brain injury disrupts this tight control by allowing the influx from extracellular pools through various channels as well as release from endoplasmic reticulum (ER) stores to lead enormous increases in intracellular Ca2+ (47). An increased intracellular free Ca2+ levels activate multiple Ca2+-dependent enzymes, contributing to neuronal death and dysfunction. The calcium-dependent neutral cysteine proteases, calpains, are major Ca2+-dependent proteases. There are two major isozymes, μ- and m-calpains, which require micromolar or milimolar levels of Ca2+ for activation in vitro, respectively. Calpain is a heterodimer comprising a 30-kDa small regulatory subunit and a 80-kDa catalytic subunit. During activation, the 30-kDa subunit is cleaved to yield a final 17-kDa form, while the 80-kDa subunit is converted to 76-kDa enzymatically active form. Calpains function under physiological conditions to mediate many normal homeostatic functions but when activated under brain ischemia-reperfusion condition (48), contributes to neuronal cell death by cleaving multiple substrates including cytoskeletal and associated proteins, kinases and phosphatases, membrane receptors and transporters. Excessive activation of calpain due to an increase in free Ca2+ leads to cytoskeletal protein breakdown, subsequent loss of structural integrity and disturbances of axonal transport, and finally to necrotic cell death (49). Early studies show that a calpain inhibitor, Cbz-Val-Phe-H, was able to reduce cerebral ischemic infarction induced by MCAO model (50). No calpain inhibitors, however, enter human clinical trials for stroke. As calpains are also cysteine proteases, at least one of the challenges is also the ability to make specific calpain inhibitors with appropriate bioavailability compatible with inhibiting cell death induced by ischemic brain injury.
Necroptosis
Although death receptor mediated apoptosis represents a canonical apoptotic pathway, stimulation of death receptors under apoptotic deficient conditions is now known to activate necrotic cell death, termed necroptosis (51). The activation of death receptors by their respective ligands, such as FasL (CD95L) and TNFα, respectively, leads to the formation of DISC (death-inducing signaling complex) that includes the adaptor protein FADD (Fas-associated death domain), caspase-8 and death domain-containing kinase RIP1. In apoptotic proficient condition, the recruitment of caspase-8 leads to its activation which in turn activates downstream caspases, such as caspase-3, and mitochondrial damage by cleaving Bid (1). In apoptotic deficient cells when caspases cannot be activated, however, stimulation of death receptors leads to the activation of RIP1 kinase and necroptosis (52, 53).
Following brain ischemia, expressions of TNFα, FasL and Fas are increased in the ischemic penumbra. The role of TNFα in ischemic brain injury in vivo is controversial. On the one hand, neutralization of endogenous TNFα is reported to reduce infarct volume. However, mice lacking TNFR1/p55 or both TNFR1 and TNFR2/p75 showed enhanced ischemic damage which might be attributable to the role of TNFRs in mediating the activation of NF-κB which is pro-survival. On the other hand, lpr mice, lacking a functional Fas, exhibit a profound reduction of infarct size. FasL and TNFα may act coordinately in mediating ischemic brain injury as mice that are doubly deficient for both TNFα (tnf-/-) or FasL (gld) are protected against brain ischemia (54). Furthermore, treatment of wild-type mice after induction of ischemia with neutralizing antibodies against TNFα and FasL diminished infarct volumes and significantly improve survival of the animals.
Ser/Thr kinase RIP1 has been found to be essential for death receptor mediated necroptosis (52). RIP1 is a death-domain containing kinase associated with the death receptors but the kinase activity of RIP1 is dispensable for the induction of apoptosis. FADD-mediated activation of either caspase-8 or RIP1 represents a bifurcation point between apoptotic and necroptotic programs in the Fas signaling pathway. When caspases are inactive, death-inducing ligands may activate caspase-independent pathways regulated by specific kinase signaling pathways. A family of small molecule inhibitors of necroptosis, termed necrostatins, were identified using high throughput cell-based screens (51, 52, 55, 56). Necrostatins selectively block necroptosis in cells exposed to the co-treatment of FasL or TNFα and the pan-caspase inhibitor, z-VAD.fmk, but have no effect on apoptosis. Interestingly, necrostatin-1 (Nec-1) was found to be a potent allosteric inhibitor of RIP1 by selectively targeting the inactive conformation of the kinase (52). Nec-1 was effective in protecting against ischemic brain damage induced by MCAO model with an extended time window. Nec-1 is also effective in protecting against traumatic brain injury (57) and ischemic heart injury (58). Thus, necrostatins represent a class of promising lead neuroprotective compounds.
The mechanism by which the activation of RIP1 kinase leads to necroptosis is still not clear. Recently, Hitomi et al. identified multiple key players of necroptosis in a genome-wide siRNA screen (59). Interestingly, Bmf, a member of BH3-only member of Bcl-2 family, was found to be one of the key mediators of necroptosis, which raised the possibility that the activation of RIP1 kinase might lead to necroptosis by inducing necrotic damage of mitochondria.
The concept of “chemical genetics” is to use small molecules as a tool to explore biological problems (60). Necroptosis may provide the first example where a novel cellular pathway is explored by “saturation chemical genetics” (Figure 1) to identify all the key biochemical steps that can be chemically targeted using a cell-based screen. In contrast to that of caspases, which still lack effective chemical inhibitors after extensive research for fifteen years, the “translatability” of targeting necroptosis for treatments of human diseases may be significantly accelerated by the availability of necrostatins.
Figure 1.
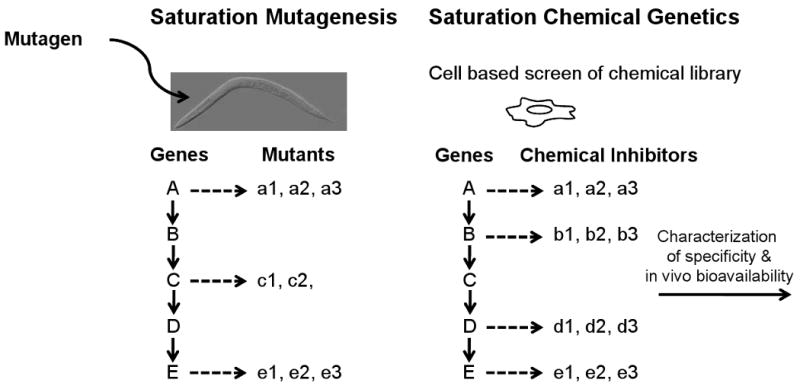
Analogy of classical saturation mutagenesis and saturation chemical genetics. Mutagenesis of a model genetic organism, such as nematode C. elegans, may be carried out exhaustively in order to isolate mutants for all the genes in an interested pathway that can be isolated. In an analogous fashion, we may develop a specific cell based screen to isolate inhibitors for all the proteins in the pathway so as to identify all the targets that can be chemically inhibited. Early identification of such chemical inhibitors may allow us to select targets that are compatible with the treatment of relevant disease in vivo.
Summary
Extensive researches in the cell death field in the past fifteen years have provided rich insights into the mechanisms by which neurons die under ischemic conditions. Stroke is a severe and acute condition: even a brief ischemic insult of brain may trigger complex cellular events that lead to both apoptotic and necrotic cell death in a progressive manner. Although we have already discovered several important targets that play important roles in mediating apoptotic and necrotic cell death induced by stroke, the challenge is if we can develop specific small molecule inhibitors that can be delivered effectively into the brain in a manner compatible with the acute nature of stroke.
How can we meet this challenge? The past fifteen years of cell death research have taught us that a key factor determining if a major biological discovery can be translated into a treatment for human diseases is whether it can be effectively targeted chemically in a manner compatible with treatment of the diseases. In the past, we have always emphasized the need to fully explore the biological pathways and validate biological targets before even considering the development of chemical inhibitors. While such approaches have high rates of success in developing direct small molecule inhibitors in vitro, the in vivo bioavailabilities of compounds developed by such approaches have not always met the requirement. Development of necrostatins represents an exception against this tradition as these inhibitors were isolated from a large-scale chemical screen using a cell-based assay. Such large-scale “black-box” chemical screens are analogous to saturation mutagenesis in classical genetic analysis which has been frequently used to fully interrogate a genetic pathway and thus, can be termed “saturation chemical genetics”. Necrostatins provide an example demonstrating that with carefully designed screens, it may be possible to fully interrogate a novel biological pathway by chemical approaches for understanding biological mechanisms. Although defining the biological targets of small molecules represents a challenge by itself, it has become much more feasible with the availability of genome wide analytical tools including RNAi, proteomics and microarray analysis. The target validation can be further facilitated by the availability of chemical probes and other genomic tools. While no one wishes to deemphasize the importance of thoroughly interrogating important biological pathways and disease mechanisms, systematic exploration of biological pathways using “saturation chemical genetics” might provide a new direction for selecting important sites that are suitable for targeting chemically. With devastating diseases such as stroke, we clearly need to think “out-of-box” for developing new therapies that hopefully one day will reach the clinics.
Acknowledgments
The author expresses deep gratitude to Dr. Mike Moskowitz for insightful comments and suggestions. This work was supported in part by a NIH Director's Pioneer Award (DP1OD000580), grants from the National Institute of Neurological Disorders and Stroke (UO1 NS050560) and the National Institute on Aging (R37-AG012859 and PO1 AG027916).
References
- 1.Li J, Yuan J. Caspases in apoptosis and beyond. Oncogene. 2008;27:6194–6206. doi: 10.1038/onc.2008.297. [DOI] [PubMed] [Google Scholar]
- 2.Cao G, Pei W, Ge H, et al. In Vivo Delivery of a Bcl-xL Fusion Protein Containing the TAT Protein Transduction Domain Protects against Ischemic Brain Injury and Neuronal Apoptosis. J Neurosci. 2002;22:5423–5431. doi: 10.1523/JNEUROSCI.22-13-05423.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Danial NN, Korsmeyer SJ. Cell death: critical control points. Cell. 2004;116:205–219. doi: 10.1016/s0092-8674(04)00046-7. [DOI] [PubMed] [Google Scholar]
- 4.Plesnila N, Zinkel S, Amin-Hanjani S, Qiu J, Korsmeyer SJ, Moskowitz MA. Function of BID -- a molecule of the bcl-2 family -- in ischemic cell death in the brain. Eur Surg Res. 2002;34:37–41. doi: 10.1159/000048885. [DOI] [PubMed] [Google Scholar]
- 5.Plesnila N, Zinkel S, Le DA, et al. BID mediates neuronal cell death after oxygen/glucose deprivation and focal cerebral ischemia. Proc Natl Acad Sci U S A. 2001;98:15318–15323. doi: 10.1073/pnas.261323298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Becattini B, Sareth S, Zhai D, et al. Targeting apoptosis via chemical design: inhibition of bid-induced cell death by small organic molecules. Chem Biol. 2004;11:1107–1117. doi: 10.1016/j.chembiol.2004.05.022. [DOI] [PubMed] [Google Scholar]
- 7.Hara H, Friedlander RM, Gagliardini V, et al. Inhibition of interleukin 1beta converting enzyme family proteases reduces ischemic and excitotoxic neuronal damage. Proc Natl Acad Sci U S A. 1997;94:2007–2012. doi: 10.1073/pnas.94.5.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hara H, Fink K, Endres M, et al. Attenuation of transient focal cerebral ischemic injury in transgenic mice expressing a mutant ICE inhibitory protein. J Cereb Blood Flow Metab. 1997;17:370–375. doi: 10.1097/00004647-199704000-00002. [DOI] [PubMed] [Google Scholar]
- 9.Schielke GP, Yang GY, Shivers BD, Betz AL. Reduced ischemic brain injury in interleukin-1 beta converting enzyme-deficient mice. J Cereb Blood Flow Metab. 1998;18:180–185. doi: 10.1097/00004647-199802000-00009. [DOI] [PubMed] [Google Scholar]
- 10.Le DA, Wu Y, Huang Z, et al. Caspase activation and neuroprotection in caspase-3- deficient mice after in vivo cerebral ischemia and in vitro oxygen glucose deprivation. Proc Natl Acad Sci U S A. 2002;99:15188–15193. doi: 10.1073/pnas.232473399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Hayashi Y, Jikihara I, Yagi T, et al. Immunohistochemical investigation of caspase-1 and effect of caspase-1 inhibitor in delayed neuronal death after transient cerebral ischemia. Brain Res. 2001;893:113–120. doi: 10.1016/s0006-8993(00)03307-2. [DOI] [PubMed] [Google Scholar]
- 12.Relton JK, Rothwell NJ. Interleukin-1 receptor antagonist inhibits ischaemic and excitotoxic neuronal damage in the rat. Brain Res Bull. 1992;29:243–246. doi: 10.1016/0361-9230(92)90033-t. [DOI] [PubMed] [Google Scholar]
- 13.Betz AL, Yang GY, Davidson BL. Attenuation of stroke size in rats using an adenoviral vector to induce overexpression of interleukin-1 receptor antagonist in brain. J Cereb Blood Flow Metab. 1995;15:547–551. doi: 10.1038/jcbfm.1995.68. [DOI] [PubMed] [Google Scholar]
- 14.Lawrence CB, Allan SM, Rothwell NJ. Interleukin-1beta and the interleukin-1 receptor antagonist act in the striatum to modify excitotoxic brain damage in the rat. Eur J Neurosci. 1998;10:1188–1195. doi: 10.1046/j.1460-9568.1998.00136.x. [DOI] [PubMed] [Google Scholar]
- 15.Tehranian R, Andell-Jonsson S, Beni SM, et al. Improved recovery and delayed cytokine induction after closed head injury in mice with central overexpression of the secreted isoform of the interleukin-1 receptor antagonist. J Neurotrauma. 2002;19:939–951. doi: 10.1089/089771502320317096. [DOI] [PubMed] [Google Scholar]
- 16.Boutin H, LeFeuvre RA, Horai R, Asano M, Iwakura Y, Rothwell NJ. Role of IL-1alpha and IL-1beta in ischemic brain damage. J Neurosci. 2001;21:5528–5534. doi: 10.1523/JNEUROSCI.21-15-05528.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Emsley HC, Smith CJ, Georgiou RF, et al. A randomised phase II study of interleukin-1 receptor antagonist in acute stroke patients. J Neurol Neurosurg Psychiatry. 2005;76:1366–1372. doi: 10.1136/jnnp.2004.054882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Barres BA. The mystery and magic of glia: a perspective on their roles in health and disease. Neuron. 2008;60:430–440. doi: 10.1016/j.neuron.2008.10.013. [DOI] [PubMed] [Google Scholar]
- 19.Friedlander RM, Gagliardini V, Rotello RJ, Yuan J. Functional role of interleukin 1 beta (IL-1 beta) in IL-1 beta-converting enzyme-mediated apoptosis. J Exp Med. 1996;184:717–724. doi: 10.1084/jem.184.2.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Keller M, Ruegg A, Werner S, Beer HD. Active caspase-1 is a regulator of unconventional protein secretion. Cell. 2008;132:818–831. doi: 10.1016/j.cell.2007.12.040. [DOI] [PubMed] [Google Scholar]
- 21.Benchoua A, Guegan C, Couriaud C, et al. Specific caspase pathways are activated in the two stages of cerebral infarction. J Neurosci. 2001;21:7127–7134. doi: 10.1523/JNEUROSCI.21-18-07127.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Li H, Colbourne F, Sun P, Zhao Z, Buchan AM, Iadecola C. Caspase inhibitors reduce neuronal injury after focal but not global cerebral ischemia in rats. Stroke. 2000;31:176–182. doi: 10.1161/01.str.31.1.176. [DOI] [PubMed] [Google Scholar]
- 23.Manabat C, Han BH, Wendland M, et al. Reperfusion differentially induces caspase-3 activation in ischemic core and penumbra after stroke in immature brain. Stroke. 2003;34:207–213. doi: 10.1161/01.STR.0000047101.87575.3C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Irving EA, Bamford M. Role of mitogen- and stress-activated kinases in ischemic injury. J Cereb Blood Flow Metab. 2002;22:631–647. doi: 10.1097/00004647-200206000-00001. [DOI] [PubMed] [Google Scholar]
- 25.Borsello T, Clarke PG, Hirt L, et al. A peptide inhibitor of c-Jun N-terminal kinase protects against excitotoxicity and cerebral ischemia. Nat Med. 2003;9:1180–1186. doi: 10.1038/nm911. [DOI] [PubMed] [Google Scholar]
- 26.Bennett BL, Sasaki DT, Murray BW, et al. SP600125, an anthrapyrazolone inhibitor of Jun N-terminal kinase. Proc Natl Acad Sci U S A. 2001;98:13681–13686. doi: 10.1073/pnas.251194298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao Y, Signore AP, Yin W, et al. Neuroprotection against focal ischemic brain injury by inhibition of c-Jun N-terminal kinase and attenuation of the mitochondrial apoptosis-signaling pathway. J Cereb Blood Flow Metab. 2005;25:694–712. doi: 10.1038/sj.jcbfm.9600062. [DOI] [PubMed] [Google Scholar]
- 28.Guan QH, Pei DS, Liu XM, Wang XT, Xu TL, Zhang GY. Neuroprotection against ischemic brain injury by SP600125 via suppressing the extrinsic and intrinsic pathways of apoptosis. Brain Res. 2006;1092:36–46. doi: 10.1016/j.brainres.2006.03.086. [DOI] [PubMed] [Google Scholar]
- 29.Kuan CY, Whitmarsh AJ, Yang DD, et al. A critical role of neural-specific JNK3 for ischemic apoptosis. Proc Natl Acad Sci U S A. 2003;100:15184–15189. doi: 10.1073/pnas.2336254100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Okuno S, Saito A, Hayashi T, Chan PH. The c-Jun N-terminal protein kinase signaling pathway mediates Bax activation and subsequent neuronal apoptosis through interaction with Bim after transient focal cerebral ischemia. J Neurosci. 2004;24:7879–7887. doi: 10.1523/JNEUROSCI.1745-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Putcha GV, Le S, Frank S, et al. JNK-mediated BIM phosphorylation potentiates BAX-dependent apoptosis. Neuron. 2003;38:899–914. doi: 10.1016/s0896-6273(03)00355-6. [DOI] [PubMed] [Google Scholar]
- 32.Herdegen T, Claret FX, Kallunki T, et al. Lasting N-terminal phosphorylation of c-Jun and activation of c-Jun N-terminal kinases after neuronal injury. J Neurosci. 1998;18:5124–5135. doi: 10.1523/JNEUROSCI.18-14-05124.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wei L, Ying DJ, Cui L, Langsdorf J, Yu SP. Necrosis, apoptosis and hybrid death in the cortex and thalamus after barrel cortex ischemia in rats. Brain Res. 2004;1022:54–61. doi: 10.1016/j.brainres.2004.06.080. [DOI] [PubMed] [Google Scholar]
- 34.Unal-Cevik I, Kilinc M, Can A, Gursoy-Ozdemir Y, Dalkara T. Apoptotic and necrotic death mechanisms are concomitantly activated in the same cell after cerebral ischemia. Stroke. 2004;35:2189–2194. doi: 10.1161/01.STR.0000136149.81831.c5. [DOI] [PubMed] [Google Scholar]
- 35.Liu CL, Siesjo BK, Hu BR. Pathogenesis of hippocampal neuronal death after hypoxia-ischemia changes during brain development. Neuroscience. 2004;127:113–123. doi: 10.1016/j.neuroscience.2004.03.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang J, Dawson VL, Dawson TM, Snyder SH. Nitric oxide activation of poly(ADP-ribose) synthetase in neurotoxicity. Science. 1994;263:687–689. doi: 10.1126/science.8080500. [DOI] [PubMed] [Google Scholar]
- 37.Beckman JS. The double-edged role of nitric oxide in brain function and superoxide-mediated injury. J Dev Physiol. 1991;15:53–59. [PubMed] [Google Scholar]
- 38.Amé JC, Jacobson EL, Jacobson MK. ADP-ribose polymer metabolism. Oxford University Press; New York: 2000. [Google Scholar]
- 39.Lazebnik YA, Kaufmann SH, Desnoyers S, Poirier GG, Earnshaw WC. Cleavage of poly(ADP-ribose) polymerase by a proteinase with properties like ICE. Nature. 1994;371:346–347. doi: 10.1038/371346a0. [DOI] [PubMed] [Google Scholar]
- 40.Yap E, Tan WL, Ng I, Ng YK. Combinatorial-approached neuroprotection using pan-caspase inhibitor and poly (ADP-ribose) polymerase (PARP) inhibitor following experimental stroke in rats; is there additional benefit? Brain Res. 2008;1195:130–138. doi: 10.1016/j.brainres.2007.12.024. [DOI] [PubMed] [Google Scholar]
- 41.Yu SW, Wang H, Poitras MF, et al. Mediation of poly(ADP-ribose) polymerase-1-dependent cell death by apoptosis-inducing factor. Science. 2002;297:259–263. doi: 10.1126/science.1072221. [DOI] [PubMed] [Google Scholar]
- 42.Eliasson MJ, Sampei K, Mandir AS, et al. Poly(ADP-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat Med. 1997;3:1089–1095. doi: 10.1038/nm1097-1089. [DOI] [PubMed] [Google Scholar]
- 43.Endres M, Wang ZQ, Namura S, Waeber C, Moskowitz MA. Ischemic brain injury is mediated by the activation of poly(ADP-ribose)polymerase. J Cereb Blood Flow Metab. 1997;17:1143–1151. doi: 10.1097/00004647-199711000-00002. [DOI] [PubMed] [Google Scholar]
- 44.McCullough LD, Zeng Z, Blizzard KK, Debchoudhury I, Hurn PD. Ischemic nitric oxide and poly (ADP-ribose) polymerase-1 in cerebral ischemia: male toxicity, female protection. J Cereb Blood Flow Metab. 2005;25:502–512. doi: 10.1038/sj.jcbfm.9600059. [DOI] [PubMed] [Google Scholar]
- 45.Krishnakumar R, Gamble MJ, Frizzell KM, Berrocal JG, Kininis M, Kraus WL. Reciprocal binding of PARP-1 and histone H1 at promoters specifies transcriptional outcomes. Science. 2008;319:819–821. doi: 10.1126/science.1149250. [DOI] [PubMed] [Google Scholar]
- 46.Ju BG, Lunyak VV, Perissi V, et al. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science. 2006;312:1798–1802. doi: 10.1126/science.1127196. [DOI] [PubMed] [Google Scholar]
- 47.Xu K, Tavernarakis N, Driscoll M. Necrotic cell death in C. elegans requires the function of calreticulin and regulators of Ca(2+) release from the endoplasmic reticulum. Neuron. 2001;31:957–971. doi: 10.1016/s0896-6273(01)00432-9. [DOI] [PubMed] [Google Scholar]
- 48.Yamashima T, Saido TC, Takita M, et al. Transient brain ischaemia provokes Ca2+, PIP2 and calpain responses prior to delayed neuronal death in monkeys. Eur J Neurosci. 1996;8:1932–1944. doi: 10.1111/j.1460-9568.1996.tb01337.x. [DOI] [PubMed] [Google Scholar]
- 49.Yamashima T. Ca2+-dependent proteases in ischemic neuronal death: a conserved ‘calpain-cathepsin cascade’ from nematodes to primates. Cell Calcium. 2004;36:285–293. doi: 10.1016/j.ceca.2004.03.001. [DOI] [PubMed] [Google Scholar]
- 50.Hong SC, Goto Y, Lanzino G, Soleau S, Kassell NF, Lee KS. Neuroprotection with a calpain inhibitor in a model of focal cerebral ischemia. Stroke. 1994;25:663–669. doi: 10.1161/01.str.25.3.663. [DOI] [PubMed] [Google Scholar]
- 51.Degterev A, Huang Z, Boyce M, et al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol. 2005;1:112–119. doi: 10.1038/nchembio711. [DOI] [PubMed] [Google Scholar]
- 52.Degterev A, Hitomi J, Germscheid M, et al. Identification of RIP1 kinase as a specific cellular target of necrostatins. Nat Chem Biol. 2008;4:313–321. doi: 10.1038/nchembio.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holler N, Zaru R, Micheau O, et al. Fas triggers an alternative, caspase-8-independent cell death pathway using the kinase RIP as effector molecule. Nat Immunol. 2000;1:489–495. doi: 10.1038/82732. [DOI] [PubMed] [Google Scholar]
- 54.Martin-Villalba A, Hahne M, Kleber S, et al. Therapeutic neutralization of CD95-ligand and TNF attenuates brain damage in stroke. Cell Death Differ. 2001;8:679–686. doi: 10.1038/sj.cdd.4400882. [DOI] [PubMed] [Google Scholar]
- 55.Teng X, Degterev A, Jagtap P, et al. Structure-activity relationship study of novel necroptosis inhibitors. Bioorg Med Chem Lett. 2005;15:5039–5044. doi: 10.1016/j.bmcl.2005.07.077. [DOI] [PubMed] [Google Scholar]
- 56.Wang K, Li J, Degterev A, Hsu E, Yuan J, Yuan C. Structure-activity relationship analysis of a novel necroptosis inhibitor, Necrostatin-5. Bioorg Med Chem Lett. 2007;17:1455–1465. doi: 10.1016/j.bmcl.2006.11.056. [DOI] [PubMed] [Google Scholar]
- 57.You Z, Savitz S, Yang J, Degterev A, Yuan J, Cuny GD, Moskowitz MA, Whalen MJ. Necrostatin-1 Reduces Histopathology and Improves Functional Outcome After Controlled Cortical Impact In Mice. J Cereb Blood Flow Metab. 2008 doi: 10.1038/jcbfm.2008.44. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lim SY, Davidson SM, Mocanu MM, Yellon DM, Smith CC. The cardioprotective effect of necrostatin requires the cyclophilin-D component of the mitochondrial permeability transition pore. Cardiovasc Drugs Ther. 2007;21:467–469. doi: 10.1007/s10557-007-6067-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Hitomi J, Christofferson DE, Ng A, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell. 2008 doi: 10.1016/j.cell.2008.10.044. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Schreiber SL. Chemical genetics resulting from a passion for synthetic organic chemistry. Bioorg Med Chem. 1998;6:1127–1152. doi: 10.1016/s0968-0896(98)00126-6. [DOI] [PubMed] [Google Scholar]