

Conspectus
Water is ubiquitous in nature, but it exists as pure water infrequently. From the ocean to biology, water molecules interact with a wide variety of dissolved species. Many of these species are charged. In the ocean, water interacts with dissolved salts. In biological systems, water interacts with dissolved salts as well as with charged amino acids, the zwitterionic head groups of membranes, and other biological groups that carry charges. Water plays a central role in vast number of chemical processes because of its dynamic hydrogen bond network. A water molecule can form up to four hydrogen bonds in an approximately tetrahedral arrangement. These hydrogen bonds are continually being broken and new bonds are being formed on a picosecond time scale. The ability of water’s hydrogen bond network to rapidly reconfigure enables water to accommodate and facilitate chemical processes. Therefore, the influence of charged species on water hydrogen bond dynamics is important.
Recent advances in ultrafast coherent infrared spectroscopy have greatly expanded our understanding of water dynamics. Two dimensional infrared (2D IR) vibrational echo spectroscopy is providing new observables that yield direct information on the fast dynamics of molecules in their ground electronic state under thermal equilibrium conditions. 2D IR vibrational echoes are akin to 2D NMR but operate on time scales that are many orders of magnitude shorter. In a 2D IR vibrational echo experiment (see Conspectus figure), three IR pulses are tuned to the vibrational frequency of interest, which in this case is the frequency of the hydroxyl stretching mode of water. The first two pulses “label” the initial molecular structures by their vibrational frequencies. The system evolves between pulses two and three, and the third pulse stimulates the emission of the vibrational echo pulse, which is the signal. The vibrational echo pulse is heterodyne detected by combining it with another pulse, the local oscillator. Heterodyne detection provides phase and amplitude information, which are both necessary to perform the two Fourier transforms that take the data from the time domain to a two dimensional frequency domain spectrum. The time dependence of a series of 2D IR vibrational echo spectra provides direct information on system dynamics.
Here we use two types of 2D IR vibrational echo experiments to examine the influence that charged species have on water hydrogen bond dynamics. Solutions of NaBr and NaBF4 are studied. The NaBr solutions are studied as a function of concentration using vibrational echo measurements of spectral diffusion and polarization selective IR pump-probe measurements of orientational relaxation. Both types of measurements show the slowing of hydrogen bond network structural evolution with increasing salt concentration. NaBF4 is studied using vibrational echo chemical exchange spectroscopy. In these experiments it is possible to directly observe the chemical exchange of water molecules switching their hydrogen bond partners between and other water molecules. The results demonstrate that water interacting with ions has slower hydrogen bond dynamics than pure water, but the slowing is a factor of three or four rather than orders of magnitude.

I. Introduction
The hydrogen bond network of pure water, which is responsible for water’s unique properties, undergoes rapid structural evolution.1 A question of fundamental importance is how do the dynamics of water in the immediate vicinity of an ion or ionic group differ from those of pure water? Water dynamics in ion hydration shells play a significant role in the nature of systems such as proteins and micelles and in processes such as ion transport through transmembrane proteins.
In pure water the hydrogen bond network is constantly evolving with a range of time scales from tens of femtoseconds to picoseconds.2-6 Hydrogen bonds are continually forming and breaking through concerted hydrogen bond rearrangements.7 These dynamical processes can be observed on the timescales they occur using ultrafast infrared spectroscopy. Measurements of spectral diffusion using ultrafast two dimensional infrared (2D IR) vibrational echo spectroscopy5,8,9 as well as other ultrafast IR techniques4,10 have determined the multiple time scales for the hydrogen bond dynamics. The slowest time component of the FFCF (1.7 ps) is associated with the randomization of the hydrogen bond network through concerted hydrogen bond rearrangements. The orientational relaxation time of pure water (2.6 ps)4,6 is also assigned to concerted hydrogen bond rearrangement via jump reorientation.7
Ionic hydration and the dynamics of water in salt solutions have been studied extensively by NMR,11 Neutron diffraction,12 computer simulations,13 and infrared spectroscopy.9,14,15 There is information about the structure of ionic solvation shells from diffraction experiments. However, unraveling the dynamics of the solvation shell and the back and forth exchange of waters hydrogen bonded to ions and to water molecules is an ongoing experimental problem.
Here, two types of ultrafast 2D IR vibrational echo experiments as well as polarization selective IR pump-probe experiments are used to study water hydrogen bond dynamics in concentrated salt solutions. The two 2D IR vibrational echo observables are chemical exchange16,17 and spectral diffusion.5,8,9 The application of these two types of vibrational echo experiments is dictated by the IR absorption spectrum of the hydroxyl stretch of water. The OD hydroxyl stretch of dilute HOD in H2O is studied rather than pure H2O or D2O to eliminate vibrational excitation transfer.18,19 Vibrational excitation transfer interferes with orientational relaxation measurements, and it will artificially contribute to both chemical exchange and spectral diffusion. MD simulations have shown that dilute HOD in H2O does not change the behavior of water and that observations of the OD hydroxyl stretch report on the dynamics and local environment of water.20
Figure 1A shows background subtracted IR absorption spectra of the OD stretch of HOD in H2O salt solutions for NaBF417 for several salt concentrations. The numbers, n, are the number of water molecules per salt, e.g., n = 30 means that there are 30 water molecules for one NaBF4. As the concentration of the salt increases, a peak develops on the high frequency side of the water spectrum. This peak arises from ODs hydrogen bonded to the anions.17 In addition, the main water peak shifts to the blue with increasing concentration but essentially maintains its shape. The presence of distinct peaks for ODs hydrogen bonded to other water molecules and to anions makes it possible to perform the chemical exchange spectroscopy (CES) experiments, 17 in which hydrogen bond switching between anions and water oxygens is manifested by the growth of off-diagonal peaks in the 2D IR vibrational echo spectra. The CES experiments were performed on the highest concentration sample, 5.5 M with n = 7.
Figure 1.

FT-IR spectra of the OD stretching mode of dilute HOD in H2O as a function of salt concentration. A. NaBF4 spectra. The OD hydroxyl bound to the anion (small peak at high frequency) and the OD bound to another water’s oxygen are spectrally resolved. B. NaBr spectra. The water-anion and water-water bands are not resolved.
The NaBr spectra (Figure 1B) do not show two distinct peaks at any salt concentration. The spectra are broad21 and the peak position is increasingly blue-shifted as the NaBr concentration increases. For pure water the broad spectrum is associated with different numbers and a wide range of strengths of the hydrogen bonds.21 For pure water, subensembles of water molecules with more and/or stronger hydrogen bonds have red-shifted absorptions, while subensembles with fewer and/or weaker hydrogen bonds are blue-shifted. In the type of systems being considered here, in which the hydroxyl groups are likely to be interacting with ions, the blue-shift should not be interpreted as an indication of weakening of the hydrogen bond structure in an aqueous solutions, but rather as an overall change in the nature of the local water environments. Because the NaBr spectra do not show distinct peaks, CES cannot be employed. Instead, measurements of spectral diffusion (change in the 2D IR vibrational echo line shapes) can be used to examine the hydrogen bond dynamics.9
II. Measurements of Hydrogen Bond Dynamics
A. Chemical Exchange Spectroscopy Measurements of Water — NaBF4
Chemical exchange occurs when two species in equilibrium interconvert without changing the overall number of either species. 2D IR vibrational echo chemical exchange spectroscopy has been developed recently16,22-25 and applied to the formation and dissociation dynamics of organic solute-solvent complexes,16,22,26 the rate of isomerization around a carbon-carbon single bond,25 the switching between well defined protein structural substates,27 and hydrogen bond dynamics in NaBF4 solutions17 discussed here.
Chemical exchange between two species has a well-defined effect on the 2D IR vibrational echo spectrum.16 At times short compared to the exchange time, the peaks corresponding to the two species appear as two positive going bands on the diagonal of the 2D spectrum. A 2D contour plot spectrum like this is shown in the Conspectus figure (also see Figure 3A below). The diagonal is the dashed line. Positive going peaks are red and negative going peaks are blue. The horizontal axis is ωτ, corresponding to the frequencies that the vibrational oscillators interact with when the first pulse in the three pulse sequence is applied. The vertical axis is ωm. It corresponds to the frequencies of oscillators’ interacting with the third pulse, which gives rise to the vibrational echo pulse at the same frequencies. So ωm corresponds to the frequencies contained in the vibrational echo wave packet. The time between the first and second pulses is τ, and the time between the second and third pulses is Tw. The vibrational echo pulse is emitted at time ≤τ after the third pulse. The vibrational echo is detected through a monochromator using an MCT array detector, which measures thirty two frequencies at once (see Conspectus figure). Frequency resolving the combined echo-local oscillator wave packet performs one of the two Fourier transforms necessary to obtain a 2D spectrum. The detection of the many wavelengths in the vibrational echo pulse with the monochromator gives the ωm axis. This axis reads out the final frequencies of all of the species.
Figure 3.

2D IR vibrational echo spectra of the 5.5 M (n = 7) solution of NaBF4 at Tw = 200 fs (A) and Tw = 4 ps (B). Positive going peaks are marked by + and negative going peaks are marked by -. By 4 ps additional peaks at locations labeled A, B, and C have grown in because of chemical exchange (see text).
The first two pulses in the sequence label the initial species. As τ is scanned, the echo pulse moves temporally relative to the fixed local oscillator pulse, producing a temporal interferogram at each ωm where there is signal. Numerical Fourier transformation of these temporal interferograms, one at each ωm, gives the ωτ axis. The ωτ axis reports the initial frequencies of the species, and the ωm axis reports the final frequencies of the species after the system has evolved for time, Tw.
In the Conspectus figure (also see Figure 3A below), the high frequency peak on the diagonal arises from ODs hydrogen bonded to anions. The lower frequency peak on the diagonal comes from ODs hydrogen bonded to water oxygens. In addition, there are two negative going off-diagonal bands (blue in the figure) that arise from vibrational echo emission at the v = 1 to v = 2 (1-2) transition frequencies of each diagonal peak.28,29 The 1-2 peaks are shifted to lower frequency along the vibrational echo emission (ωm) axis of the 2D spectrum by the anharmonicities of the OD stretch of the two species.
It is useful to discuss the ideal CES case, in which the anharmonic shifts are so large that when projected onto the ωm axis the off-diagonal 1-2 peaks do not overlap with the 0-1 peaks.30 (Note that this is not the case for the salt solution spectrum shown in the Conspectus figure and Figure 3 below.) In the ideal case, we can consider only the 0-1 peaks. At short Tw, there are two peaks on the diagonal, call them A and B (see Figure 2A). The system is in equilibrium, so A’s are constantly turning into B’s, and B’s are turning into A’s. At longer Tw, some A’s have turned into B’s and some B’s into A’s (see Figure 2B). For those A’s that turn into B’s, the initial frequency on both the ωτ and ωm axes is ωA but the final frequency on ωm is ωB, producing an off-diagonal peak at (ωA,ωB). For those B’s that turn into A’s, a different off-diagonal peak is produced at (ωB,ωA). The time dependence of the growth of these off-diagonal peaks and the decay of the diagonal peaks yields the chemical exchange times.16 Figure 2C shows both the 0-1 and 1-2 portions of spectrum at long Tw. There are equivalent chemical exchange peaks in both portions.16
Figure 2.
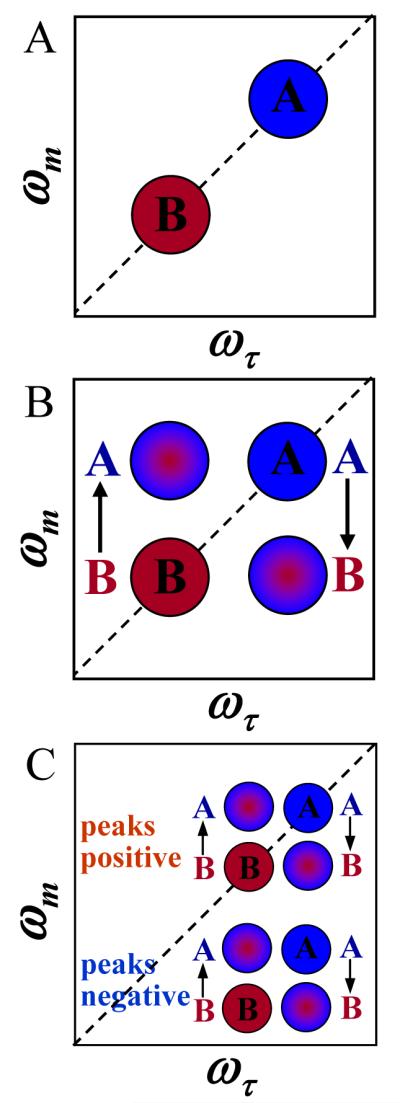
A. The 0-1 region of a hypothetical spectrum at short time. One peak for each species is on the diagonal. B. The 0-1 region at long time. The off-diagonal chemical exchange peaks have grown in. C. The full spectrum at long time showing both the 0-1 and 1-2 regions of the spectrum.
The water- chemical exchange system behaves in the same manner except that there is overlap between chemical exchange peaks and other peaks in the 2D spectrum. The overlapping peaks produce a more complex appearing spectrum as the chemical exchange peaks grow. The overlap of the peaks is taken into account in the data analysis.17
In the following, hydroxyl-water (hw) signifies hydroxyls hydrogen bonded to oxygen atoms of water molecules. Hydroxyl-anion (ha) signifies hydroxyls hydrogen bonded to anions. The first two pulses label the ha’s and hw’s. As time increases, additional peaks grow in due to hydrogen bond rearrangements that cause ha’s to change into hw’s and vice versa. The exchange dynamics are extracted from the time dependent growth of these off-diagonal peaks when the effects of the vibrational lifetimes and orientational relaxation of the two species are included in the chemical exchange kinetic model analysis.16,17 The vibrational lifetimes and orientational relaxation times were measured with polarization selective IR pump-probe experiments.17
Figure 3 displays the 2D IR chemical exchange spectrum at 200 fs (A) and Tw = 4 ps (B). The growth of the chemical exchange peaks at 4 ps is clear from the change in the spectrum compared to the Tw = 200 fs spectrum. In B, only the higher frequency region of the 2D spectrum is presented, because this region of the spectrum shows the influence of the growth of the off-diagonal peaks most clearly. Because the lifetime of hw is considerably shorter than ha (see below), the hw peak has decreased in amplitude relative to the ha peak when compared to the 200 fs spectrum. Chemical exchange will cause four additional off-diagonal peaks to grow in, two of which are positive going, arising from the 0-1 transitions, and two that are negative going and come from the 1-2 transitions. In Figure 3 the most evident chemical exchange peak is the 0-1 hw→ha peak, labeled A; this peak does not overlap with any other peak. The spectrum contains two other exchange peaks. The 1-2 hw→ha peak is labeled B. The 1-2 hw→ha peak eats away a strip from the diagonal 0-1 hw band because it is negative, relatively narrow along the ωm axis, and extended along the ωτ axis. This is seen very clearly by comparing the hw peak in Figure 3B to its short time counterpart in Figure 3A. The last exchange peak in Figure 3 is the 0-1 ha→hw peak, labeled C. It is positive going and is manifested as a reduction in the bottom portion of the negative going 1-2 ha peak. The shapes of the diagonal and off-diagonal CES peaks have been explicated theoretically and experimentally.23,30 The important point is that we know exactly where all the peaks are. The overlap of the peaks is handled in the data analysis.17
A series of 2D spectra over a range of Tw was collected and analyzed. The chemical exchange times can be obtained from the peak volumes as a function of Tw.16,23 The time dependence of the 0-1 and corresponding 1-2 peaks are the same.16,23 Because the system is in equilibrium, the ha→hw and hw→ha chemical exchange peaks grow in with the same time dependence.16,17,23 Therefore, all of the dynamics are reflected in the subset of peaks, the diagonal 0-1 ha peak, the diagonal 0-1 hw peak, and the off-diagonal 0-1 chemical exchange hw→ha peak.17 The volumes associated with these peaks are plotted in Figure 4A.
Figure 4.

A. Peak volumes as a function of Tw of the diagonal and chemical exchange peaks. The solid curves are the result of fitting to the chemical exchange kinetic model. B. Vibrational population decays for OD hydroxyl bound to an anion (ha) and for the OD bound to another water oxygen (hw). The solid curves are the result of simultaneously fitting the population decays and the peak volumes shown in A to the chemical exchange kinetic model.
To obtain the chemical exchange rate, the data in Figure 4A are fit with a model that includes the vibrational population decay rates and the orientational relaxation rates of hw and ha.16,17,23 Polarization selective pump-probe experiments were used to determine the apparent lifetime. The data are shown in Figure 4B. Using the measured values as an initial guess, the data in Figures 4A and B were simultaneously fit to a system of coupled differential equations.23 In the fitting, the only adjustable parameters are the two vibrational lifetimes and the exchange rate for the process, ha → hw. The simultaneous fits give an exchange rate, vibrational lifetimes, and orientational relaxation times that are internally consistent with the data measured by 2D IR CES and polarization selective pump-probe spectroscopy.
The fits to the exchange model are shown as the solid curves in Figures 4A and B. The quality of the fits in both panels is excellent. In Figure 4A, the fit reproduces the time dependence of the diagonal and chemical exchange peaks. The exchange time for ha → hw is Taw = 7 ± 1 ps. The exchange time for hw → ha, Twa, is related to Taw by the equilibrium constant.17 Using R = 0.317 gives Twa = 24 ps. This value will be dependent on the concentration of salt because hw → ha can only occur if a water molecule is very close to an anion. Therefore, the exchange dynamics are better characterized by the time constant, Taw = 7 ps, which may be relatively insensitive to salt concentration.
Figure 4B shows the vibrational population relaxation data (symbols) and the exchange model (solid curves). The process of exchange modifies the vibrational population decays such that the experimentally measured decays do not provide the true lifetimes. Initially, the data are fit with single exponential decays to extract a first guess for the lifetimes. The hw decay is substantially faster than the ha decay. Because the system is in equilibrium, at t = 0, equal population is exchanged between the two species. However, as time proceeds, the hw population decays more than the ha population, and the exchange process will serve to bolster the population of hw, making the apparent lifetime longer than the true lifetime. The converse is true for ha. Note that the number of molecules undergoing exchange is always constant, but the fraction of excited molecules of each species depends on the vibrational lifetime of that species. The shapes of the population decay curves, particularly the ha curve, are substantially influenced by the chemical exchange.
Simultaneously fitting the 2D IR CES data and the pump probe data yields the true values for the lifetimes of τhw = 2.2 ± 0.1 ps and τha = 9.4 ± 1 ps. More important is that the fits using the chemical exchange model reproduce the functional form and time dependence of all of the curves in Figures 4A and B. Measurements of the orientational anisotropy decay following procedures described previously6 yields values of τrha = 5.0 ps and τrhw = 4.1 ps where τrha and τrhw are the long time orientational correlation times for ha and hw, respectively. Because the orientational time constants are close in value, the measured values are not significantly modified by the chemical exchange.
The time constants for orientational motion are faster than the time constant for chemical exchange. The ha→hw exchange time is 7 ± 1 ps. The orientational relaxation times and the chemical exchange times are not directly comparable. Nonetheless, it is reasonable to expect that orientational relaxation will be faster than chemical exchange. In pure water, orientational relaxation is modeled as jump reorientation. Here, the jumps can be from ha to hw and hw to ha, as well as ha to ha, and hw to hw. The first two types of jumps produce both chemical exchange and orientational relaxation while the last two produce only orientational relaxation. Thus, there are more pathways contributing to orientational relaxation than to chemical exchange, in accord with the observation of faster reorientation. The orientational relaxation times are less than a factor of two slower than the orientational relaxation time of pure water, τr = 2.6 ps. Thus, the chemical exchange time and the orientational relaxation times show that dynamics of water in this concentrated salt solution are not tremendously slower than in pure water.
B. Spectral Diffusion Measurements of Water — NaBr
The CES method depends on having two resolvable peaks (see Figure 1A) so that the growth of off-diagonal chemical exchange peaks can be observed. The spectra of NaBr solutions (Figure 1B) display a single OD stretching band because the hw and ha peaks overlap to such a great extent that they are not resolved. Therefore, spectral diffusion is used to investigate the water-ion dynamics rather than chemical exchange.
Spectral diffusion in pure water was briefly mentioned in the Introduction. The OD hydroxyl stretch frequency is very sensitive to the hydrogen bond configuration. Stronger hydrogen bonds and more hydrogen bonds shift the frequency to the red (low energy). Weaker and fewer hydrogen bonds shift the frequency to the blue. As the hydrogen bond structure evolves, the frequency of the OD stretch changes. The time dependent evolution of the frequency is called spectral diffusion. Tracking the time dependence of the frequency provides information on the time dependence of the hydrogen bond structural rearrangement.5,8 Experiments and simulations of spectral diffusion have shown that there are a variety of time scales for hydrogen bond dynamics in pure water.5,8 The very fast dynamics (tens to hundreds of femtoseconds) are associated with very local motions of the hydrogen bonds. The slowest component of the dynamics (1.7 ps) corresponds to the complete randomization of the network structure through concerted hydrogen bond rearrangements.
Spectral diffusion can be understood using the ideas introduced to discuss chemical exchange. For pure water, rather than two resolved peaks on the diagonal, imagine a continuous overlapping set of peaks along the diagonal. At short time before significant dynamics have occurred, the 2D spectrum will be elongated along the diagonal because of the many overlapping peaks. As the hydrogen bond structure changes, off-diagonal peaks will grow in. However, instead of two peaks at two specific frequencies in the 0-1 region, there will be a continuum of peaks spanning all accessible frequencies. These “off-diagonal” peaks overlap, and their effect is to change the shape of the 2D spectrum. As Tw increases, the spectrum broadens, and goes from being elongated along the diagonal to increasingly symmetrical. The change in shape can be analyzed using a variety of methods to give the frequency-frequency correlation function. Basically, the FFCF describes the time evolution of an initial frequency ω(0) to other frequencies ω(t) at later time t, which is directly related to an initial local hydrogen bonding structure experience by an OD at t = 0 evolving to other structures at time t.
In the NaBr solutions, in addition to spectral diffusion, there is also chemical exchange, in which OD hydroxyls bound to water oxygens (hw) and OD hydroxyls bound to Br- (ha) switch. The overall dynamics are illustrated schematically in Figure 5. The left well represents hw and the right well represents ha. Based on the experiments on NaBF4 we anticipate that the switching between ha and hw will occur on the many picosecond time scale. On a much shorter time scale, spectral diffusion will cause sampling of frequencies without switching between species. Because the ha and hw peaks overlap strongly, spectral diffusion in each of the wells shown in Figure 5 will sample a large range of frequencies. The diagram has the two types of species separated with distinct regions of spectral diffusion. While this is the case for NaBF4, it is not the physical situation for NaBr. In the NaBr solutions, the ranges of spectral diffusion strongly overlap. However, without chemical exchange interconverting ha’s and hw’s, full spectral diffusion, that is sampling of all structures and therefore all frequencies, cannot occur.
Figure 5.

A schematic illustration of the processes of spectral diffusion and chemical exchange.
To analyze the Tw dependence of the 2D lineshapes, we employ the Center Line Slope (CLS) method, which is an accurate approach for obtaining the FFCF.31,32 Figure 6 shows 2D IR vibrational echo spectra for the 6 M (n = 8) NaBr solution at three Tws, 200 fs, 1 ps, and 9 ps. The dashed line on the top panel is the diagonal. The positive going peaks (red) on the diagonal are from the OD stretch 0-1 transition and the off-diagonal negative going peaks (blue) are from the 1-2 transition. We will focus on the 0-1 transition peaks. At 200 fs, the 0-1 band is elongated along the diagonal. By 1 ps, the band is less elongated, and by 9 ps, the band is virtually symmetrical. At sufficiently long time, the 0-1 band will become round in the absence of the 1-2 band. However, the negative 1-2 band overlaps the bottom of the positive 0-1, flattening it out. The blue lines are the center lines. They are calculated as has been described previously.31,32
Figure 6.

2D IR vibrational echo data for the 6 M (n = 8) NaBr solution at three Tws. The red peaks are positive going 0-1 transitions, and the blue peaks are negative going 1-2 transitions. The diagonal is shown in the top panel. As Tw increases, the shapes of the bands change. The solid lines are the center lines used to obtain the frequency-frequency correlation function.
The basic idea of the CLS method is that a line can be constructed from the data at each Tw that has a slope which is directly related to the shape of the spectrum. In the analysis used here, the FFCF is related to the inverse of the slope. At short time, the center line slope approaches 1, so the inverse is 1. At long time, the center line is vertical (infinite slope, see Figure 6 bottom panel), so the inverse is 0. The FFCF can be calculated from the Tw dependence of the inverse of the CLS.31
Figure 7 displays the inverse of the CLS, C(Tw), for pure water and three NaBr solutions.9,33 It is immediately clear that as the NaBr concentration increases the water dynamics slow. Extensive simulations of pure water5,8 and comparisons to vibrational echo data3,5,8 show that the FFCF decays on multiple time scales. The slowest time scale for pure water, 1.7 ps, arises from the complete randomization of the hydrogen bond network. We find that FFCFs for the NaBr solutions also decay with multiple time scales with the faster components being similar to those found for pure water.9 The slowest component of the FFCF shows the most dramatic change. In analogy to pure water, we assume that this component reflects the final complete randomization of the hydrogen bond network. However, in NaBr solutions, there will be hydrogen bonds between water molecules as well as hydrogen bonding of water to ions. The slowest FFCF component for pure water and the 1.5 M, 3 M, and 6 M NaBr solutions are 1.7 ±0.5 ps, 2.6 ±0.5 ps, 3.5 ±0.5 ps, and 4.8 ±0.6 ps, respectively. For the highest concentration there are only eight water molecules per NaBr (n = 8), which is insufficient to complete even a single solvation shell for the cation and anion. Therefore, all of the molecules will be interacting directly with ions. Even for this sample, the hydrogen bond randomization is only approximately a factor of three slower than occurs in pure water.
Figure 7.

The data (symbols) reflect the change in shape of the 2D IR spectra as a function of Tw using the center line slope (CLS) representation for pure water and three NaBr concentrations. As the ion concentration increases, the water hydrogen bond dynamics slow.
Figure 8 displays decays of the orientational anisotropy, r(t), of the OD hydroxyl for pure water and the three NaBr concentrations.9,34 r(t) is the second Legendre polynomial orientational correlation function divided by 2.5. The data describe the randomization of the direction of the OD bond vector. As with the vibrational echo spectral diffusion, the orientational relaxation slows as the NaBr concentration increases. The slowest component of r(t) for pure water and the 1.5 M, 3 M, and 6 M NaBr solutions are 2.6 ±0.1 ps, 3.9 ±0.3 ps, 5.1 ±0.2 ps, and 6.7 ±0.3 ps, respectively. The complete randomization of the bond vector occurs through jump reorientation rather than Gaussian orientational diffusion.7,35 The orientations of water molecules are restricted because of hydrogen bonds to other water molecules. Concerted hydrogen bond network rearrangement is required to randomize the orientation. The switching of hydrogen bonds between partners causes jumps in the orientation of ∼60°. Therefore, the slowest component of the orientational relaxation is closely related to the slowest component of the spectral diffusion. Both require hydrogen bond network randomization.
Figure 8.

Decay of the orientational anisotropies of pure water and three concentrations of NaBr. As the NaBr concentration increases, the orientational relaxation slows.
In the n = 8 NaBr solution, the slowest component of the orientational relaxation is almost three times longer than in pure water. In the model we are using, both the slowest component of the spectral diffusion and the complete randomization of the OD orientation require a global reorganization of the hydrogen bond structure. The FFCF and the orientational correlation function (r(t)) cannot be directly compared because they are different correlation functions. It is interesting to compare the effect of NaBr concentration on the slowest components of the FFCF and r(t). This can be done by taking the ratios, Ri, of the time constants of the slowest components for NaBr solutions with respect to the value for pure water. For the FFCF, RFFCF = 1.5, 2.1, and 2.8 going from low to high concentration. For r(t), Ror = 1.5, 2.0, and 2.6. Within experimental error, the two sets of ratios are identical, which supports the picture that the slowest components of both experimental observables are related to the same global hydrogen bond rearrangement.
Previously, two-color IR pump-probe experiments were performed by pumping the OH stretch of HOD in D2O salt solutions (6 M NaCl, NaBr, or NaI) near the center of the OH stretching band and measuring vibrational population relaxation at different frequencies.15 The results were interpreted as spectral diffusion and were analyzed in terms of a correlation time constants τc . The correlation times were reported to be 20 to 50 times longer than those of pure water. Both the direct chemical exchange measurements on NaBF4 and the vibrational echo spectral diffusion measurements on NaBr discussed here demonstrate that water dynamics are not slowed as much as suggested by the earlier pump-probe experiments.
III. Discussion
In both the NaBF4 and NaBr solutions, the hydrogen bond rearrangement dynamics can be divided into four subsets based on the initial and final hydrogen bonding partner of the OD hydroxyl, which is the species under observation in all of the experiments. The four processes are: hw→hw, ha→ha, hw→ha, and ha→hw. In the NaBF4 chemical exchange experiments, we observe only the hw→ha and ha→hw processes, and therefore we obtain the time for switching from a water hydroxyl bound to an anion to a hydroxyl bound to a water oxygen, and vice versa. We found that the time for the ha→hw process is 7 ps. In the NaBr solutions, all four processes contribute to spectral diffusion and orientational relaxation. As the NaBr concentration goes up, the water concentration goes down, and the number of hw→hw processes decreases. For the lower NaBr concentrations, the number of ha→ha processes is probably negligible because the anions are separated. For the highest NaBr concentration, there are 8 water molecules with 16 water oxygen accepting sites per NaBr. Br- will accept ∼6 hydroxyls to solvate it in dilute solution. Therefore, even at the highest concentration, the number water accepting sites substantially out number the Br- accepting sites. So, it is likely that the ha→ha process is not substantial even at this high concentration. If this is the case, then increasing the NaBr concentration increases the importance of the chemical exchange processes ha→hw and hw→ha relative to the other two processes. Based on the results of the NaBF4 experiments, we can reasonably assume that the chemical exchange processes are slower than the hw→hw process, which in bulk water gives a spectral diffusion time of 1.7 ps and an orientational relaxation time of 2.6 ps.
In light of these ideas, the NaBr concentration dependent data can be viewed as follows. The identical concentration dependence of the ratios RFFCF and Ror show that the spectral diffusion observable and the orientational relaxation observable are measuring different aspects of the same phenomena. In contrast to the direct chemical exchange measurements, both of these observables have contributions from the chemical exchange pathways, ha→hw and hw→ha, and the non-chemical exchange pathways, hw→hw and ha→ha. As the concentration of NaBr increases, the chemical exchange pathways will become increasingly important relative to the non-chemical exchange pathways. The observed slowing of the dynamics with increasing NaBr concentration (see Figures 7 and 8) shows that the chemical exchange pathways are slower than the non-chemical exchange pathways. Even at the highest NaBr concentration (6 M, n = 8), there will still be non-chemical exchange pathways participating in the spectral diffusion and orientational relaxation dynamics. However, at all of the concentrations used in the NaBr experiments, the hw→hw process will not be the same as it is in bulk. The slowest components of the spectral diffusion and the orientational relaxation involve the randomization of the hydrogen bond network, which requires concerted motions of a number of water molecules.7,36 The OD hydroxyl under observation can switch from one water oxygen to another, but the other waters participating in the process may be associated with ions. Therefore, the slowing of the dynamics with increased NaBr concentration is not necessarily a superposition of a decreasing contribution of normal bulk water dynamics and an increasing contribution of slower chemical exchange dynamics. Because of the concerted multi-water nature of the dynamics that randomize the hydrogen bond structure, the hw→hw process is also likely to become slower as the NaBr concentration increases. At the highest concentration, the hw→hw process may not be able to occur without chemical exchange participating as part of the concerted process. Therefore, the dynamics measured at the highest concentration are likely to be dominated by and close to the chemical exchange dynamics.
The take home message is that the interaction of water with ions slows hydrogen bond network dynamics. However, even at very high salt concentrations, the lengthening of the time scale for a water to switch between being hydrogen bonded to an ion and being hydrogen bonded to another water molecule is slower by a factor of three or four, not orders of magnitude. The results present here indicate that water interacting with other types of charged species, for example, a charged amino acid at the surface of a protein, will result in a longer time to reorganize the local hydrogen bond network than pure water, but the increase in time scale should be significantly less than an order of magnitude.
Acknowledgements
This work was supported by the Department of Energy (DE-FG03-84ER13251), the National Institutes of Health (2-R01-GM061137-09), the Air Force Office of Scientific Research (F49620-01-1-0018), and the National Science Foundation (DMR 0652232). EEF thanks the Stanford Graduate Fellowship program for a research fellowship. DEM thanks the NSF for a Graduate Research Fellowship. DER thanks the Fannie and John Hertz Foundation and a Stanford Graduate Fellowship program for graduate research fellowships.
Biographical Information
Michael D. Fayer was born in Los Angeles, CA in 1947. He received his B.S. (1969) and his Ph. D. (1974) from the University of California at Berkeley. He joined the faculty at Stanford University in 1974, where he is currently the David Mulvane Ehrsam and Edward Curtis Franklin Professor of Chemistry.
David E. Moilanen was born in Redmond, WA in 1980. He received his B.S. from the University of Washington in 2003 and is currently a graduate student at Stanford University.
Daryl Wong was born in San Jose, CA in 1983. He received his B.S. from the University of California at Berkeley in 2006 and is currently a graduate student at Stanford University..
Daniel E. Rosenfeld was born in Los Angeles, CA in 1984. He received his B.S. and M.S. from Yale University in 2007 and is currently a graduate student at Stanford University.
Emily E. Fenn was born in Boston, MA in 1984. She received her B.S. from the Massachusetts Institute of Technology in 2006 and is currently a graduate student at Stanford University.
Sungnam Park was born in GangJin, South Korea in 1973. He received his B.S. from the Korea University in 1997 and his Ph. D. from the University of Chicago in 2005. He was a Post Doctoral Research Associate in the Department of Chemistry, Stanford University. He is presently an Assistant Professor at Korea University.
References
- (1).Schuster P, Zundel G, Sandorfy C. The Hydrogen Bond. Recent Developments in Theory and Experiments. Amsterdam; North Holland: 1976. [Google Scholar]
- (2).Rey R, Møller KB, Hynes JT. Hydrogen Bond Dynamics in Water and Ultrafast Infrared Spectroscopy. J. Phys. Chem. A. 2002;106:11993–11996. doi: 10.1021/jp064846m. [DOI] [PubMed] [Google Scholar]
- (3).Fecko CJ, Eaves JD, Loparo JJ, Tokmakoff A, Geissler PL. Ultrafast Hydrogen-Bond Dynamics in the Infrared Spectroscopy of Water. Science. 2003;301:1698–1702. doi: 10.1126/science.1087251. [DOI] [PubMed] [Google Scholar]
- (4).Rezus YLA, Bakker HJ. On the Orientational Relaxation of Hdo in Liquid Water. J. Chem. Phys. 2005;123:114502. doi: 10.1063/1.2009729. [DOI] [PubMed] [Google Scholar]
- (5).Asbury JB, Steinel T, Stromberg C, Corcelli SA, Lawrence CP, Skinner JL, Fayer MD. Water Dynamics: Vibrational Echo Correlation Spectroscopy and Comparison to Molecular Dynamics Simulations. J. Phys.Chem. A. 2004;108:1107–1119. [Google Scholar]
- (6).Moilanen DE, Fenn EE, Lin Y-S, Skinner JL, Bagchi B, Fayer MD. Water Inertial Reorientation: Hydrogen Bond Strength and the Angular Potential. Proc. Natl. Acad. Sci. U.S.A. 2008;105:5295–5300. doi: 10.1073/pnas.0801554105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Laage D, Hynes JT. A Molecular Jump Mechanism of Water Reorientation. Science. 2006;311:832–835. doi: 10.1126/science.1122154. [DOI] [PubMed] [Google Scholar]
- (8).Asbury JB, Steinel T, Kwak K, Corcelli SA, Lawrence CP, Skinner JL, Fayer MD. Dynamics of Water Probed with Vibrational Echo Correlation Spectroscopy. J. Chem. Phys. 2004;121:12431–12446. doi: 10.1063/1.1818107. [DOI] [PubMed] [Google Scholar]
- (9).Park S, Fayer MD. Hydrogen Bond Dynamics in Aqueous Nabr Solutions. Proc. Nat. Acad. Sci. U.S.A. 2007;104:16731–16738. doi: 10.1073/pnas.0707824104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Fecko CJ, Loparo JJ, Roberts ST, Tokmakoff A. Local Hydrogen Bonding Dynamics and Collective Reorganization in Water: Ultrafast Infrared Spectroscopy of Hod/D2o. J. Chem. Phys. 2005;122:054506. doi: 10.1063/1.1839179. (18) [DOI] [PubMed] [Google Scholar]
- (11).Shimizu A, Taniguchi Y. Nmr Studies of Reorientational Motion of Hydrated D2o Molecules of Halide Ions (F-, Cl-, Br-, and I-) in Dilute Aqueous Solutions. Bull. Chem. Soc. Jpn. 1991;64:1613–1617. [Google Scholar]
- (12).Mancinelli R, Botti A, Bruni F, Ricci MA, Soper AK. Hydration of Sodium, Potassium, and Chloride Ions in Solution and the Concept of Structure Maker/Breaker. J. Phys. Chem. B. 2007;111:13570–13577. doi: 10.1021/jp075913v. [DOI] [PubMed] [Google Scholar]
- (13).Laage D, Hynes JT. On the Residence Time for Water in a Solute Hydration Shell: Application to Aqueous Halide Solutions. J. Phys. Chem. B. 2008;112:7697–7701. doi: 10.1021/jp802033r. [DOI] [PubMed] [Google Scholar]
- (14).Smith JD, Saykally RJ, Geissler PL. The Effect of Dissolved Halide Anions on Hydrogen Bonding in Liquid Water. J. Am. Chem. Soc. 2007;129:13847–13856. doi: 10.1021/ja071933z. [DOI] [PubMed] [Google Scholar]
- (15).Kropman MF, Bakker HJ. Vibrational Relaxation of Liquid Water in Ionic Solvation Shells. Chem. Phys. Lett. 2003;370:741–746. [Google Scholar]
- (16).Zheng J, Kwak K, Asbury JB, Chen X, Piletic I, Fayer MD. Ultrafast Dynamics of Solute-Solvent Complexation Observed at Thermal Equilibrium in Real Time. Science. 2005;309:1338–1343. doi: 10.1126/science.1116213. [DOI] [PubMed] [Google Scholar]
- (17).Moilanen DE, Wong D, Rosenfeld DE, Fenn EE, Fayer MD. Ion-Water Hydrogen Bond Switching Observed with 2d Ir Vibrational Echo Chemical Exchange Spectroscopy. Proc. Nat. Acad. Sci. U.S.A. 2009;106:375–380. doi: 10.1073/pnas.0811489106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Woutersen S, Bakker HJ. Resonant Intermolecular Transfer of Vibrational Energy in Liquid Water. Nature (London) 1999;402:507–509. [Google Scholar]
- (19).Gaffney KJ, Piletic IR, Fayer MD. Orientational Relaxation and Vibrational Excitation Transfer in Methanol - Carbon Tetrachloride Solutions. J. Chem. Phys. 2003;118:2270–2278. [Google Scholar]
- (20).Corcelli S, Lawrence CP, Skinner JL. Combined Electronic Structure/Molecular Dynamics Approach for Ultrafast Infrared Spectroscopy of Dilute Hod in Liquid H2o and D2o. J. Chem. Phys. 2004;120:8107. doi: 10.1063/1.1683072. [DOI] [PubMed] [Google Scholar]
- (21).Lawrence CP, Skinner JL. Vibrational Spectroscopy of Hod in Liquid D20. Ii. Infrared Line Shapes and Vibrational Stokes Shift. J. Chem. Phys. 2002;117:8847–8854. [Google Scholar]
- (22).Kim YS, Hochstrasser RM. Chemical Exchange 2d Ir of Hydrogen-Bond Making and Breaking. Proc. Natl. Acad. Sci. 2005;102:11185–11190. doi: 10.1073/pnas.0504865102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Kwak K, Zheng J, Cang H, Fayer MD. Ultrafast 2d Ir Vibrational Echo Chemical Exchange Experiments and Theory. J. Phys. Chem. B. 2006;110:19998–20013. doi: 10.1021/jp0624808. [DOI] [PubMed] [Google Scholar]
- (24).Zheng J, Kwak K, Chen X, Asbury JB, Fayer MD. Formation and Dissociation of Intra-Intermolecular Hydrogen Bonded Solute-Solvent Complexes: Chemical Exchange 2d Ir Vibrational Echo Spectroscopy. J. Am. Chem. Soc. 2006;128:2977–2987. doi: 10.1021/ja0570584. [DOI] [PubMed] [Google Scholar]
- (25).Zheng J, Kwak K, Xie J, Fayer MD. Ultrafast Carbon-Carbon Single Bond Rotational Isomerization in Room Temperature Solution. Science. 2006:1951–1955. doi: 10.1126/science.1132178. [DOI] [PubMed] [Google Scholar]
- (26).Kwak K, Rosenfeld DE, Chung JK, Fayer MD. Solute-Solvent Complex Switching Dynamics of Chloroform between Acetone and Dimethylsulfoxide Two-Dimensional Ir Chemical Exchange Spectroscopy. J. Phys. Chem. B. 2008;112:13906–13915. doi: 10.1021/jp806035w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Ishikawa H, Kwak K, Chung JK, Kim S, Fayer MD. Direct Observation of Fast Protein Conformational Switching. Proc. Natl. Acad. Sci. U.S.A. 2008;105:8619–8624. doi: 10.1073/pnas.0803764105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Rector KD, Kwok AS, Ferrante C, Tokmakoff A, Rella CW, Fayer MD. Vibrational Anharmonicity and Multilevel Vibrational Dephasing from Vibrational Echo Beats. J. Chem. Phys. 1997;106:10027. [Google Scholar]
- (29).Tokmakoff A, Kwok AS, Urdahl RS, Francis RS, Fayer MD. Multilevel Vibrational Dephasing and Vibrational Anharmonicity from Infrared Photon Echo Beats. Chem. Phys. Lett. 1995;234:289. [Google Scholar]
- (30).Zheng J, Fayer MD. Solute-Solvent Complex Kinetics and Thermodynamics Probe by 2d-Ir Vibrational Echo Chemical Exchange Spectroscopy. J. Phys. Chem. B. 2008;112:10221–10227. doi: 10.1021/jp804087v. [DOI] [PubMed] [Google Scholar]
- (31).Kwak K, Park S, Finkelstein IJ, Fayer MD. Frequency-Frequency Correlation Functions and Apodization in 2d-Ir Vibrational Echo Spectroscopy, a New Approach. J. Chem. Phys. 2007 doi: 10.1063/1.2772269. accepted. [DOI] [PubMed] [Google Scholar]
- (32).Kwak K, Rosenfeld DE, Fayer MD. Taking Apart 2d-Ir Vibrational Echo Spectra: More Information and Elimination of Distortions. J. Chem. Phys. 2008;128:204505. doi: 10.1063/1.2927906. [DOI] [PubMed] [Google Scholar]
- (33).Park S, Kwak K, Fayer MD. Ultrafast 2d-Ir Vibrational Echo Spectroscopy: A Probe of Molecular Dynamics. Laser Phys. Lett. 2007;4:704–718. [Google Scholar]
- (34).Tan H-S, Piletic IR, Fayer MD. Polarization Selective Spectroscopy Experiments: Methodology and Pitfalls. J. Op. Soc. Am. B: Optical Physics. 2005;22:2009–2017. [Google Scholar]
- (35).Laage D, Hynes JT. Reorientational Dynamics of Water Molecules in Anionic Hydration Shells. Proc. Nat. Acad. Sci. 2007;104:11167–11172. doi: 10.1073/pnas.0701699104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Laage D, Hynes JT. On the Molecular Mechanism of Water Reorientation. J. Phys. Chem. B. 2008;112:14230–14242. doi: 10.1021/jp805217u. [DOI] [PubMed] [Google Scholar]