

Abstract
Background
Electroporation is a versatile method for in vitro or in vivo delivery of different molecules into cells. However, no study so far has analysed the effects of electric pulses used in electrochemotherapy (ECT pulses) or electric pulses used in electrogene therapy (EGT pulses) on malignant cells. We studied the effect of ECT and EGT pulses on human malignant melanoma cells in vitro in order to understand and predict the possible effect of electric pulses on gene expression and their possible effect on cell behaviour.
Methods
We used microarrays with 2698 different oligonucleotides to obtain the expression profile of genes involved in apoptosis and cancer development in a malignant melanoma cell line (SK-MEL28) exposed to ECT pulses and EGT pulses.
Results
Cells exposed to ECT pulses showed a 68.8% average survival rate, while cells exposed to EGT pulses showed a 31.4% average survival rate. Only seven common genes were found differentially expressed in cells 16 h after exposure to ECT and EGT pulses. We found that ECT and EGT pulses induce an HSP70 stress response mechanism, repress histone protein H4, a major protein involved in chromatin assembly, and down-regulate components involved in protein synthesis.
Conclusion
Our results show that electroporation does not significantly change the expression profile of major tumour suppressor genes or oncogenes of the cell cycle. Moreover, electroporation also does not changes the expression of genes involved in the stability of DNA, supporting current evidence that electroporation is a safe method that does not promote tumorigenesis. However, in spite of being considered an isothermal method, it does to some extent induce stress, which resulted in the expression of the environmental stress response mechanism, HSP70.
Background
Electroporation, as a physical method for the delivery of molecules into cells, was developed in 1982 [1]. However, since then it has been developed not only for in vitro use but also for in vivo use in a variety of applications [2]. Electroporation is of interest as a gene delivery method because, unlike transduction with viruses, it eliminates the risks and limitations linked to the use of viruses. In addition, in spite of extensive research, efficient and safe chemical vectors have not yet been developed for in vivo gene delivery [3]. Using appropriate electrical parameters, destabilization of the membrane is reversible, ensuring a high survival of permeabilized cells and the delivery of non-permeant molecules inside the cell, bypassing the normal internalisation route of these molecules [4].
The advantages of electroporation have recently been used by different groups for a novel approach to introducing chemotherapeutics in a variety of tumours, called electrochemotherapy [5-7]. Electrochemotherapy facilitates chemotherapeutic drug delivery into cells by increasing cell membrane permeability under specific electric pulses [4]. It is an effective local treatment for patients with cutaneous and subcutaneous tumour nodules, on the basis of the synergistic association of locally applied electric pulses and low permeant chemotherapeutics such as bleomycin and cisplatin. Moreover, several clinical trials with the same chemotherapeutics showed a good response of melanoma tumour nodules, as well as of other tumour types [5,6,8-10].
As mentioned earlier, electrochemotherapy is not the only application of electroporation. There are an increasing number of applications in which electroporation might be used. Electroporation is frequently used as a method of in vitro transfection of genetic materials into prokaryotic or eukaryotic cells. With the development of electric pulse generators, the method has also been used in vivo for naked DNA transfection in various rodent tissues, in order to treat various diseases and for vaccination [11-13]. The first clinical trial has also been reported for the treatment of melanoma nodules in patients with plasmid DNA encoding interleukin-12 [14].
The effect of electroporation on the level of cell genetic response has only been studied in muscle cells [15,16]. However, the effect of ECT and EGT pulses on malignant cells have not yet been analysed. In the present work, therefore, we studied the effect of ECT and EGT pulses on human malignant melanoma cells in vitro, in order to understand and predict the possible effect of electric pulses on gene expression and their possible effect on cell behaviour.
Methods
Cell line
Human malignant melanoma cells SK-MEL28 (HBT-72; American Type Culture Collection, USA) were grown as a monolayer in minimum essential medium (MEM) with Glutamax (Gibco, Paisley, UK), supplemented with 10% fetal bovine serum (FBS; Gibco) and gentamicin (30 μg/mL) (Gibco). Cells were routinely subcultured twice a week and incubated in an atmosphere with 5% CO2 at 37°C.
Electroporation protocol
Confluent cell cultures were trypsinized, washed in MEM with FBS for trypsin inactivation and once in electroporation buffer (125 mM saccharose; 10 mM K2HPO4; 2.5 mM KH2PO4; 2 mM MgCl2·6H2O) at 4°C. The final cell suspension was prepared in electroporation buffer at 4°C, at a concentration of 22 × 106 cells/mL. Aliquots of the final cell suspension (3 × 106 cells) were placed between two parallel electrodes with a 2 mm gap and subjected to eight electric pulses for ECT pulses (electric field intensity 1300 V/cm, pulse duration 100 μs and frequency 1 Hz) or eight electric pulses for EGT pulses (electric field intensity 600 V/cm, pulse duration 5 ms and frequency 1 Hz). Electric pulses were generated by a GT-1 electroporator (Faculty of Electrical Engineering, Ljubljana, Slovenia). One aliquot of cell suspension was not subjected to any electric pulses and served as the control treatment. After electroporation, cells were incubated at room temperature for 5 minutes, diluted in MEM with FBS and then plated in culture flasks for 16 h for microarray assay.
Cell survival after electroporation
Clonogenic assay was used to determine cell survival after electroporation. After exposure to ECT and EGT pulses, SK-MEL28 were plated at a concentration of 500 cells/dish. After 16 days, colonies were fixed, stained with crystal violet and counted. The plating efficiency and the surviving fraction were calculated. The experiments were performed in triplicate and repeated three times.
RNA extraction
RNA from cells was isolated using TRI REAGENT™ (Sigma Aldrich, St. Louis, USA) and the PureLink™ Micro-to-Midi Total RNA Purification System (Invitrogen, Carlsbad, USA), according to the manufacturer's instructions. Briefly, 16 hours after electroporation, cells were trypsinized, washed in MEM with FBS for trypsin inactivation and resuspended in PBS. After centrifugation at 1500 × g for 5 min, all excess liquid was removed and 1 mL of TRI REAGENT™ was added to each sample. Samples were mixed by hand for 15 s and allowed to stand for 2 – 15 min at room temperature. The resulting mixture was centrifuged at 12000 × g for 15 min at 4°C. The aqueous phase was transferred to a fresh microcentrifuge tube and an equal amount of 70% ethanol was added. Samples were transferred to a PureLink™ Micro-to-Midi Total RNA Purification System column (Invitrogen) and processed according to the manufacturer's protocol. All samples were washed from the column with 75 μl of RNAse free water.
Analysis of RNA
The quality of RNA was checked on a Bioanalyzer 2100 (Agilent, Santa Clara, USA) using RNA 6000 Nano Labchip (Agilent, Santa Clara, USA) and 6000 RNA ladder as reference (Ambion, Austin, USA). The concentration and quantity of RNA were determined with ND-1000 (Nanodrop, Wilmington, USA).
Preparation of aaRNA
Preparation of aaRNA was performed with an Amino Allyl MessageAmp™ II aRNA Amplification Kit (Ambion) according to the manufacturer's recommendations. For each hybridization, we labelled 5 μg of non-exposed cells (Cy3) and 5 μg of cells exposed to either ECT or EGT pulses (Cy5) mRNA. After removing the excess dye, the RNAs were dissolved in Nexterion Hybridization solution (Schott Nexterion, Jena, Germany).
Microarrays
Microarrays were prepared with Human Apoptosis Subset v2.0 and Human Cancer Subset v3.0 (Operon, Ebersberg, Germany) 70 mer oligonucleotides and Nexterion 70 mer Oligo Microarraying Kit (Schott Nexterion) slides. A single array contained 2698 different genes, each gene being replicated at least 4 times on each array. Oligonucleotides were spotted using an MG1000 spotter (MicroGrid, Boston, USA), immobilised and stored according to the manufacturer's instructions (Schott Nexterion). All hybridisations were performed on HS400 in duplicate (Tecan, Salzburg, Austria) according to the manufacturer's instructions (Schott Nexterion). We used an LS200 scanner (Tecan) at 6 μm resolution for scanning the microarrays.
Data analysis
We used Array-Pro Analyzer 4.5 (Media Cybernetics, Bethesda, USA) for feature extraction after imaging of microarrays. Acuity (Molecular devices, USA) was used for the filtration of bad signals, LOWESS fit and microarray data analysis. Features showing a signal intensity of more than 65000 were flagged as bad. Features with a signal less than 2 times the intensity of the background or coefficient of variation (CV, ratio between standard deviation of the background and the median feature intensity) greater than 0.3 were considered not significantly expressed and were filtered out. Log2 ratios were normalized using LOWESS fit [17] and the median of four replicates was used to calculate the average gene expression for a single sample. We filtered out genes that were not expressed in all replicate samples at least 1.5 times.
The Gene Ontology Tree Machine [18] program was used for gene enrichment analysis. All other statistical analyses were done using SPSS 16 (SPSS inc., Chicago, USA).
Results
Cell survival after electroporation
After electroporation of cells, cell viability was assessed by clonogenic assay. Using this method, we determined a 68.8% average survival rate for cells exposed to ECT pulses and a 31.4% average survival rate for cells exposed to EGT pulses.
Microarrays
The difference in expression of genes involved in cancer development was obtained by comparison of malignant melanoma cells exposed to EGT or ECT pulses against the same untreated malignant melanoma cells. In our experimental design, microarrays with 2698 different genes were used as a dual colour system in which exposed and non-exposed cells' mRNA were separately labelled, mixed and hybridised together on each array. Only microarrays expressing at least 50% of genes were used for further analysis. All oligonucleotides on the same array were spotted in quadruplicate and each microarray analysis was performed in duplicate, thus obtaining eight measurements of the same oligonucleotide. The acquired data were analysed with Acuity 4.0 to select reliable signals. Only genes, 1266 for ECT pulses and 1805 for EGT pulses, present in both duplicated microarrays were considered for further processing. We next checked the variability of replicate measurements on Operon's microarray platform. The average standard deviation of the Log2 ratio (treated/untreated) of replicates exposed to ECT pulses was 0.21, and 0.17 for replicates exposed to EGT pulses. This gives a standard deviation of 1.16 fold and 1.12 fold from the median value of replicates for ECT pulses and EGT pulses, respectively. Out of 2698 different genes, 7 genes showed differential expression (Table 1), when the groups of ECT and EGT pulses were combined. This represents roughly 0.26% of all genes present on a microarray. However, looking at specifically different pulsation parameters, ECT pulses yielded 34 differentially expressed genes, which is roughly 1% of the interrogated genes, whereas EGT pulses yielded 26 differentially expressed genes, again accounting for roughly 1% of all interrogated genes. When using a cut of value of 2.0, we found only 3 deregulated genes in the treatment with ECT pulses and EGT pulses (Tables 2 and 3).
Table 1.
Genes differentially expressed in both ECT and EGT.
| Name | RefSeq | Description | Median Log2 ratio |
|---|---|---|---|
| Down | |||
| RPL31 | NM_000993 | RIBOSOMAL PROTEIN L31 | -1.42 |
| CD28 | NM_006139 | T CELL SPECIFIC SURFACE GLYCOPROTEIN | -0.78 |
| H4FN | NM_175054 | HISTONE H4 | -0.72 |
| Up | |||
| NM_014486 | NEURONAL THREAD PROTEIN | 0.58 | |
| HSPA1B | NM_005346 | HEAT SHOCK 70 KDA PROTEIN 1 | 0.83 |
| CDC25C | NM_001790 | M PHASE INDUCER PHOSPHATASE 3 | 0.70 |
| CCNF | NM_001761 | G2/MITOTIC SPECIFIC CYCLIN F | 0.79 |
Table 2.
Genes differentially expressed using ECT.
| Name | RefSeq | Description | Median Log2 ratio |
|---|---|---|---|
| Down | |||
| RPL31 | NM_000993 | RIBOSOMAL PROTEIN L31 | -1.52 |
| RPS17 | NM_001021 | 40S RIBOSOMAL PROTEIN S17 | -1.09 |
| TBCA | NM_004607 | TUBULIN SPECIFIC CHAPERONE A | -1.08 |
| PPIA | NM_021130 | PEPTIDYLPROLYL CISTRANS ISOMERASE A | -0.97 |
| S100B | NM_006272 | S100 PROTEIN, BETA CHAIN | -0.94 |
| NM_006471 | MYOSIN REGULATORY LIGHT CHAIN 2 | -0.93 | |
| RPA3 | NM_002947 | REPLICATION PROTEIN A 14 KDA SUBUNIT | -0.89 |
| NQO1 | NM_000903 | NAD(P)H DEHYDROGENASE [QUINONE] 1 | -0.88 |
| RPS6 | NM_001010 | 40S RIBOSOMAL PROTEIN S6 | -0.81 |
| H4FN | NM_175054 | HISTONE H4 | -0.80 |
| EEF1A1 | NM_001402 | ELONGATION FACTOR 1 ALPHA 1 | -0.77 |
| ITGB4 | NM_000213 | INTEGRIN BETA4 PRECURSOR | -0.76 |
| CD28 | NM_006139 | T CELL SPECIFIC SURFACE GLYCOPROTEIN CD28 | -0.75 |
| H3F3A | NM_002107 | HISTONE H3.3 | -0.74 |
| CASP9 | NM_001229 | CASPASE 9 PRECURSOR | -0.73 |
| TNFRSF14 | NM_003820 | TUMOR NECROSIS FACTOR RECEPTOR SUPERFAMILY MEMBER 14 | -0.65 |
| CGB5 | NM_033142 | CHORIOGONADOTROPIN BETA CHAIN PRECURSOR | -0.64 |
| RPH3AL | NM_006987 | RABPHILIN 3 ALIKE | -0.61 |
| TFDP1 | NM_007111 | TRANSCRIPTION FACTOR DP-1 | -0.60 |
| CST3 | NM_000099 | CYSTATIN C PRECURSOR | -0.59 |
| Up | |||
| RB1 | NM_000321 | RETINOBLASTOMA 1 | 0.55 |
| RIN2 | NM_018993 | RAS ASSOCIATION (RALGDS/AF6) DOMAIN CONTAINING PROTEIN | 0.56 |
| DNAJB1 | NM_006145 | DNAJ HOMOLOG SUBFAMILY B MEMBER 1 | 0.56 |
| MATR3 | NM_018834 | MATRIN 3 | 0.57 |
| HOXA4 | NM_002141 | HOMEOBOX PROTEIN HOXA4 | 0.65 |
| RBBP4 | NM_005610 | CHROMATIN ASSEMBLY FACTOR 1 SUBUNIT C | 0.70 |
| CDC25C | NM_001790 | M PHASE INDUCER PHOSPHATASE 3 | 0.70 |
| NM_014486 | NEURONAL THREAD PROTEIN | 0.73 | |
| GLIPR1 | NM_006851 | GLIOMA PATHOGENESIS RELATED PROTEIN | 0.75 |
| CRABP2 | NM_001878 | RETINOIC ACID BINDING PROTEIN II | 0.77 |
| RBL2 | NM_005611 | RETINOBLASTOMA LIKE PROTEIN 2 | 0.78 |
| CCNF | NM_001761 | G2/MITOTIC SPECIFIC CYCLIN F | 0.79 |
| HSPA1B | NM_005346 | HEAT SHOCK 70 KDA PROTEIN 1 | 0.89 |
| IL6 | NM_000600 | INTERLEUKIN 6 PRECURSOR (IL6) | 0.98 |
Table 3.
Genes differentially expressed using EGT.
| Name | RefSeq | Description | Median Log2 ratio |
|---|---|---|---|
| Down | |||
| RET | NM_020975 | PROTOONCOGENE TYROSINEPROTEIN KINASE RECEPTOR | -1.364 |
| RPL31 | NM_000993 | RIBOSOMAL PROTEIN L31 | -1.218 |
| PC | NM_022172 | PYRUVATE CARBOXYLASE | -1.009 |
| NM_006590 | SNRNP ASSEMBLY DEFECTIVE 1 HOMOLOG | -0.931 | |
| IGFALS | NM_004970 | INSULIN-LIKE GROWTH FACTOR BINDING PROTEIN | -0.895 |
| CD28 | NM_006139 | T CELL SPECIFIC SURFACE GLYCOPROTEIN | -0.845 |
| CYP2A7 | NM_000764 | CYTOCHROME P450 2A7 | -0.844 |
| POLR2F | NM_021974 | DNA DIRECTED RNA POLYMERASE II | -0.840 |
| NM_007013 | WW DOMAIN CONTAINING PROTEIN 1 | -0.806 | |
| NM_004881 | QUINONE OXIDOREDUCTASE HOMOLOG | -0.795 | |
| TIMP2 | NM_003255 | METALLOPROTEINASE INHIBITOR 2 PRECURSOR | -0.795 |
| NM_005851 | DOC1 RELATED PROTEIN (DOC1R) | -0.753 | |
| H4FN | NM_175054 | HISTONE H4 | -0.648 |
| FGFR1 | NM_023111 | BASIC FIBROBLAST GROWTH FACTOR RECEPTOR 1 PRECURSOR | -0.599 |
| LIG3 | NM_013975 | DNA LIGASE III | -0.582 |
| SKP2 | NM_032637 | S PHASE KINASE ASSOCIATED PROTEIN 2 | -0.550 |
| MUC4 | NM_004532 | MUCIN 4, ISOFORM D | -0.549 |
| PSMB7 | NM_002799 | PROTEASOME SUBUNIT BETA TYPE 7 PRECURSOR | -0.544 |
| PTPN21 | NM_007039 | PROTEIN TYROSINE PHOSPHATASE | -0.539 |
| SULT1C1 | NM_001056 | SULFOTRANSFERASE | -0.520 |
| HSPB2 | NM_001541 | HEATSHOCK PROTEIN, BETA2 | -0.519 |
| Up | |||
| NM_014486 | NEURONAL THREAD PROTEIN | 0.575 | |
| HSPA1B | NM_005346 | HEAT SHOCK 70 KDA PROTEIN 1 | 0.747 |
| CDC25C | NM_001790 | MPHASE INDUCER PHOSPHATASE 3 | 0.759 |
| TTYH1 | NM_020659 | TWEETY HOMOLOG 1 | 0.775 |
| CCNF | NM_001761 | G2/MITOTICSPECIFIC CYCLIN F | 0.777 |
To find the possible biological processes involved in the response to the electroporation procedure, we used the Gene Ontology Tree Machine [18] program for gene enrichment analysis. Our original dataset of genes was compared against differentially expressed genes given in Tables 2 and 3 to check whether there was any significant gene enrichment in comparison to the original gene set. Interestingly, we found significant enrichment of down-regulated genes involved in biosynthesis, regulation of viral genome replication and viral genome replication and significant enrichment of deregulated genes involved in cytokine production in melanoma cells exposed to ECT pulses (Figure 1). Deregulated genes involved in cell division, response to unfolded proteins and response to protein stimulus, were enriched in melanoma cells exposed to EGT pulses (Figure 2).
Figure 1.
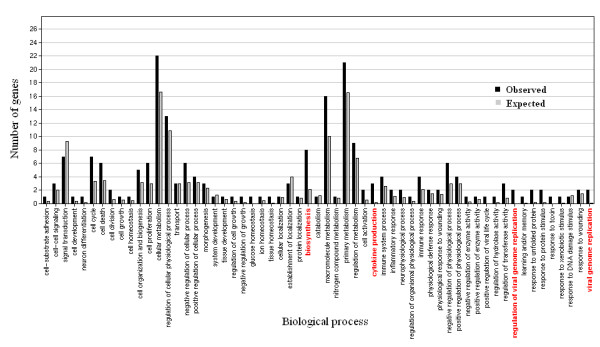
Gene enrichment analysis for cell line exposed to ECT pulses. Expected – number of genes expected to be differentially expressed. Observed – number of genes differentially expressed. In red are GO biological functions significantly enriched in the cell line exposed to ECT pulses.
Figure 2.
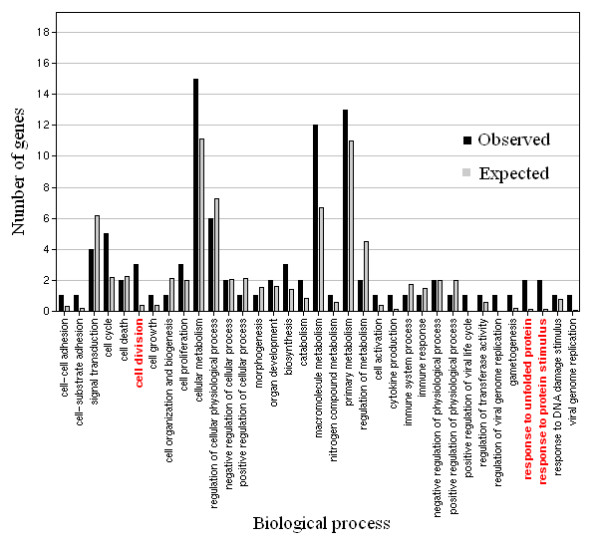
Gene enrichment analysis for cell line exposed to EGT pulses. Expected – number of genes expected to be differentially expressed. Observed – number of genes differentially expressed. In red are GO biological functions significantly enriched in the cell line exposed to EGT pulses.
In order to enable other users comprehensively to interpret and evaluate our results, the original tables of complete microarray results are available in the supplementary data (see the GEO website at http://www.ncbi.nlm.nih.gov/projects/geo Series entry: GSE15420).
Discussion
In this study, we analysed the expression profile of malignant melanoma cells for genes known to be involved in the development of cancer. This was done in order to assess whether electroporation could lead to an altered expression profile of cells, possibly making them more detrimental to patients. Our results show only minor differences in the expression of genes involved in cancer development. Overall, microarrays showed differential expression of only 7 genes, when using a threshold value of 1.5 fold and only 1 gene when using a threshold value of 2.0 fold (Table 1). When calculating the standard deviation of measurements across the microarray, we found it to be very low (1.16 and 1.12 fold for ECT and EGT pulses replicates) showing that the 1.5 fold threshold change is a reasonable and reliable cut-off value. The results obtained are also in agreement with studies performed so far [15,16]. However, these studies used mouse muscle cells to account for any damage to tissue or difference in expression profile made by electroporation for immunization purposes. Hojman et al. observed only minor histological changes and no changes in muscle performance or the gene expression profile of genes involved in cell death, inflammation or muscle regeneration [15]. Similar results were also obtained by Rubenstrunk et al. when they used Stress/Toxicology Atlas cDNA expression arrays. The group found only 2 genes out of 140 to be differentially expressed and concluded that electroporation does not induce expression of genes involved in stress and toxic response [16]. Therefore, despite the fact that only single cell line was used in our study, it is reasonable to expect that other cell lines would behave in similar way.
Interestingly, one of the seven differentially expressed genes is the stress response gene HSPA1B. HSPA1B is a member of the HSPA family of HSP70 proteins and is the strongest stress inducible member of the HSPA family [19]. It has been proposed that HSPA1B is a part of the molecular chaperon network that protects the proteome against environmental stress [20]. This shows that cells exposed to either of the electroporation protocols are exposed to the stress arising to some extent from a protein denaturation, and therefore over-express HSP70. This observation is also supported by the overexpression of DNAJB1 in cells treated with ECT pulses. DNAJB1, containing a conserved sequence motif (HPD) in the J domain is known to be critical for the acceleration of the ATPase activity of HSP70 [19].
Another interesting observation was downregulation of the histone protein H4 in both treatment protocols and a significant enrichment of downregulated genes involved in protein synthesis. Both results indicate a stall in DNA assembly to chromosomes and biosynthesis of proteins, which could arise from stress.
Conclusion
Overall, our results clearly show that electroporation does not significantly changes the expression profile of major tumour suppressor genes or oncogenes of the cell cycle. Electroporation also does not change the expression of genes involved in the stability of DNA, therefore supporting the notion that electroporation is a safe method that does not promote tumorigenesis. However, in the present study, we showed that to some extent electroporation induces HSP70, resulting in the activation of the environmental stress response mechanism.
Competing interests
The authors declare that they have no competing interests.
Authors' contributions
VM carried out the isolation of RNA, microarray experiments, data analysis and drafted the manuscript. VT and MC performed cultivation of cells, electroporation and cell survival. VT also helped to draft the manuscript. MC, DG and GS conceived the study and participated in its design and coordination and critically revised the draft. All authors read and approved the final manuscript.
Pre-publication history
The pre-publication history for this paper can be accessed here:
Contributor Information
Vid Mlakar, Email: vid.mlakar@mf.uni-lj.si.
Vesna Todorovic, Email: vesna.todorovic@vszi.upr.si.
Maja Cemazar, Email: mcemazar@onko-i.si.
Damjan Glavac, Email: damjan.glavac@mf.uni-lj.si.
Gregor Sersa, Email: gsersa@onko-i.si.
Acknowledgements
This work was supported by Slovenian Research Agency in program P3-0003 and P3-054.
References
- Neuman E, Schaefer-Ridder M, Wang Y, Hofschneider PH. Gene transfer in mouse lyoma cells by electroporation in high electric fields. EMBO J. 1982;1:841–845. doi: 10.1002/j.1460-2075.1982.tb01257.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehl J. Electroporation: theory and methods, perspectives for drug delivery, gene therapy and research. Acta Physiol Scand. 2003;177:437–47. doi: 10.1046/j.1365-201X.2003.01093.x. [DOI] [PubMed] [Google Scholar]
- Barteau B, Chèvre R, Letrou-Bonneval E, Labas R, Lambert O, Pitard B. Physicochemical parameters of non-viral vectors that govern transfection efficiency. Curr Gene Ther. 2008;8:313–323. doi: 10.2174/156652308786070961. [DOI] [PubMed] [Google Scholar]
- Mir LM, Orlowski S, Belehradek J Jr. Biomedical applications of electric pulses with special emphasis on antitumor electrochemotherapy. Bioelectrochem Bioenerg. 1995;38:203–207. doi: 10.1016/0302-4598(95)01823-W. [DOI] [Google Scholar]
- Heller R, Jaroszeski M, Glass F, Messina JL, Rapaport DP, DeConti RC, Fenske NA, Gilbert RA, Mir LM, Reintgen DS. Phase I/II trial for the treatment of cutaneous and subcutaneous tumors using electrochemotherapy. Cancer. 1996;77:964–971. doi: 10.1002/(SICI)1097-0142(19960301)77:5<964::AID-CNCR24>3.0.CO;2-0. [DOI] [PubMed] [Google Scholar]
- Marty M, Serša G, garbay JR, Gehl J, Collins CG, Snoj M, Billard V, Geertsen PF, Larkin JO, Miklavčič D, Pavlović I, Paulin-Kosir SM, Čemažar M, Morsli N, Soden DM, Rudolf Z, Robert C, O'Sullivan GC, Mir LM. Electrochemotherapy – An easy, highly effective and safe treatment of cutaneous and subcutaneous metastases: Results of ESOPE (European Standard Operating Procedures of Electrochemotherapy) study. EJC Suppl. 2006;4:3–13. [Google Scholar]
- Sersa G, Miklavcic D, Cemazar M, Rudolf Z, Pucihar G, Snoj M. Electrochemotherapy in treatment of tumours. Eur J Surg Oncol. 2008;34:232–240. doi: 10.1016/j.ejso.2007.05.016. [DOI] [PubMed] [Google Scholar]
- Quaglino P, Mortera C, Osella-Abate S, Barberis M, Illengo M, Rissone M, Savoia P, Bernengo MG. Electrochemotherapy with intravenous bleomycin in the local treatment of skin melanoma metastases. Ann Surg Oncol. 2008;15:2215–2222. doi: 10.1245/s10434-008-9976-0. [DOI] [PubMed] [Google Scholar]
- Campana LG, Mocellin S, Basso M, Puccetti O, De Salvo GL, Chiarion-Sileni V, Vecchiato A, Corti L, Rossi CR, Nitti D. Bleomycin-based electrochemotherapy: clinical outcome from a single institution's experience with 52 patients. Ann Surg Oncol. 2009;16:191–199. doi: 10.1245/s10434-008-0204-8. [DOI] [PubMed] [Google Scholar]
- Sersa G, Cemazar M, Miklavcic D, Rudolf Z. Electrochemotherapy of tumours. Radiol Oncol. 2006;40:163–174. [Google Scholar]
- Heller LC, Heller R. In vivo electroporation for gene therapy. Hum Gene Ther. 2006;17:890–897. doi: 10.1089/hum.2006.17.890. [DOI] [PubMed] [Google Scholar]
- Cemazar M, Sersa G. Electrotransfer of therapeutic molecules into tissues. Curr Opin Mol Ther. 2007;9:554–562. [PubMed] [Google Scholar]
- Bodles-Brakhop AM, Heller R, Draghia-Akli R. Electroporation for the Delivery of DNA-based Vaccines and Immunotherapeutics: Current Clinical Developments. Mol Ther. 2009;17:585–592. doi: 10.1038/mt.2009.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daud AI, DeConti RC, Andrews S, Urbas P, Riker AI, Sondak VK, Munster PN, Sullivan DM, Ugen KE, Messina JL, Heller R. Phase I trial of interleukin-12 plasmid electroporation in patients with metastatic melanoma. J Clin Oncol. 2008;26:5896–5903. doi: 10.1200/JCO.2007.13.9048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hojman P, Zibert JR, Gissel H, Eriksen J, Gehl J. Gene expression profiles in skeletal muscle after gene electrotransfer. BMC Mol Biol. 2007;8:56. doi: 10.1186/1471-2199-8-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubenstrunk A, Mahfouldi A, Scherman D. Delivery of electric pulses for DNA electrotransfer to mouse muscle does not induce the expression of stress related genes. Cell Biol Toxicol. 2004;20:25–31. doi: 10.1023/B:CBTO.0000021079.69089.4f. [DOI] [PubMed] [Google Scholar]
- Cleveland WS, Devlin SJ. Locally weighted regression: an approach to regresion analysis by local fitting. J Am Stat Assoc. 1988;83:596–610. doi: 10.2307/2289282. [DOI] [Google Scholar]
- Zhang B, Schmoyer D, Kirov S, Snoddy J. GOTree Machine (GOTM): a web-based platform for interpreting sets of interesting genes using Gene Ontology hierarchies. BMC Bioinformatics. 2004;5:16. doi: 10.1186/1471-2105-5-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vos MJ, Hageman J, Carra S, Kampinga HH. Structural and functional diversities between members of the human HSPB, HSPA, and DNAJ chaperone families. Biochemistry. 2008;47:7001–7011. doi: 10.1021/bi800639z. [DOI] [PubMed] [Google Scholar]
- Albanese V, Yam AY-W, Baughman J, Parnot C, Frydman J. System analyses reveal two chaperone networks with distinct functions in eukaryotic cells. Cell. 2006;124:75–88. doi: 10.1016/j.cell.2005.11.039. [DOI] [PubMed] [Google Scholar]