

Abstract
X-linked lymphoproliferative disease (XLP) is an immunodeficiency caused by defects in the adaptor molecule SAP. The manifestations of XLP generally occur following Epstein-Barr virus (EBV) infection and include fulminant mononucleosis, hypogammaglobulinemia and lymphoma. In this report, we describe two unrelated patients with fatal T cell-mediated central nervous system vasculitis for whom repeated serologic and molecular testing for EBV was negative. In both patients, clonal T cell populations were observed, but neither demonstrated evidence of lymphoma. Thus, loss of SAP function can lead to dysregulated immune responses characterized by the uncontrolled expansion and activation of T cells independent of EBV infection.
Keywords: X-linked lymphoproliferative disease, central nervous system, vasculitis
Introduction
X-linked lymphoproliferative disease (XLP) is a rare immunodeficiency characterized by abnormal immune responses following primary Epstein-Barr virus (EBV) infection. This disorder is caused in most patients by mutations in the SH2D1A gene, which encodes the adaptor molecule Signaling Lymphocytic Activation Molecule-associated protein (SAP). Lymphocytic vasculitis has been rarely observed in XLP; however, most reported patients have a history of ongoing or prior EBV infection[1-5]. Here we describe two unrelated XLP patients who developed an aggressive vasculitis involving the central nervous system (CNS). Interestingly, neither patient had evidence of EBV infection at presentation or during progression of disease. In both patients, clonal but non-malignant T cell populations were present within affected tissues. These findings indicate that even in the absence of detectable EBV infection, SAP-deficient CD8+ lymphocytes can be aberrantly activated following certain antigenic triggers, leading to their expansion, accumulation within, and destruction of vessels of the CNS.
Materials and Methods
Patient 1
Patient No.1 was an 18-year old male who presented with headache and blurred vision one month after receiving the meningococcal vaccine. MRI of the brain showed bitemporal lesions with enhancement and hemorrhage consistent with inflammatory vasculitis (Figures 1A and B). Brain biopsy revealed vascular infiltration by CD8+ T lymphocytes with necrosis of vessel walls. In situ hybridization for EBER was negative. Quantitative EBV polymerase chain reaction (PCR) analyses of brain, cerebrospinal fluid (CSF), blood and bone marrow (BM) were negative. Serum EBV serologies indicated that the patient was EBV-naïve. Studies for Herpes Simplex Virus (HSV), Varicella Zoster Virus (VZV), Cytomegalovirus (CMV), enterovirus and Human Immunodeficiency Virus (HIV) were negative. Immunologic workup revealed a normal IgG level, but elevated IgM and reduced IgA. Mutational analysis revealed an SH2D1A C163T substitution, introducing a stop codon at arginine 55.
Figure 1.
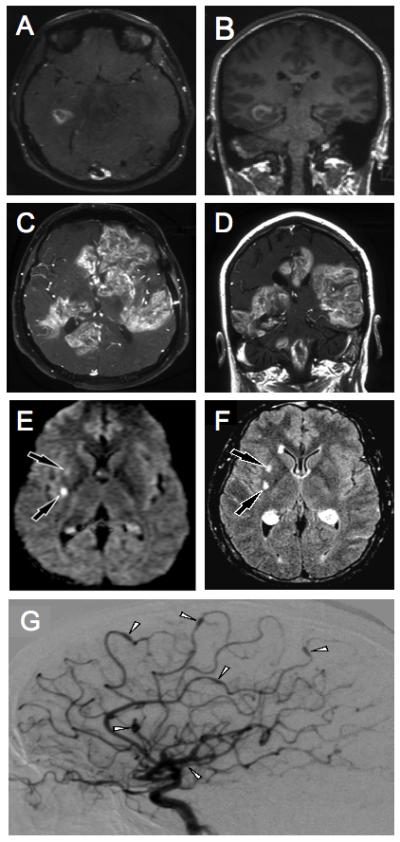
Radiologic findings demonstrating vasculitis in the described patients. Brain MRI findings of Patient No. 1 at presentation (A) shows abnormal signal intensity and enhancement in the right temporal lobe on axial T1 post contrast imaging. The coronal T1 post contrast image (B) shows the abnormality to be inferior and medial in the right temporal lobe. Eleven months later, axial T1 post contrast images (C, D) show multiple areas of contrast enhancement in the cerebral hemispheres, corpus callousum, right cerebellum and the brain stem. Brain MRI of Patient No. 2 at presentation demonstrates hyperintensities on diffusion and FLAIR weighted images in the right basal ganglia consistent with acute small vessel lacunar infarcts (E, F). Two weeks later, a catheter angiogram of the right internal carotid (G) was performed which showed multiple sites of ectasia and aneurysmal dilation (white arrowheads) representing vasculitis.
The patient’s disease progressed (Figures 1C and D) and he expired eleven months after presentation, despite treatment with intravenous methylprednisolone, immunoglobulin, dexamethasone, methotrexate, rituximab and alemtuzumab; oral etoposide, sirolimus and cyclophosphamide; and cranial radiation. At autopsy, evaluation of the brain revealed invasion of vessels by CD3+, CD8+, CD4- lymphocytes with necrosis of vessel walls and brain parenchyma (Figures 2A-F). There was no vasculitis outside of the CNS. TCR γ chain gene rearrangement of involved tissues from the brain revealed a clonal T cell population; however, there was no morphologic evidence of lymphoma. Affected material was negative for EBV by EBER staining and EBV PCR.
Figure 2.
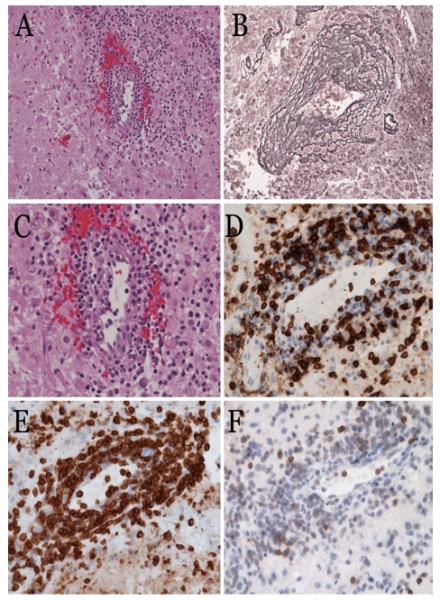
Histopathologic evaluation of vasculitic tissue from the brain. H & E stained sections of autopsy material from the brain of Patient No. 1 demonstrate perivascular lymphohistiocytic infiltrates (A). The lymphocytes are mostly small and round, with benign appearance; scattered cells are medium to large sized (C). Reticulin staining highlights the mononuclear infiltrate within vessel walls and adventitia in a “tree ring” fashion (B). The inflammatory infiltrate is composed of predominantly CD8+ (D), and CD3+ (E) T-lymphocytes. The T-cells show aberrantly decreased or loss of CD5 staining (F).
Patient 2
Patient No. 2 was a 31-year old male who presented with headaches and fevers. Past medical history revealed an upper respiratory infection complicated by jaundice, anemia and fever four years prior to presentation. He was diagnosed with hereditary spherocytosis based on osmotic fragility testing and underwent splenectomy with resolution of symptoms. Splenic pathology showed hemophagocytosis and eosinophilia in the red pulp with absence of germinal centers. A diagnosis of XLP was made one year later when a maternal uncle developed EBV-related non-Hodgkin lymphoma. Genetic testing revealed a C163T SH2D1A mutation.
Three years prior to presentation, the patient developed eosinophilia. Work-up was negative for malignancy or infection. EBV serology and PCR of BM were negative. Immunologic evaluation revealed reduced B cells; however, immunoglobulin levels were normal. Two years later, evaluation of blood showed a clonal TCR γ chain rearrangement with a population of TCRα/β+, CD4+, CD8-, CD25- CD7+, CD5low, CD2low, CD3low/neg cells comprising approximately 30% of lymphocytes. BM evaluation revealed scattered lymphocytic aggregates with presence of clonal cells but no leukemia. EBV PCR of blood and BM were negative.
The patient developed headaches, fever and neurologic deficits. Brain MRI demonstrated changes consistent with acute small vessel lacunar infarcts (Figures 1E and F). MR angiograms did not reveal vasculitis; however, a cerebral angiogram showed diffuse abnormalities and multiple small aneurysms (Figure 1G). CSF culture and PCR were negative for CMV and VZV. Hepatitis A, B, C and HIV serologies were negative. EBV PCR of CSF and blood were negative. The patient was started on high dose methylprednisolone and methotrexate with improvement.
He subsequently developed nodular pulmonary infiltrates and biopsy showed lymphocytic vasculitis with presence of non-malignant CD3+, CD8+, CD4- cells. Although the majority of lymphocytes infiltrating vessel walls were CD8+, the previously identified clonal TCR γ gene rearrangement was found in lung tissue by PCR suggesting the presence of the same CD4+ CD3low/neg cells detected 3 years earlier by flow cytometry. EBV in situ hybridization and PCR of lung tissue were negative. Three months later, the patient expired from a subarachnoid hemorrhage. At autopsy there was marked CD8+ T cell infiltration of small and medium sized vessels of the CNS as well as involvement of vessels in the heart, lungs and liver.
Results and Discussion
CNS vasculitis was first reported in XLP in 1985 with the description of a 12-year old boy who died from a cerebral hemorrhage[4]. At autopsy, CD8+ T lymphocytes, monocytes and plasma cells were found invading arteries of the meninges, right cerebrum, circle of Willis and spinal cord. DNA hybridization confirmed presence of EBV in BM, spleen, thymus and liver; however, it is not stated whether EBV was present within the vascular inflammatory infiltrate in the brain. Since this report, other cases of CNS vasculitis have been described, including patients with and without associated systemic vasculitis, with many reports documenting presence of EBV within infiltrating B lymphocytes or vascular structures[1-3,5,6]. Although the pathogenesis of vasculitis in XLP is poorly understood, it is proposed that vessel damage may result as a bystander effect due to the aberrant activation of CD8+ lymphocytes by EBV+ B cells. Alternatively, vessel walls might themselves become directly infected by EBV due to altered viral clearance. Infected vascular cells then become the target of attack by CD8+ T cells[1].
Two case reports suggest that CNS vasculitis can develop in XLP in the absence of EBV. The first report describes a 22-year old male with a history of mononucleosis, who presented with vasculitis involving the brain, skin and lung. EBV was not found by PCR in blood, and affected tissues from lung and skin were negative for EBV by staining for LMP1, EBNA2 and EBER. While this case suggests that vasculitis may be unrelated to EBV, this patient had a clinical history of EBV infection and vasculitic tissues were not examined for the presence of EBV by PCR[7]. The second case describes a SAP-deficient patient with a CD8+ T cell limbic encephalitis. In situ hybridization of brain and PCR of CSF were negative for EBV but PCR of affected vessels was not performed[8]. Thus, while both cases suggest that vasculitis occurs independently of EBV, affected tissues were not directly examined by PCR, the most sensitive assay to detect presence of viral genomes. The patients described here were tested for EBV repeatedly, and on all occasions, there was no detectable viral DNA by PCR within inflamed vessels. These observations, which confirm that vasculitis can occur in the absence of EBV, are consistent with findings from the XLP Registry[9], which show that 12% of patients develop disease features, such as lymphoma, without evidence of a past or ongoing EBV infection.
Although the antigen specificity of the T cell clones in our patients is not known, it is conceivable that these clonal cells contributed to the vasculitis or other features of disease in our patients. Indeed, T cell clones producing cytokines such as IL-5 are known to cause eosinophilia and the most common phenotype is CD3-, CD4+ [10], such as that observed in Patient No. 2. Thus, the presence of eosinophilia in a patient with XLP should prompt an evaluation for a clonal T cell population, as well as an underlying infection or malignancy.
Patient No. 1 developed disease following meningococcal vaccination. Cases of vasculitis have been described in patients receiving Hepatitis B and influenza vaccines[11-13]; however, an association between the meningococcal vaccine and development of CNS vasculitis has not been reported. Nonetheless, rare reports exist of meningococcal vaccine recipients developing Guillain-Barre syndrome[14] and optic neuritis[15]. The temporal relationship between the administration of vaccines and the onset of immunologic disorders raises the possibility that for genetically predisposed individuals, specific immunizations might trigger development of an autoimmune disorder.
Similar to the cases in this report, most XLP patients in the literature died from their CNS vasculitis [1,4,5,8]. Thus, CNS vasculitis in XLP differs from CNS vasculitis occurring in the setting of other inflammatory conditions such as rheumatoid arthritis[16] or systemic lupus erythematosis[17], where it is possible to achieve remissions through the use of methylprednisone and cyclophosphamide or agents targeting B cells or Tumor Necrosis Factor-α. Given its poor outcome, it is critical that XLP CNS vasculitis be recognized promptly and treated aggressively. For those patients who do not respond to conventional treatments, alternative strategies should be considered, such as myeloabative or reduced intensity allogeneic hematopoietic stem cell transplantation (aHSCT), which would target the activated CD8+ T cells and correct the underlying immune defect. Indeed, aHSCT is curative for patients with XLP[3,18], as well as patients with the related condition hemophagocytic lymphohistiocytosis[19,20], including those with CNS involvement.
Acknowledgements
We thank John Butman MD for assistance in formatting the radiology figures of Patient No. 2. We also thank all of the people who helped to take excellent care of these patients.
Footnotes
Conflict of Interest Statement
The authors have no conflicts of interest to disclose. This work was supported in part by the intramural program of the National Institutes of Health.
References
- 1.Dutz JP, Benoit L, Wang X, et al. Lymphocytic vasculitis in X-linked lymphoproliferative disease. Blood. 2001;97(1):95–100. doi: 10.1182/blood.v97.1.95. see comment. [DOI] [PubMed] [Google Scholar]
- 2.Grossniklaus HE, Aaberg TM, Purnell EW, et al. Retinal necrosis in X-linked lymphoproliferative disease. Ophthalmology. 1994;101(4):705–709. doi: 10.1016/s0161-6420(94)31275-9. [DOI] [PubMed] [Google Scholar]
- 3.Hershberger VS, Hutchins RK, Witte DP, et al. Epstein-Barr virus-related bilateral acute retinal necrosis in a patient with X-linked lymphoproliferative disorder. Archives of Ophthalmology. 2003;121(7):1047–1049. doi: 10.1001/archopht.121.7.1047. [DOI] [PubMed] [Google Scholar]
- 4.Loeffel S, Chang CH, Heyn R, et al. Necrotizing lymphoid vasculitis in X-linked lymphoproliferative syndrome. Archives of Pathology & Laboratory Medicine. 1985;109(6):546–550. [PubMed] [Google Scholar]
- 5.Weeks JK, Helton KJ, Conley ME, et al. Diffuse CNS vasculopathy with chronic Epstein-Barr virus infection in X-linked lymphoproliferative disease. Ajnr: American Journal of Neuroradiology. 2006;27(4):884–886. [PMC free article] [PubMed] [Google Scholar]
- 6.Grosieux C, Amoric JC, Mechinaud F, et al. Cutaneous and neurologic vasculitis disclosing EBV-selective immunodeficiency. Annales de Dermatologie et de Venereologie. 1996;123(6-7):387–392. [PubMed] [Google Scholar]
- 7.Kanegane H, Ito Y, Ohshima K, et al. X-linked lymphoproliferative syndrome presenting with systemic lymphocytic vasculitis. American Journal of Hematology. 2005;78(2):130–133. doi: 10.1002/ajh.20261. [DOI] [PubMed] [Google Scholar]
- 8.Verhelst H, Van Coster R, Bockaert N, et al. Limbic encephalitis as presentation of a SAP deficiency. Neurology. 2007;69(2):218–219. doi: 10.1212/01.wnl.0000265597.56202.6c. [DOI] [PubMed] [Google Scholar]
- 9.Sumegi J, Huang D, Lanyi A, et al. Correlation of mutations of the SH2D1A gene and epstein-barr virus infection with clinical phenotype and outcome in X-linked lymphoproliferative disease. Blood. 2000;96(9):3118–3125. see comment. [PubMed] [Google Scholar]
- 10.Simon HU, Plotz SG, Dummer R, et al. Abnormal clones of T cells producing interleukin-5 in idiopathic eosinophilia. N Engl J Med. 1999;341(15):1112–1120. doi: 10.1056/NEJM199910073411503. [DOI] [PubMed] [Google Scholar]
- 11.Perez C, Loza E, Tinture T. Giant cell arteritis after influenza vaccination. Arch Intern Med. 2000;160(17):2677. doi: 10.1001/archinte.160.17.2677. [DOI] [PubMed] [Google Scholar]
- 12.Zaas A, Scheel P, Venbrux A, et al. Large artery vasculitis following recombinant hepatitis B vaccination: 2 cases. J Rheumatol. 2001;28(5):1116–1120. [PubMed] [Google Scholar]
- 13.Begier EM, Langford CA, Sneller MC, et al. Polyarteritis nodosa reports to the vaccine adverse event reporting system (VAERS): implications for assessment of suspected vaccine-provoked vasculitis. J Rheumatol. 2004;31(11):2181–2188. [PubMed] [Google Scholar]
- 14.Haber P, Sejvar J, Mikaeloff Y, et al. Vaccines and guillain-barre syndrome. Drug Saf. 2009;32(4):309–323. doi: 10.2165/00002018-200932040-00005. [DOI] [PubMed] [Google Scholar]
- 15.Laria C, Alio J, Rodriguez JL, et al. Optic neuritis after meningococcal vaccination. Arch Soc Esp Oftalmol. 2006;81(8):479–482. doi: 10.4321/s0365-66912006000800009. [DOI] [PubMed] [Google Scholar]
- 16.Mrabet D, Meddeb N, Ajlani H, et al. Cerebral vasculitis in a patient with rheumatoid arthritis. Joint Bone Spine. 2007;74(2):201–204. doi: 10.1016/j.jbspin.2006.08.004. [DOI] [PubMed] [Google Scholar]
- 17.Nikolov NP, Smith JA, Patronas NJ, et al. Diagnosis and treatment of vasculitis of the central nervous system in a patient with systemic lupus erythematosus. Nat Clin Pract Rheumatol. 2006;2(11):627–633. doi: 10.1038/ncprheum0337. quiz 634. [DOI] [PubMed] [Google Scholar]
- 18.Lankester AC, Visser LF, Hartwig NG, et al. Allogeneic stem cell transplantation in X-linked lymphoproliferative disease: two cases in one family and review of the literature. Bone Marrow Transplant. 2005;36(2):99–105. doi: 10.1038/sj.bmt.1705016. [DOI] [PubMed] [Google Scholar]
- 19.Horne A, Trottestam H, Arico M, et al. Frequency and spectrum of central nervous system involvement in 193 children with haemophagocytic lymphohistiocytosis. Br J Haematol. 2008;140(3):327–335. doi: 10.1111/j.1365-2141.2007.06922.x. [DOI] [PubMed] [Google Scholar]
- 20.Cooper N, Rao K, Goulden N, et al. The use of reduced-intensity stem cell transplantation in haemophagocytic lymphohistiocytosis and Langerhans cell histiocytosis. Bone Marrow Transplant. 2008;42(Suppl 2):S47–50. doi: 10.1038/bmt.2008.283. [DOI] [PubMed] [Google Scholar]