

Abstract
Insulin-like growth factor 1 receptor (IGF-1R) signaling has been implicated in several human neoplasms. However, the role of serum levels of insulin-like growth factors (IGFs) in lung cancer risk is controversial. We assessed the role of tissue-derived IGFs in lung carcinogenesis. We found that IGF-1 and IGF-2 levels in bronchial tissue specimens containing high-grade dysplasia were significantly higher than in those containing normal epithelium, hyperplasia, and squamous metaplasia. Derivatives of human bronchial epithelial cell lines with activation mutation in KRAS (V12) or loss of p53 overexpressed IGF-1 and IGF-2. Transformed characteristics of these cells were significantly suppressed by inactivation of IGF-1R or inhibition of IGF-1 or IGF-2 expression but enhanced by ovrexpresion of insulin-like growth factor receptor (IGF-1R) or exposure to the tobacco carcinogens (TC) 4-(methylnitrosamino)-1-(3-pyridyl)-1-butanone (NNK) and benzo[a]pyrene (BaP). We further determined the role of IGF-1R signaling in lung tumorigenesis by determining antitumor activities of the selective IGF-1R tyrosine kinase inhibitor cis-3-[3-(4-methyl-piperazin-l-yl)-cyclobutyl] -1-(2-phenyl-quinolin-7-yl) –imidazo [1,5-a]pyrazin-8-ylamine (PQIP) using an in vitro progressive cell system and an in vivo mouse model with a lung-specific IGF-1 transgene after exposure to TCs, including NNK plus BaP. Our results demonstrate that airway epithelial cells produce IGFs in an autocrine or paracrine manner, and these IGFs act jointly with TCs to enhance lung carcinogenesis. Further, the use of selective IGF-1R inhibitors may be a rational approach to controlling lung cancer.
Keywords: IGF, IGF-1R, lung cancer, cigarette smoking, carcinogenesis
Introduction
Lung cancer is the most commonly diagnosed cancer in the United States and causes the most mortality (1). Thus, we must investigate the cause of lung cancer and develop specific and effective chemopreventive and therapeutic approaches for the disease. Tobacco smoking confers the greatest risk for lung cancer (2); however, not all smokers develop lung cancer (3), and genetic factors are thought to play a role in lung cancer susceptibility in smokers.
The metabolites of tobacco carcinogens (TCs) induce DNA mutations in the initiation stage (4, 5), resulting in the accumulation of multiple genetic or epigenetic changes in tumor suppressors and oncogenes (6-12). Human bronchial epithelial (HBE) cells with damaged DNA are normally eliminated through apoptosis. However, deregulated apoptosis is thought to protect abnormal cells with damaged DNA and contribute to the development of various human cancers, including lung cancer. Hence, it is reasonable to assume that certain survival pathways protect TC-exposed HBE cells from apoptosis, leading to clonal outgrowth of initiated cells during the promotion stage of lung cancer. Such pathways could be effective targets for controlling the development and progression of lung cancer.
Many studies have focused on the epidermal growth factor receptor (EGFR) and its cognate ligands, which have been found to be overexpressed in 40%–80% of non–small cell lung cancers (NSCLCs). However, erlotinib and gefitinib, the two available tyrosine kinase inhibitors (TKIs) that target EGFR, have yielded poor responses in patients with NSCLC and a history of smoking (13) compared with patients who have never smoked. Those findings suggest that EGFR may not be the appropriate target in NSCLC patients with a history of smoking.
The insulin-like growth factor 1 receptor (IGF-1R) signaling pathway, which plays a key role in cell survival and transformation (14, 15), could be an effective chemopreventive target in lung cancer. IGF-1R is primarily activated by its cognate ligands (IGF-1 and IGF-2) with 1/2 to 1/15 the affinity for IGF-2 that it has for IGF-1. Insulin receptor (IR) also binds to IGFs, with roughly 1/40 to 1/100 the affinity for IGF-1 that it has for insulin (16), while insulin and IGF-2 are roughly equipotent for the IR-A isoform (17). The deregulated activation of IGF-1R has been implicated in the development of various cancers in mouse models (18-20). Inactivation of IGF-1R by gene disruption, antisense oligonucleotides, neutralizing antibodies, or dominant-negative mutants has yielded antitumor activities (21, 22). Those findings led us to hypothesize that IGF-1R signaling is involved in the malignant transformation of lung epithelial cells, rendering the resultant premalignant and malignant HBE cells depend on the signaling. However, prior studies have had inconsistent findings regarding the link between the circulating level of IGFs and lung cancer risk (23-25).
The purpose of this study was to determine the role of tissue-derived IGFs in lung carcinogenesis. We measured the expression of IGFs and the subsequent activation of IGF-1R in human preneoplastic HBE cell lines and tissue specimens. We further assessed the impact of IGF-1R signaling in the transformation of HBE cells in vitro and lung tumor progression in vivo. Finally, we determined the efficacy of genomic and pharmacologic approaches targeting IGF-1R in TC-exposed premalignant and malignant HBE cell lines and in IGF-1 transgenic mice carrying lung tumors induced by the TCs 4-(methylnitrosamino)- 1-(3-pyridyl)-1-butanone (NNK) and benzo[a]pyrene (BaP), given concurrently. On the basis of our findings, we suggest that IGF-1R is a potential target in preventing TC-induced lung cancer.
Materials and Methods
Cells and reagents
Human bronchial epithelial cell lines, 1799, 1198, 1170-I were described previously (26, 27). HBEC, HBEC/RASv12 HBEC/p53i and HBEC/p53i-Ras were provided by Dr. Minna (University of Texas Southwestern Medical Center) (28). IR+/R- cell line was a gift from Dr. Baserga (Thomas Jefferson University) (29). A549, H460, H596 and H1993 were obtained from the American Type Culture Collection. Urethane and BaP were purchased from Sigma-Aldrich. The IGF-1R TKI cis-3-[3-(4-methyl-piperazin-l-yl)-cyclobutyl]-1-(2-phenyl-quinolin-7-yl)-imidazo[1,5-a]pyrazin-8-ylamine (PQIP) was obtained from OSI Pharmaceuticals, Inc. Ad-dnIGF-1R was described previously (30). α-IR3 was purchased from Calbiochem.
Tissue microarray immunohistochemical analyses and biochemical analyses
Immunohistochemical analyses of tissue microarray comprising 367 biopsy specimens of normal, hyperplasic, squamous metaplastic and low- or high-grade dysplastic bronchial tissue specimens (Supplementary Table S1 shows patient demographics) were performed as described previously (26). The detailed description of the methods is in supplemental information. To analyze IGF-1R or insulin receptor tyrosine phosphorylation status, we immunoprecipitated total lysates with specific antibodies (1 μg) against IGF-1R, IR or phosphorylated tyrosine (pTyr) (all from Santa Cruz Biotech). Protein G-sepharose (Millipore) was added, and then the lysates were analyzed by Western blotting using Abs detecting IGF-1R, IR or pTyr (26, 31).
Colony and foci forming assay and retroviral transduction
Anchorage-dependent and -independent colony formation assays were done as previously described using complete medium (28). For the foci-forming assay, cells were grown in supplemented K-SFM medium (Invitrogen) up to 95% density in 24-well plates and then the medium were changed to unsupplemented medium. Fourteen days later the foci were counted after H&E staining (28). Retrovirus was generated and transduced to the cells as described previously (28). Further details are in supplemental information.
Quantitative real-time reverse transcription-polymerase chain reaction
Quantitative real-time reverse transcription polymerase chain reaction (QRTPCR) was performed using SYBR Green with an ABI PRISM 7700 sequence detection system (Applied Biosystems). Relative quantification of gene expression was performed using the comparative cycle threshold (CT) method according to the manufacturer's protocol (User Bulletin #2, ABI PRISM 7700 Sequence Detection System; PerkinElmer). The relative expression of the target genes was calculated as 2-(ΔCT) with difference (Δ) between CT for the target gene and that for the control gene (L32, ribosomal protein).
Animal treatment and histopathologic analysis
The IGF-1 Tg mice (C57/BL6 background) (32) were backcrossed with FVB/N mice (Jackson Laboratory) until more than 98% of the genome was identical to that in the FVB/N strain (tested by The University of Texas M. D. Anderson Cancer Center Genetically Engineered Mouse Facility). The mice were given food and water ad libitum and housed at 22 ± 2°C on a 12-hour:12-hour; light:dark cycle in a conventionally maintained facility. All mouse experiments were performed according to protocols approved by the Institutional Animal Care and Use Committee.
The mice were exposed to urethane (1 μmol in PBS) or NNK and BaP (3 μmol each in 0.1 mL of dimethylsulfoixide/corn oil) by means of a single intra-peritoneal injection or oral gavage for 8 weeks, respectively. To test the antitumor activities of PQIP, we treated the mice orally with 75 mg/kg PQIP dissolved in 25 mM l(+)-tartaric acid or vehicle only daily for 5 months after the first dose of NNK and BaP. The mice were monitored for weight changes twice a week.
The lungs were isolated after perfusion of PBS for protein extraction or pathological evaluation. Formalin-fixed mouse lungs were embedded in paraffin with the dorsal aspect facing downward so that most tumors would be present in 80 serial sections. Five slides (one from each of serial sections 1, 20, 40, 60 and 80) from each sample were stained with H&E for histopathologic assessment by a veterinary pathologist. The lung sections were microscopically evaluated to measure mean tumor number (N) and volume (V) with the pathologist blinded to the treatment conditions or genotypes. Tumor volume in mm3 was calculated as (long diameter × short diameter2)/2.
Serum glucose measurement
Intra-cardiac blood samples were collected for glucose evaluation. Blood samples were stored at 4°C overnight, and then the components were separated by centrifugation after the blood clotted. The supernatant was used for measuring the serum glucose concentration with an Ascensia Contour blood glucose monitoring system (Bayer Health Care LLC).
Statistical analysis
The Student's t-test, Mann-Whitney test or Fisher's exact test analyses were performed with Excel (Microsoft) or SPSS (SPSS Inc.) software. Differences between means with P < 0.05 were accepted as statistically significant; differences between means with 0.05 < P < 0.10 were accepted as representing tendencies.
Results
Expression of IGFs increases during lung carcinogenesis
We measured the expression levels of IGF-1, IGF-2, IGF-1R and phospho-IGF-1R in tissue micro arrays comprising 367 biopsy specimens of normal, hyperplasic, squamous metaplastic (SQM), and low- (L-DYS) or high (H-DYS)-grade dysplastic bronchial tissue specimens (Supplementary Table S1 shows patient demographics). IGF-1, IGF-2, and IGF-1R expressions were primarily cytoplasmic and less frequently nuclear (Figure 1A). We observed significantly higher expression of IGF-1 (P < 0.0001) and IGF-2 (P = 0.004) in H-DYS than in normal bronchial specimens (Figure 1B). We next assessed whether increased levels of the IGFs were associated with activation of IGF-1R by performing immunohistochemical analysis using an antibody that detects pIGF-1R/IR (Tyr1162,1163/Tyr1150,1151); the staining appeared in the cell membranes, cytoplasm, and nuclei. The staining in the membrane was significantly higher in H-DYS than in normal, hyperplastic, SQM, and L-DYS bronchial specimens (Figure 1B) and correlated well with the levels of IGF-1 and IGF-2 (Figure 1C). These results suggested that expression of IGFs is induced at an early stage of lung carcinogenesis, leading to activation of IGF-1R signaling.
Figure 1. Immunohistochemical (IHC) scoring of insulin-like growth factor (IGF) signaling molecules in normal and preneoplastic lesions in patients with non–small cell lung cancer.
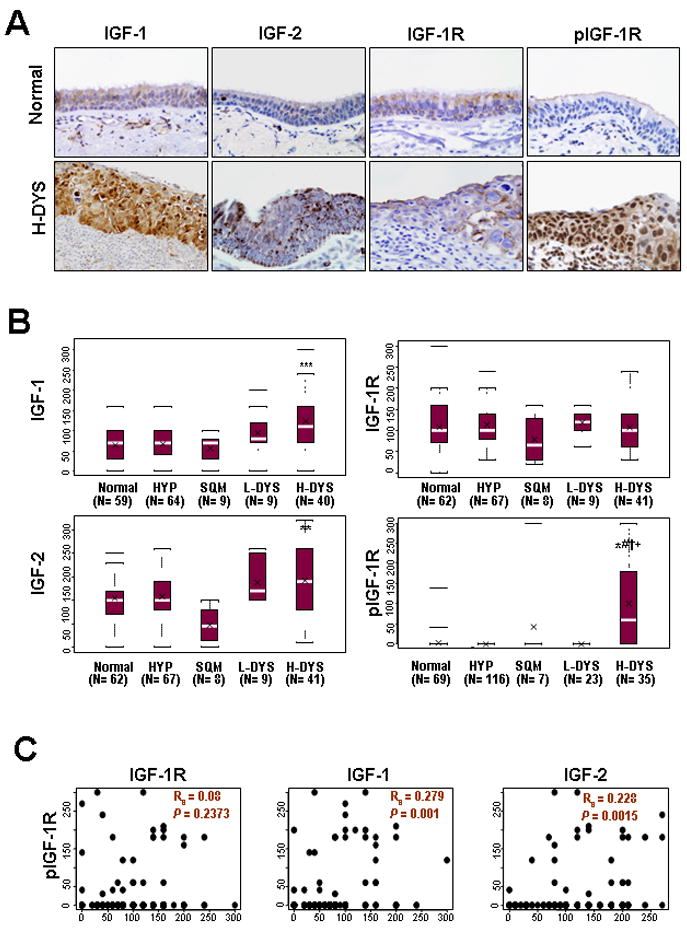
A) Representative results of IHC staining of IGF signaling biomarkers in normal and high-grade dysplasia (H-DYS) specimens. B) Comparison of IHC scoring of IGF-1, IGF-2, IGF-1R, or pIGF-1R by histologic evaluation of normal, hyperplasic, squamous metaplastic (SQM), and low- (L-DYS) or H-DYS bronchial tissue specimens. Box plots show the medians and the 25th and 75th percentiles; the bars give the ranges. Statistical significance of differences was determined with a repeated-measures model, and differences between groups were considered significant when; *, P<0.05, **, P<0.01, ***, P<0.001, compared with normal tissue; #, P<0.05, compared with HYP; †, P<0.05, compared with SQM; +, P<0.05, compared with L-DYS. C) Linear regression analysis performed to evaluate the significance of any association between the scoring of IGF-1R, IGF-1, or IGF-2 and the scoring of pIGF-1R in smokers and nonsmokers. The associations between pIGF-1R and IGF-1R, IGF-1 and IGF-2 were evaluated by using Spearman's rank correlation coefficient. The correlations were considered significant when P<0.05.
Dependence of HBE cells on autocrine IGFs for cell transformation
Because increased IGF expression was detected in human preneoplastic bronchial tissues, in which mutations of p53 or K-Ras frequently occur (33), we analyzed the immortalized HBE cell line (HBEC) and its derivatives expressing p53 short interfering RNA (HBEC/p53i), RASv12 (HBEC/Rasv12), or both (HBEC/p53i-Ras). These cell lines harbor several features of transformation, but are not yet fully malignant and mimic the premalignant stage in human lung carcinogenesis (28). QRT-PCR revealed that both HBEC/RASv12 and HBEC/p53i cells had markedly higher IGF-1 and IGF-2 mRNA expression than HBEC (Fig. 2A, left). H460 and A549 cell lines, of which the relative mRNA expressions of IGF-1 and IGF-2 were previously reported (34), were used as controls (Supplementary Fig. S1), and the relative amount of mRNA expression compared to in A549 are shown. These HBEC cell lines showed similar levels of IGF-1R but no detectable levels of IR or pIR expression (Fig. 2A, right; Supplementary Fig. 2 and 3). In contrast, pIGF-1R (Tyr1131), pIGF-1R (Tyr1135/1136, data not shown), and pAkt (Ser473) levels were obviously higher in HBEC cells expressing p53i and/or RASv12 than in parental HBEC cells (Fig. 2A, right), suggesting that loss of p53 expression and/or mutation of KRAS (V12) led the HBE cells to produce IGFs and activate IGF-1R.
Figure 2. Activated signaling of insulin-like growth factors (IGFs) in an in vitro lung carcinogenesis model that was established with defined genetic changes.
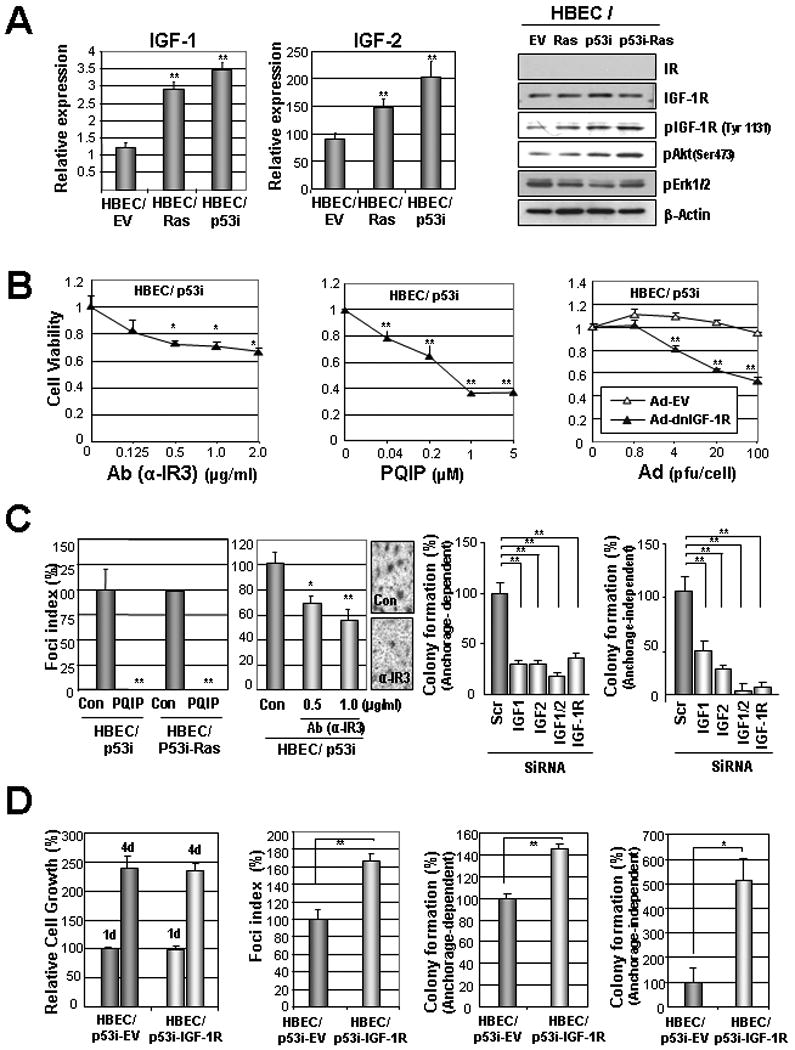
A) Real-time RT-PCR analysis of IGF-1 and IGF-2 mRNA levels in the indicated HBEC derivatives cultured in the absence of growth factor (left). Relative amounts of mRNAs normalized to those of A549 (Supplemental Figure 1) are shown. IGF-1R signaling cascades in the indicated HBEC derivatives (right). B) MTT analysis for the effects of α-IR3 (IGF-1R Ab), PQIP (IGF-1R TKI), adenovirus expressing dominant negative IGF-1R (Ad-dnIGF-1R), or empty adenovirus (Ad-EV) on the viability of HBEC/p53i cells cultured for 3 days in the absence of growth factor. C) Foci formation (left) and anchorage-dependent (middle) and -independent colony formation (right) of HBEC/p53i-RASv12 cells treated with PQIP or α-IR3 (left) or transfected with IGF-1-, IGF-2-, or IGF-1R-specific siRNAs (right). The relative numbers of colonies and foci in defined area are shown. Data are means ± standard error of means (SEM). D) Retroviral overexpression of IGF-1R enhanced tumorigenicity but not growth/survival of HBEC/p53i cells. Student's t test; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
We then investigated whether activation of IGF-1R contributes to HBEC transformation. We first examined the effects of IGF-1R inactivation on the survival potential of HBEC/p53i cells when cultured in the absence of EGF. We found that HBEC/p53i cells treated with IGF-1R-targeting monoclonal antibody (α-IR3) or a tyrosine kinase inhibitor (PQIP) targeting IGF-1R or infection with adenovirus expressing dominant negative IGF-1R (Ad-dnIGF-1R), all of which effectively suppressed the activation of IGF-1R signaling (Supplementary Fig. 3 and 4), induced a dose-dependent decrease in the viability of HBEC/p53i cells (Fig. 2B). We also found significantly decreased abilities of PQIP- and α-IR3-treated HBEC/p53i cells to form foci in confluent conditions compared to vehicle-treated control cells (Fig. 2C, left). Furthermore, both the anchorage-dependent and -independent colony-forming abilities of HBEC/p53i cells were significantly decreased when IGF or IGF-1R expression was abrogated by transfection with specific small interfering RNAs against IGF-1, IGF-2, or IGF-1R (Fig. 2C, right). HBEC/p53i-Ras showed similar response to the pharmacological and genetic approaches to suppress IGF-1R signaling (data not shown). To further investigate the role of IGF-1R signaling in the transformation of HBE cells, we infected HBEC/p53i cells with a retroviral vector carrying the human IGF-1R cDNA (HBEC/p53i-IGF-1R) or pBabe-hyg (HBEC/p53i-EV) as a control. When cultured in normal growth condition, the two cell lines showed no significant difference in growth. In contrast, HBEC/p53i-IGF-1R cells demonstrated significantly more foci formation and anchorage-dependent and -independent colony-forming ability than did HBEC/p53i-EV cells. However, the HBEC/p53i-IGF-1R cells were still not able to develop tumors in nude mice until 6 weeks after the subcutaneous injection (data not shown). These results suggest activation of IGF-1R contributes to HBE cellular transformation. However, more genetic changes in addition to IGF-1R activation and loss of p53 are needed to acquire a full malignant phenotype in HBE cells.
Despite the fact that TCs are genotoxic and may induce cell death, HBE cells have shown increased viability when exposed to NNK (26). Thus, we sought to determine whether activated IGF-1R can exert protective effects on TC-exposed HBE cells. HBEC derivatives showed markedly increased viability when exposed to NNK in the absence of EGF, with the greatest increase seen in HBEC/p53i-Ras cells (Fig. 3A). Moreover, HBEC/p53i-Ras cells showed the greatest increase in the numbers of foci (Fig. 3B, left) and anchorage-independent colony formation (Fig. 3B, right). These transformed phenotypes induced by NNK exposure in HBEC/p53i-Ras cells were almost completely suppressed by PQIP, indicating the role of IGF-1R signaling in TC-induced HBE cell transformation.
Figure 3. Insulin-like growth factors (IGFs) and tobacco carcinogens synergizing to stimulate the transformation of human bronchial epithelium cells (HBEC).
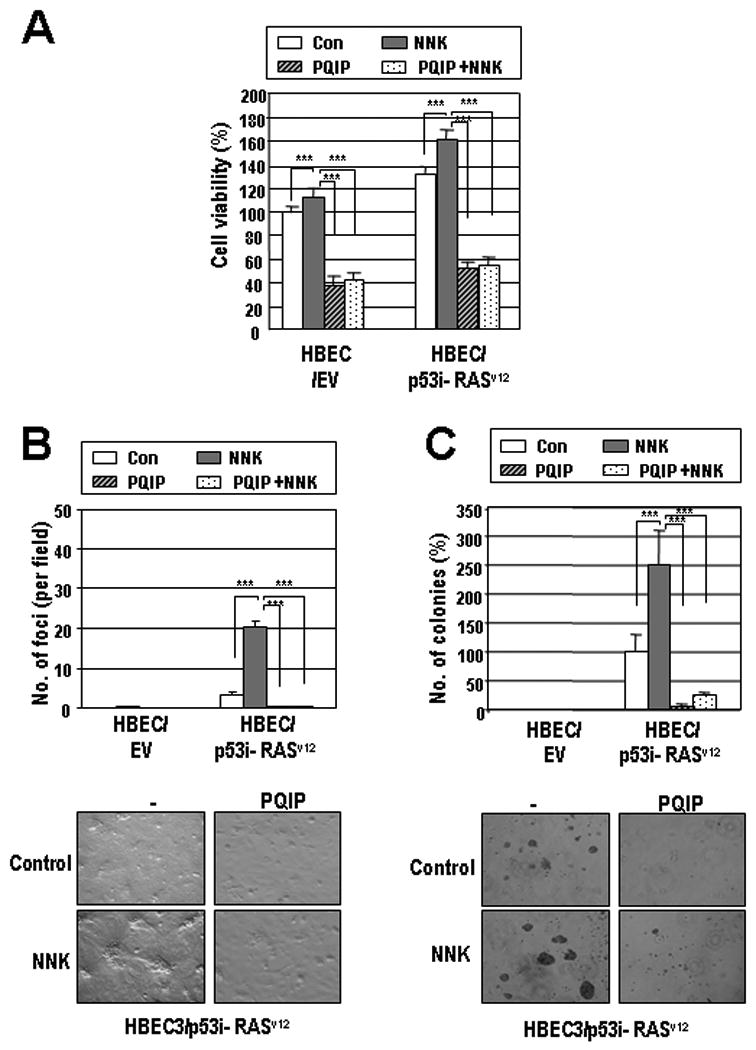
HBEC/EV and HBEC/p53i- RASv12 were treated with NNK (5 μM) or vehicle with or without PQIP (1 μM). MTT (A), foci-forming (B), and soft agar assays (C) were performed. Representative pictures of foci and soft agar colony of HBEC/p53i-RASv12 are shown. Data are means ± SEM. Student's t test; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
Dependence of TC-exposed HBE cells on IGF-1R signaling for maintaining transformed phenotypes
The findings above showed an important role for IGF-1R-mediated signaling in HBE cell transformation. Hence, we evaluated the effects of inhibiting IGF-1R signaling on an in vitro model of progressive lung carcinogenesis, which was composed of immortalized 1799, premalignant 1198, and malignant 1170-I HBE cells (27, 35). The 1198 and 1170-I cells exhibited greater levels of pIGF-1R and pIRS-1 (Fig. 4A) than the 1799 cells. The 1198 and 1170-I cells showed dose-dependent decreases in cell viability (Fig. 4B) and anchorage-dependent colony formation (Fig. 4C) when infected with Ad-dnIGF-1R whereas they showed no such decreases when infected with Ad-EV. In contrast, 1799 cells were minimally affected by the Ad-dnIGF-1R infection. Furthermore, 1198 cells (data not shown) and 1170-I cells (Fig. 4D) showed significantly decreased viability in response to the IGF-1R TKI (PQIP), but not to EGFR TKI (erlotinib) when treated in the absence of a growth factor. Neither PQIP nor erlotinib affected 1799 cells' viability. These findings indicate that IGF-1R activation is required for the transformation of TC-exposed HBE cells.
Figure 4. Effects of insulin-like growth factor-1 receptor (IGF-1R) signaling blocking on the survival of tobacco carcinogens (TC)-induced in vitro lung carcinogenesis model cell lines.
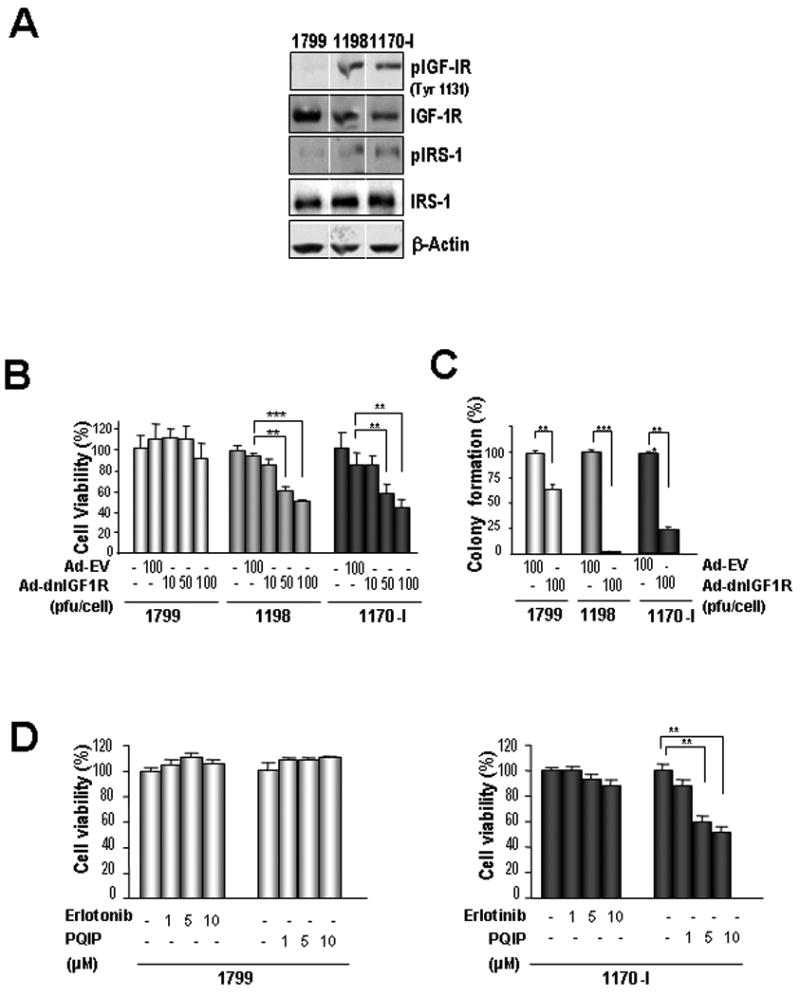
A) Expression of the IGFR signaling molecules in 1788, 1198, and 1170-I cells. B-D) MTT (B, D) and anchorage-dependent colony formation (C) analyses of 1799, 1198, and 1170-1 cells after the IGF-1R blocking with Ad-dnIGF-1R or with PQIP or the EGFR blocking with erlotinib in the absence of growth factor. (B, D) The cells treated with the drugs for 3 days were subjected to the MTT assay. Data are means ± SEM. Student's t test; *, P < 0.05; **, P < 0.01; ***, P < 0.001.
IGF-induced spontaneous lung tumor formation and facilitation of TC-induced lung tumorigenesis
To determine the role of IGF-1R signaling in lung tumorigenesis in vivo, we analyzed IGF-1 Tg mice (FVB/N background), in which human IGF-1 is selectively expressed in the type II alveolar cells of the lung under the control of surfactant protein C promoter (32), after exposure to either vehicle (Con) or NNK/BaP. At 15 months of age, the IGF-1 Tg mice showed an increase in spontaneous lung tumor incidence (Fig. 5A and Supplementary Table 2). Although the difference was not statistically significant (P=0.136), probably due to the small sample size of the WT mice and the low tumor incidence in the IGF-1 Tg mice, this result suggests that overexpression of IGF-1 may facilitate spontaneous tumor formation in the lung. When exposed to NNK/BaP, the IGF-1 Tg mice showed significantly greater lung tumor parameters than did the WT mice (Fig. 5A and Supplementary Table 2). The TC exposure induced markedly greater increases in tumor incidence, multiplicity and volume, compared to vehicle-treated controls, in IGF-1 Tg mice than in WT mice. The TC expedited lung tumor formation more in IGF-1 Tg than in WT mice in younger ages, with another carcinogen or in another genetic background (Supplementary Fig. 5 and 6). To assess the difference in the degree of lung carcinogenesis between IGF-1 Tg and WT mice, we performed histological evaluations of the lung tissues of the 9-month-old mice (7 months after the first dose of NNK and BaP at 2 months old) (Fig. 5B). The IGF-1 Tg mice exhibited advanced carcinogenesis (Fig. 5B), incidence, multiplicity, and volume of lung adenocarcinoma compared to WT mice. Representative pictures of the lungs of NNK/BaP-exposed WT and IGF-1 Tg mice are shown in Figure 5C. Typically, epithelial hyperplasia of the bronchioles, atypical adenomatous hyperplasia, adenomas, and adenocarcinomas were observed, suggesting that the IGF-1 transgene facilitated lung cancer development initiated by TCs. All together, these data strongly indicate that IGF-1 overexpression synergizes with TCs to accelerate lung carcinogenesis.
Figure 5. Tissue-specific expression of insulin-like growth factor-1 (IGF-1) accelerating tobacco carcinogen induced lung tumorigenesis in mice.
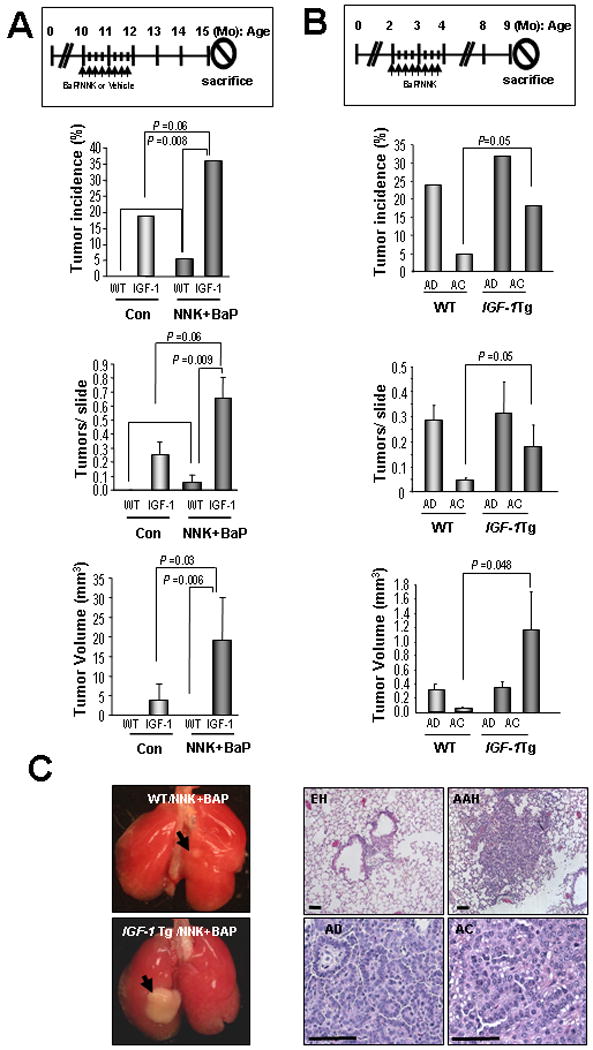
A) Spontaneous or NNK/BaP-induced lung tumor formation in WT and IGF-1 Tg mice. Ten-month-old WT or IGF-1 Tg mice were exposed to NNK/BaP or vehicle for 8 weeks, sacrificed at the age of 15 months. Data are shown with means ± SEM. B) Two-month-old WT or IGF-1 Tg mice were exposed to NNK/BaP for 8 weeks, sacrificed at age 9 months, and histologically evaluated. C) The whole-mount images of representative tumors and the histology of H&E stained sections. EH, epithelial hyperplasia; AAH, atypical adenohyperplasia. Fisher's exact test was used for tumor incidence. Mann-Whitney test was used for the others.
Antitumor activity of IGF-1R-targeted therapy for TC-induced tumors in IGF-1 Tg mice
We then examined whether inactivation of IGF-1R can prevent TC-stimulated lung cancer development by assessing the antitumor activity of PQIP in the IGF-1 Tg mice exposed to TCs. We exposed 2-month-old IGF-1 Tg mice to TCs and started treating them with PQIP at 7 months of age, when the tumors were already present (Supplementary Fig. 6). The lungs were examined when the mice were 10 months old, at which time the mice treated with PQIP had a lower tumor incidence than the control group, although the difference was not statistically significant (Fig. 6B, left). PQIP treatment significantly decreased the tumor multiplicities and the tumor volume in IGF-1 Tg mice. The histological evaluation revealed that PQIP treatment reduced the incidence of both adenomas and adenocarcinomas, but the reduction was not significant. However, the NNK/BaP-exposed mice had marginally significantly fewer adenocarcinomas after PQIP treatment than did the untreated mice. Most important, we observed a significant difference in the mean volume of the AD and AC between the NNK/BaP group and the NNK/BaP + PQIP group. These findings indicate that PQIP had an inhibitory effect on TC/IGF-1-induced lung tumor progression. The observed antitumor activities of PQIP were also associated with the inhibition of pIGF-1R and pAkt in lung tissues while not inhibiting pErk1/2 (Fig. 6C). PQIP treatment also inhibited IR activation in vivo (Fig. 6C). However, in the treated mice, it induced only moderate hyperglycemia, which was not statistically significant (Fig. 6D). No obvious changes in body weight (Fig. 6D) and no detectable levels of organ toxicity (data not shown) were found after PQIP treatment. Overall, these results indicate that the antitumor activities of PQIP are likely responsible for the observed decrease in lung tumor multiplicity and volume.
Figure 6. Effect of PQIP on lung tumorigenesis induced by insulin-like growth factor 1 and NNK/BaP.
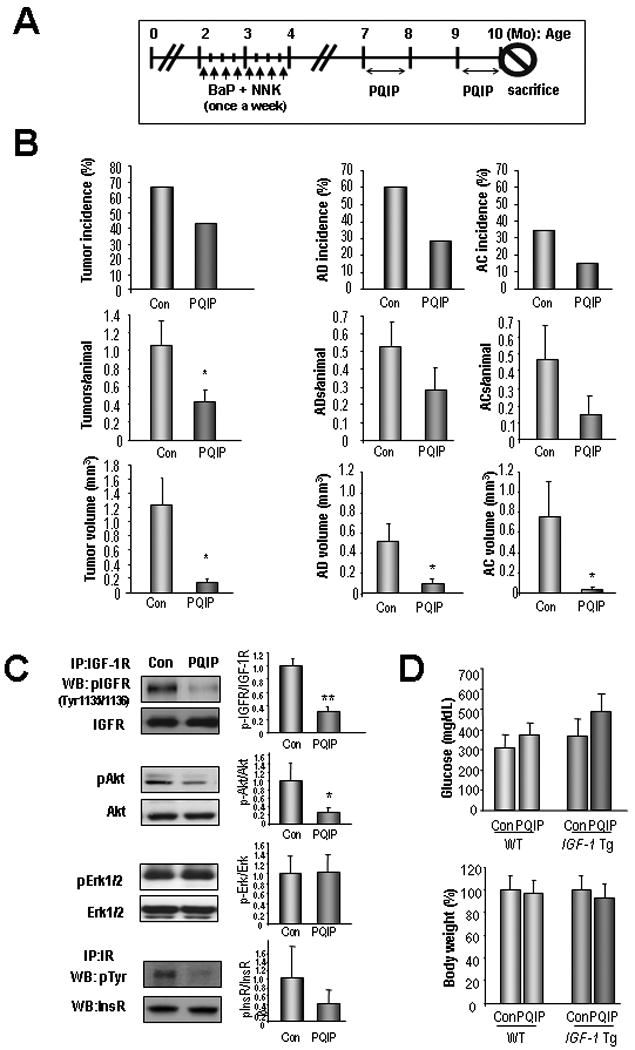
A) Treatment schedule for NNK/BaP and PQIP treatment. The vehicle (Con) or PQIP were administrated daily for 2 months with 1 month interval. B) Histological evaluation of the lung tumors induced by TCs; adenomas, AD; adenocarcinomas, AC. Student's t-test or Mann-Whitney statistical test were used. C) Inhibition of IGF signaling in the mice treated with PQIP. Total lung lysates from the IGF-1 Tg mice were subjected to immunoprecipitation. Densitometry analyses result of phosphorylated protein vs total protein intensity in 3 or more samples is shown. D) Serum glucose level was measured. A comparison of the body weight of the mice before and after the treatment is given. Student's t test; *, P < 0.05.
Discussion
Several prospective epidemiological studies have implicated high levels of circulating IGF-1 levels in an increased risk of many human cancers, and components of the IGF axis have been variably associated with tumor grade and prognosis (14, 36). We previously demonstrated that down-regulation of IGF-binding protein 3 occurs frequently in patients with NSCLC and head and neck squamous cell carcinoma (HNSCC) (37-39), suggesting that IGF-1R signaling plays a role in aerodigestive tract cancer. However, prior studies have had controversial findings on the role of IGFs in lung cancer (23-25). The present studies revealed significant overexpression of IGFs and an association between activation of IGF-1R in human bronchial preneoplastic tissue samples, suggesting that increases in tissue-derived IGFs and subsequent activation of IGF-1R are common features of early lung carcinogenesis.
Focusing on the role of the overexpression of IGFs in bronchial preneoplastic epithelium, we noted that HBEC/RASv12 or HBEC/p53i had increased expression of IGF-1 and IGF-2 genes and increased activation of IGF-1R compared to the parental cells. Previous studies demonstrated that the knockdown of p53 and the introduction of K-RASV12 did not permit increased EGFR activity or overexpression but did permit a partial bypass of EGFR dependence (28). In the current study, retroviral overexpression of IGF-1R conferred enhanced tumorigenicity to HBEC cells. We also demonstrated that the inhibition of IGF-1R signaling through pharmacologic approaches or genomic approaches decreased the survival and tumorigenic potentials of HBEC/p53i cells. It is noteworthy that KRAS and p53 mutations are frequently found in bronchial metaplasia and dysplasia (33) and are implicated in the expression of IGFs (40-44). Therefore, it is likely that genetic changes involved in carcinogenesis, such as mutation of KRAS and loss of p53 expression, induce expression of IGFs and activation of IGF-1R, permitting the transformation of HBE cells. Although the mechanism by which KRAS-mutant-expressed and p53-suppressed HBE cells induce IGF expression remains to be explored, our in vitro and in vivo findings support the idea that IGF-1R signaling is activated by autocrine IGFs in early lung carcinogenesis and jointly act with TCs in the etiology of cancer. We also assessed the role of tissue-derived IGFs in lung cancer by taking advantage of previously described, lung-specific IGF-I Tg mice (32). It has been demonstrated that overexpression of IGFs can enhance spontaneous or carcinogen-induced tumor formation (18, 20, 45) and that disrupting IGF-1R function results in tumor growth inhibition in several model systems (46, 47). Consistent with these findings, the expression of IGF-1 in the alveolar epithelium resulted in increased spontaneous tumors, showing that IGF-1R activated by persistent IGF-1 expression can induce lung tumor development in mice. The effect of IGF overexpression in tumor development was more evident when the development was induced by exposure to TCs. Thus, it is plausible to say that constitutive expression of IGF-1 in lung epithelial cells makes the cells more susceptible to TC-induced lung carcinogenesis.
TC-induced human lung cancer evolves through a multi-step process that is driven by genetic and epigenetic changes. However, premalignant and malignant changes in lung epithelial cells could depend on specific cell-survival pathways that protect them from apoptosis. Because chemoprevention is the use of chemical agents to reverse, suppress, or prevent the progress of carcinogenesis (48), our findings that IGF-1R signaling inhibition increases tumor latency suggests that targeting IGF-1R has the potential for clinical lung cancer chemoprevention. In our study, we also noticed that PQIP inhibited IR and caused moderate but not significant hyperglycemia in the mice. PQIP increased insulin levels in animal models (personal communication with OSI Pharmaceuticals) as shown for the IGF-1R/IR tyrosine kinase inhibitor BMS-554417 (49). Therefore, the mild hyperglycemia may have been maintained by hyper-secreted insulin in response to PQIP as a compensatory mechanism. Further studies are warranted to assess the potential involvement of the compensatory mechanism.
Taken together, our results provide the first evidence, to our knowledge, that preneoplastic HBE cells produce IGFs that enhance TC-stimulated lung cancer development through biochemical pathways (IGF-1R activation) in addition to genetic (DNA mutation) effect. Our data also suggest that IGFs could serve as markers for the early identification of individuals predisposed to smoking-induced lung cancers. It is important to remember, however, that our animal model, in which lung-specific IGF overexpression combined with TC treatment led to lung cancer development, is similar but not identical to human lung cancer, which is due to multiple accumulated mutations. Therefore, our results do not necessarily prove the efficacy of IGF-1R inhibitors against human NSCLC. However, our findings are proof-of-principle that targeted inactivation of IGF-1R may yield chemopreventive benefits in individuals with a history of smoking. Extensive and complete chronic toxicity profile testing in clinical trials is warranted before further development of IGF-1R TKIs as lung cancer chemopreventive agents.
Supplementary Material
Acknowledgments
We thank OSI Pharmaceuticals for providing PQIP.
Grant support: NIH grants R01 CA109520 and CA100816-01A1 (H.-Y. L.), and in part DOD grant W81XWH-04-1-0142-01-VITAL (W.K. H.), NIH grant P50 CA58187 (Y.E.M.) and a Department of Veterans Affairs Merit Review Grant (Y.E.M.).
References
- 1.Jemal A, Siegel R, Ward E, Murray T, Xu J, Thun MJ. Cancer statistics, 2007. CA Cancer J Clin. 2007;57:43–66. doi: 10.3322/canjclin.57.1.43. [DOI] [PubMed] [Google Scholar]
- 2.Hecht SS. Cigarette smoking and lung cancer: chemical mechanisms and approaches to prevention. Lancet Oncol. 2002;3:461–9. doi: 10.1016/s1470-2045(02)00815-x. [DOI] [PubMed] [Google Scholar]
- 3.Peto R, Darby S, Deo H, Silcocks P, Whitley E, Doll R. Smoking, smoking cessation, and lung cancer in the UK since 1950: combination of national statistics with two case-control studies. BMJ. 2000;321:323–9. doi: 10.1136/bmj.321.7257.323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tam IY, Chung LP, Suen WS, et al. Distinct epidermal growth factor receptor and KRAS mutation patterns in non-small cell lung cancer patients with different tobacco exposure and clinicopathologic features. Clin Cancer Res. 2006;12:1647–53. doi: 10.1158/1078-0432.CCR-05-1981. [DOI] [PubMed] [Google Scholar]
- 5.Hagiwara N, Mechanic LE, Trivers GE, et al. Quantitative detection of p53 mutations in plasma DNA from tobacco smokers. Cancer Res. 2006;66:8309–17. doi: 10.1158/0008-5472.CAN-06-0991. [DOI] [PubMed] [Google Scholar]
- 6.Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M. Transcriptional repression of the anti-apoptotic survivin gene by wild type p53. J Biol Chem. 2002;277:3247–57. doi: 10.1074/jbc.M106643200. [DOI] [PubMed] [Google Scholar]
- 7.Xu HJ, Quinlan DC, Davidson AG, et al. Altered retinoblastoma protein expression and prognosis in early-stage non-small-cell lung carcinoma. J Natl Cancer Inst. 1994;86:695–9. doi: 10.1093/jnci/86.9.695. [DOI] [PubMed] [Google Scholar]
- 8.Floyd HS, Jennings-Gee JE, Kock ND, Miller MS. Genetic and epigenetic alterations in lung tumors from bitransgenic Ki-rasG12C expressing mice. Mol Carcinog. 2006;45:506–17. doi: 10.1002/mc.20181. [DOI] [PubMed] [Google Scholar]
- 9.Camps C, Sirera R, Bremnes R, et al. Is there a prognostic role of K-ras point mutations in the serum of patients with advanced non-small cell lung cancer? Lung Cancer. 2005;50:339–46. doi: 10.1016/j.lungcan.2005.06.007. [DOI] [PubMed] [Google Scholar]
- 10.Sugio K, Uramoto H, Ono K, et al. Mutations within the tyrosine kinase domain of EGFR gene specifically occur in lung adenocarcinoma patients with a low exposure of tobacco smoking. Br J Cancer. 2006;94:896–903. doi: 10.1038/sj.bjc.6603040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Toyooka S, Tokumo M, Shigematsu H, et al. Mutational and epigenetic evidence for independent pathways for lung adenocarcinomas arising in smokers and never smokers. Cancer Res. 2006;66:1371–5. doi: 10.1158/0008-5472.CAN-05-2625. [DOI] [PubMed] [Google Scholar]
- 12.Mao L, Lee JS, Kurie JM, et al. Clonal genetic alterations in the lungs of current and former smokers. J Natl Cancer Inst. 1997;89:857–62. doi: 10.1093/jnci/89.12.857. [DOI] [PubMed] [Google Scholar]
- 13.Pao W, Miller V, Zakowski M, et al. EGF receptor gene mutations are common in lung cancers from “never smokers” and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc Natl Acad Sci U S A. 2004;101:13306–11. doi: 10.1073/pnas.0405220101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pollak MN, Schernhammer ES, Hankinson SE. Insulin-like growth factors and neoplasia. Nat Rev Cancer. 2004;4:505–18. doi: 10.1038/nrc1387. [DOI] [PubMed] [Google Scholar]
- 15.Sell C, Dumenil G, Deveaud C, et al. Effect of a null mutation of the insulin-like growth factor I receptor gene on growth and transformation of mouse embryo fibroblasts. Mol Cell Biol. 1994;14:3604–12. doi: 10.1128/mcb.14.6.3604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boute N, Pernet K, Issad T. Monitoring the activation state of the insulin receptor using bioluminescence resonance energy transfer. Mol Pharmacol. 2001;60:640–5. [PubMed] [Google Scholar]
- 17.Denley A, Carroll JM, Brierley GV, et al. Differential activation of insulin receptor substrates 1 and 2 by insulin-like growth factor-activated insulin receptors. Mol Cell Biol. 2007;27:3569–77. doi: 10.1128/MCB.01447-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.DiGiovanni J, Bol DK, Wilker E, et al. Constitutive expression of insulin-like growth factor-1 in epidermal basal cells of transgenic mice leads to spontaneous tumor promotion. Cancer Res. 2000;60:1561–70. [PubMed] [Google Scholar]
- 19.Moorehead RA, Fata JE, Johnson MB, Khokha R. Inhibition of mammary epithelial apoptosis and sustained phosphorylation of Akt/PKB in MMTV-IGF-II transgenic mice. Cell Death Differ. 2001;8:16–29. doi: 10.1038/sj.cdd.4400762. [DOI] [PubMed] [Google Scholar]
- 20.Moorehead RA, Sanchez OH, Baldwin RM, Khokha R. Transgenic overexpression of IGF-II induces spontaneous lung tumors: a model for human lung adenocarcinoma. Oncogene. 2003;22:853–7. doi: 10.1038/sj.onc.1206188. [DOI] [PubMed] [Google Scholar]
- 21.Bahr C, Groner B. The insulin like growth factor-1 receptor (IGF-1R) as a drug target: novel approaches to cancer therapy. Growth Horm IGF Res. 2004;14:287–95. doi: 10.1016/j.ghir.2004.02.004. [DOI] [PubMed] [Google Scholar]
- 22.Garcia-Echeverria C. Medicinal chemistry approaches to target the kinase activity of IGF-1R. IDrugs. 2006;9:415–9. [PubMed] [Google Scholar]
- 23.Spitz MR, Barnett MJ, Goodman GE, Thornquist MD, Wu X, Pollak M. Serum insulin-like growth factor (IGF) and IGF-binding protein levels and risk of lung cancer: a case-control study nested in the beta-Carotene and Retinol Efficacy Trial Cohort. Cancer Epidemiol Biomarkers Prev. 2002;11:1413–8. [PubMed] [Google Scholar]
- 24.London SJ, Yuan JM, Travlos GS, et al. Insulin-like growth factor I, IGF-binding protein 3, and lung cancer risk in a prospective study of men in China. J Natl Cancer Inst. 2002;94:749–54. doi: 10.1093/jnci/94.10.749. [DOI] [PubMed] [Google Scholar]
- 25.Lukanova A, Toniolo P, Akhmedkhanov A, et al. A prospective study of insulin-like growth factor-I, IGF-binding proteins-1, -2 and -3 and lung cancer risk in women. Int J Cancer. 2001;92:888–92. doi: 10.1002/ijc.1265. [DOI] [PubMed] [Google Scholar]
- 26.Jin Q, Feng L, Behrens C, et al. Implication of AMP-activated protein kinase and Akt-regulated survivin in lung cancer chemopreventive activities of deguelin. Cancer Res. 2007;67:11630–9. doi: 10.1158/0008-5472.CAN-07-2401. [DOI] [PubMed] [Google Scholar]
- 27.Klein-Szanto AJ, Iizasa T, Momiki S, et al. A tobacco-specific N-nitrosamine or cigarette smoke condensate causes neoplastic transformation of xenotransplanted human bronchial epithelial cells. Proc Natl Acad Sci U S A. 1992;89:6693–7. doi: 10.1073/pnas.89.15.6693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sato M, Vaughan MB, Girard L, et al. Multiple oncogenic changes (K-RAS(V12), p53 knockdown, mutant EGFRs, p16 bypass, telomerase) are not sufficient to confer a full malignant phenotype on human bronchial epithelial cells. Cancer Res. 2006;66:2116–28. doi: 10.1158/0008-5472.CAN-05-2521. [DOI] [PubMed] [Google Scholar]
- 29.Prisco M, Romano G, Peruzzi F, Valentinis B, Baserga R. Insulin and IGF-I receptors signaling in protection from apoptosis. Horm Metab Res. 1999;31:80–9. doi: 10.1055/s-2007-978703. [DOI] [PubMed] [Google Scholar]
- 30.Lee CT, Wu S, Gabrilovich D, et al. Antitumor effects of an adenovirus expressing antisense insulin-like growth factor I receptor on human lung cancer cell lines. Cancer Res. 1996;56:3038–41. [PubMed] [Google Scholar]
- 31.Han JY, Oh SH, Morgillo F, et al. Hypoxia-inducible factor 1alpha and antiangiogenic activity of farnesyltransferase inhibitor SCH66336 in human aerodigestive tract cancer. J Natl Cancer Inst. 2005;97:1272–86. doi: 10.1093/jnci/dji251. [DOI] [PubMed] [Google Scholar]
- 32.Frankel SK, Moats-Staats BM, Cool CD, Wynes MW, Stiles AD, Riches DW. Human insulin-like growth factor-IA expression in transgenic mice promotes adenomatous hyperplasia but not pulmonary fibrosis. Am J Physiol Lung Cell Mol Physiol. 2005;288:L805–12. doi: 10.1152/ajplung.00420.2004. [DOI] [PubMed] [Google Scholar]
- 33.Wistuba II, Lam S, Behrens C, et al. Molecular damage in the bronchial epithelium of current and former smokers. J Natl Cancer Inst. 1997;89:1366–73. doi: 10.1093/jnci/89.18.1366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Quinn KA, Treston AM, Unsworth EJ, et al. Insulin-like growth factor expression in human cancer cell lines. J Biol Chem. 1996;271:11477–83. doi: 10.1074/jbc.271.19.11477. [DOI] [PubMed] [Google Scholar]
- 35.Chun KH, Kosmeder JW, 2nd, Sun S, et al. Effects of deguelin on the phosphatidylinositol 3-kinase/Akt pathway and apoptosis in premalignant human bronchial epithelial cells. J Natl Cancer Inst. 2003;95:291–302. doi: 10.1093/jnci/95.4.291. [DOI] [PubMed] [Google Scholar]
- 36.Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100:57–70. doi: 10.1016/s0092-8674(00)81683-9. [DOI] [PubMed] [Google Scholar]
- 37.Chang YS, Kong G, Sun S, et al. Clinical significance of insulin-like growth factor-binding protein-3 expression in stage I non-small cell lung cancer. Clin Cancer Res. 2002;8:3796–802. [PubMed] [Google Scholar]
- 38.Chang YS, Wang L, Liu D, et al. Correlation between insulin-like growth factor-binding protein-3 promoter methylation and prognosis of patients with stage I non-small cell lung cancer. Clin Cancer Res. 2002;8:3669–75. [PubMed] [Google Scholar]
- 39.Papadimitrakopoulou VA, Brown EN, Liu DD, et al. The prognostic role of loss of insulin-like growth factor-binding protein-3 expression in head and neck carcinogenesis. Cancer Lett. 2006;239:136–43. doi: 10.1016/j.canlet.2005.08.009. [DOI] [PubMed] [Google Scholar]
- 40.Ali SH, DeCaprio JA. Cellular transformation by SV40 large T antigen: interaction with host proteins. Semin Cancer Biol. 2001;11:15–23. doi: 10.1006/scbi.2000.0342. [DOI] [PubMed] [Google Scholar]
- 41.Zhang L, Zhan Q, Zhan S, et al. p53 regulates human insulin-like growth factor II gene expression through active P4 promoter in rhabdomyosarcoma cells. DNA Cell Biol. 1998;17:125–31. doi: 10.1089/dna.1998.17.125. [DOI] [PubMed] [Google Scholar]
- 42.Bocchetta M, Eliasz S, De Marco MA, Rudzinski J, Zhang L, Carbone M. The SV40 large T antigen-p53 complexes bind and activate the insulin-like growth factor-I promoter stimulating cell growth. Cancer Res. 2008;68:1022–9. doi: 10.1158/0008-5472.CAN-07-5203. [DOI] [PubMed] [Google Scholar]
- 43.Cadoret A, Baron-Delage S, Bertrand F, et al. Oncogene-induced up-regulation of Caco-2 cell proliferation involves IGF-II gene activation through a protein kinase C-mediated pathway. Oncogene. 1998;17:877–87. doi: 10.1038/sj.onc.1202013. [DOI] [PubMed] [Google Scholar]
- 44.Durkin JP, Chakravarthy B, Mealing G, et al. The role of signal-transducing events in the proliferative response of cells to a mitogenic viral K-ras protein. Cell Signal. 1990;2:285–97. doi: 10.1016/0898-6568(90)90056-g. [DOI] [PubMed] [Google Scholar]
- 45.DiGiovanni J, Kiguchi K, Frijhoff A, et al. Deregulated expression of insulin-like growth factor 1 in prostate epithelium leads to neoplasia in transgenic mice. Proc Natl Acad Sci U S A. 2000;97:3455–60. doi: 10.1073/pnas.97.7.3455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Camirand A, Pollak M. Co-targeting IGF-1R and c-kit: synergistic inhibition of proliferation and induction of apoptosis in H 209 small cell lung cancer cells. Br J Cancer. 2004;90:1825–9. doi: 10.1038/sj.bjc.6601682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Haddad T, Yee D. Targeting the insulin-like growth factor axis as a cancer therapy. Future Oncol. 2006;2:101–10. doi: 10.2217/14796694.2.1.101. [DOI] [PubMed] [Google Scholar]
- 48.Sporn MB, Dunlop NM, Newton DL, Smith JM. Prevention of chemical carcinogenesis by vitamin A and its synthetic analogs (retinoids) Fed Proc. 1976;35:1332–8. [PubMed] [Google Scholar]
- 49.Haluska P, Carboni JM, Loegering DA, et al. In vitro and in vivo antitumor effects of the dual insulin-like growth factor-I/insulin receptor inhibitor, BMS-554417. Cancer Res. 2006;66:362–71. doi: 10.1158/0008-5472.CAN-05-1107. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.