

Graphical abstract
Design, synthesis and biological evaluation of a series of chloropyridyl ester-derived SARS-CoV 3CLpro Inhibitors are described Inhibitors 10 has shown potent activity in both enzyme inhibitory and antiviral assays

Keywords: Synthesis, SARS 3CLpro, Ester, Antiviral, Inhibitor
Abstract
Design, synthesis and biological evaluation of a series of 5-chloropyridine ester-derived severe acute respiratory syndrome-coronavirus chymotrypsin-like protease inhibitors is described. Position of the carboxylate functionality is critical to potency. Inhibitor 10 with a 5-chloropyridinyl ester at position 4 of the indole ring is the most potent inhibitor with a SARS-CoV 3CLpro IC50 value of 30 nM and an antiviral EC50 value of 6.9 μM. Molecular docking studies have provided possible binding modes of these inhibitors.
Since its first appearance in southern China in late 2002, severe acute respiratory syndrome (SARS) has been recognized as a global threat.1 It has affected more than 8000 individuals in 32 countries and caused nearly 800 fatalities worldwide within a few months.2 Its causative pathogen is a novel coronavirus and termed as SARS-CoV.3, 4 While SARS is contained in the world and no more cases have been reported since April 2004, there is expectation that this epidemic will strike again in an even more severe form. Furthermore, the nature of its unpredictable outbreak is a potential threat to the global economy and public health. To date, no effective therapy exists for this viral illness.
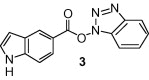
The SARS coronavirus is a positive-strand RNA virus. The 5′ two-thirds of the genome encodes two overlapping polyproteins, pp1a and pp1ab, which are processed to generate the viral replication complex. During viral replication, the replicase polyprotein undergoes extensive processing by two viral proteases namely, chymotrypsin-like protease (3CLpro) and papain-like protease (PLpro).5, 6 Because of their essential roles in viral replication, both proteases are recognized as attractive targets for development of anti-SARS therapeutics.7 The structure and activity of the active sites of both SARS-CoV 3CLpro and SARS-CoV PLpro have been elucidated. Thus far, inhibitor design efforts are mostly limited to SARS-CoV 3CLpro and numerous covalent and noncovalent inhibitors have been reported.7 In our continuing interest in the design and development of SARS-CoV 3CLpro inhibitors, we recently reported structure-based design of a number of potent peptidomimetic SARS-CoV 3CLpro inhibitors (1 and 2).8 The SARS-CoV 3CLpro active site contains a catalytic dyad where a cysteine residue acts as a nucleophile and a histidine residue acts as the general acid base.9 The inhibitors bind to SARS-CoV 3CLpro through a covalent bond with the active site Cys-145 residue. These inhibitors contain peptidomimetic scaffolds and lacked adequate potency, particularly antiviral activity suitable for drug-development. Recently, Wong and co-workers reported a new class of potent small molecule benzotriazole ester-based 3CLpro inhibitors. Compound 3 is the most potent inhibitor among the benzotriazole esters.10 The mode of action involves acylation of the active site Cys-145 assisted by the catalytic dyad. This irreversible enzyme acylation was verified by electrospray ionization mass spectrometry of the inhibited enzyme. While these inhibitors have shown very impressive SARS-CoV 3CLpro enzyme inhibitory activity, their antiviral activity required improvement.11 It seems the indole-5-carboxylate moiety plays an important role in binding with the enzyme active site. Another class of hetereoaromatic ester inhibitors was also identified and studied.12, 13 The 5-chloropyridine moiety in 4 proved to be the key unit for the activity against 3CLpro. The report however lacked antiviral data. We report herein the development of 3-chloropyridyl ester-based SARS-CoV 3CLpro inhibitors that exhibit potent enzyme inhibitory activity as well as very good SARS-CoV antiviral activity in cell culture assays. We have also carried out molecular docking studies to obtain the potential binding mode of these inhibitors.
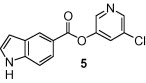
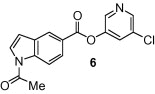
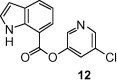
The general synthetic method for 5-chloropyridyl ester inhibitors is outlined in Scheme 1 . Various chloro-3-pyridinyl esters 5, 9, 10, 12–14 (Table 1 ) were synthesized by esterification of 5-chloro-3-pyridinol and the corresponding carboxylic acids14 mediated by DCC and DMAP at 23 °C in CH2Cl2. The synthesis of 1-acetylindolecarboxylate inhibitors were carried out by acetylation of indole 5 and 10 with acetic anhydride and pyridine under reflux to provide amide 6 and 11, respectively, in excellent yields.
Scheme 1.

Synthesis of inhibitors 5, 6, 9–14.
Table 1.
Structures and activity of inhibitors
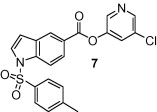
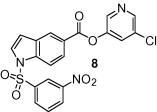
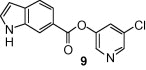
The synthesis of 1-sulfonylindolecarboxylate inhibitors is outlined in Scheme 2 . Direct sulfonamidation of the indole under regular TsCl/DMAP condition at 23 °C or higher temperatures could not provide the desired product. To increase its reactivity, the indole 15 was reduced to indoline 16 by sodium cyanoborohydride in excellent yield.15 The resulting indoline readily reacted with tosyl chloride or 3-nitrobenzenesulfonyl chloride to give sulfonamides 17 or 18 in good yields. Oxidation of indolines 17 and 18 to their corresponding indoles 19 and 20, respectively, was achieved using manganese dioxide at high temperature.16 Hydrolysis of the methyl esters to the corresponding acids 21 or 22 using sodium hydroxide followed by the general esterification method described in Scheme 1 afforded the target compounds 7 or 8.
Scheme 2.

Synthesis of inhibitors 7 and 8.
The structure and activity of inhibitors are shown in Table 1. The enzyme inhibitory activity of the active esters against SARS-CoV-3CLpro was determined using the full-length, authentic version of the enzyme in a FRET-based, microplate assay described by Grum-Tokars and co-workers.8, 17 The assays were performed in 96-well microplates using a reaction volume of 100 μL which contained 50 mM HEPES, pH 7.5, 100 nM authentic SARS-CoV-3CLpro enzyme, 1 mM DTT, 0.01 mg/mL BSA and varying concentrations of inhibitors. The reaction components, with the exception of substrate, were incubated for 20 min and the reaction was initiated by the addition of FRET-substrate HiLyte Fluor™ 488-Glu-Ser-Ala-Thr-Leu-Gln-Ser-Gly-Leu-Arg-Lys-Ala-Lys(QXL520™)-NH2, giving a final substrate concentration of 2 μM as described.8, 17 The IC50 values for inhibitors were determined by measuring the rates of reaction with increasing inhibitor concentrations.
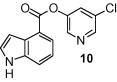
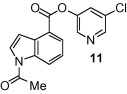
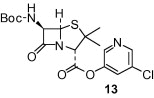
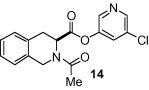
As shown in Table 1, the known10 benzotriazole ester inhibitor 3 was evaluated in our assay as a control. In inhibitor 5, the benzotriazole unit was replaced by a 5-chloropyridine unit. Inhibitor 5 has shown comparable enzymatic inhibitory potency (IC50 0.31 μM) as that of 3. However, inhibitor 3 did not exhibit any antiviral activity while 5-chloropyridyl ester 5 exhibited antiviral activity with an EC50 value of 24 μM.18 When the indole nitrogen was acetylated, the resulting compound 6 remained quite potent (IC50 of 0.40 μM) as did the tosylated indole 7 (IC50 of 0.37 μM). Interestingly, its nitrobenzenesulfonamide analog 8 shows an improved IC50 value (89 nM). We then investigated the importance of the carboxylic position on the benzene ring of indole. Accordingly, carboxylate substitution on indole rings at 5, 6, 4 and 7 positions resulted in chloropyridinyl esters 5, 9, 10 and 12, respectively. These inhibitors were evaluated and as it turned out, inhibitor 10, with a carboxylate at the 4-position, was the most potent inhibitor with an IC50 of 30 nM, a 10-fold potency enhancement over 5 containing a carboxylate at the 5-position. Compound 10 also shows the best SARS-CoV antiviral activity with an EC50 value of 6.9 μM. Acylation of 10–11 resulted in a 30-fold loss of potency. Penicillin-derived chloropyridine 13 did not show any appreciable activity. Tetrahydroisoquinoline derivative 14, however, exhibited an enzyme IC50 value of 0.14 μM. In general, an indole with a free nitrogen is more potent than its corresponding protected analogue.
To confirm that 3CLpro is covalently modified by 10, we determined enzyme modification using MALDI-TOF.19 Authentic SARS-CoV 3CLpro was incubated with compound 10 for 20 min and then analyzed in comparison with untreated enzyme. A shift of approximately 217 Da was observed after treatment of SARS-CoV 3CLpro with the inhibitor confirming covalent modification. Covalent modification by similar reactive esters has also been reported.(a), 20
To obtain molecular insight into the binding properties of these active ester-based inhibitors, we conducted docking studies in the 3CLpro active site. GOLD3.221 was used to dock our most active compound, 10 (GRL-0496), into the active site of the authentic SARS-CoV 3CLpro structure (PDBID: 2HOB).22 In search of obtaining a model of the associated complex between the unreacted ester and protein, (i.e. prior to nucleophilic attack by Cys-145), the distance between the carbonyl carbon atom of 10 and the sulfur atom of Cys-145 was constrained to be in the range of 2.5–3.5 Å. This pre-reaction or ‘collision complex’ is shown in Figures 1 and 2 , and resulted in a distance of 2.8 Å between the carbonyl carbon of 10 and the sulfur of Cys-145. This orientation of the ligand has the chloropyridyl group situated in the S1 pocket, with the chloro group pointing towards the surface of the protein. The nitrogen of the chloropyridinyl leaving group is in close proximity (2.4 Å) to the imidazole nitrogen of His-163. The carbonyl oxygen is situated between three backbone nitrogens, forming three hydrogen bonds. As shown in Figure 2: the first hydrogen bond is from Cys-145(NH) (2.3 Å), the second is from Ser-144(NH) (2.4 Å), and the third is from Gly-143(NH) (2.8 Å). This suggests that a fairly strong hydrogen bonding network is present within the active site which likely aids in positioning and stabilizing the carbonyl group of the ester for nucleophilic attack by the Cys-145. The indole group of 10 is positioned near the more hydrophobic S2 pocket, with the indole nitrogen likely interacting with the imidazole group of His-41, see Figure 2.
Figure 1.

Structures of SARS-CoV 3CLpro inhibitors.
Figure 2.

Relaxed stereoview of the GOLD conformation of an associated complex between 10 (green) and 3CLpro (PDBID: 2HOB), with residues shown in magenta.
Next, we analyzed the interaction between 10 and 3CLpro in the ‘post-reaction’ or covalently modified state. The product of the reaction of 10 with 3CLpro was docked using a more recently released 3CLpro crystal structure, (PDBID: 2V6N).20 This crystal structure contains a benzotriazole ester molecule which has reacted with the thiol of Cys-145, forming a covalently bound ligand similar to the compounds presented in this paper. We have used this crystal structure for the post-reaction complex due to the notable movement of the His-41 in 2V6N, which flips and is able to π stack with the aromatic moiety of the smaller covalently bound ligand.20 GOLD3.221 was chosen again for generating this model, as it has the capability for docking covalently bound ligands.
Figure 3 is a docked model of 10 covalently attached to the Cys-145 sulfur atom. The model suggests that the indole group of 10 shifts and positions itself where the leaving group was in the complex, more towards the S1 pocket. This positioning of inhibitor 10 in the complex is not surprising as a similar orientation has been proposed before by James’ group for similar esters.23, 24 More importantly though is the obvious π–π stacking of the indolyl of 10 with the imidazole ring of His-41, which is also seen in the benzotriazole group of the referenced crystal structure (2V6N).20 There is approximately 4 Å between the aromatic rings of the indolyl and the imidazole of His-41. This interaction clearly determines the position of the indolyl group of our compounds. Other residues shown in Figure 3, such as Asp-187 and Gln-189 are more than 5 Å away from the indolyl, but might come into play with larger substituents, such as in compound 11.
Figure 3.

GOLD docked conformation of 10 (green), covalently linked to Cys-145 of 3CLpro based on the 2V6N 3CLpro structure.20
The results of the docking studies presented here suggest that the indole group of 10 can potentially occupy two different binding pockets during the course of the reaction. Dynamic enzymatic rearrangement in the vicinity of Cys-145 have recently been suggested from the X-ray structure of SARS-CoV 3CLpro that had been reacted with 1-(4-dimethylaminobenzoyloxyl)-benzotriazole,20 and suggests that after covalently linking to Cys-145, the indolyl of 10 shifts towards the S1 pocket and stacks with the shifted imidazole ring of His-41, locking the orientation, with implications of where substitutions on the indolyl ring would be most beneficial. By having both of these models now available, structural modifications of the indole group can be tailored to enhance interactions with the S2 pocket for the complex formation to readily occur, but at the same time, consider that the ligand must be mobile enough to then occupy the S1 pocket post-reaction.
In conclusion, our design strategies by combining the key parts of two mechanism-based inhibitors led to a series of 5-chloropyridinyl indolecarboxylate inhibitors with enzymatic potency at submicromolar levels. The position of the carboxylic acid ester is critical to its potency. Indolecarboxylate 10 with a carboxylate functionality at the 4-position is the most potent inhibitor with an enzyme inhibitory activity against SARS-CoV 3CLpro with an IC50 of 30 nM and antiviral potency with an EC50 value of 6.9 μM. Further design and synthesis of more effective inhibitors are in progress in our laboratories.
Acknowledgment
The financial support of this work is provided by the National Institute of Health (NIAID, P01 A1060915).
References and notes
- 1.World Health Organization, Communicable Disease Surveillance & Response, website: http://www.who.int/csr/sars/archive/2003_05_07a/en and http://www.who.int/csr/sars/country/en/country2003_08_15.pdf.
- 2.He J.-F., Peng G.-W., Min J., Yu D.-W., Liang W.-L., Zhang S.-Y., Xu R.-H., Zheng H.-Y., Wu X.-W., Xu J., Wang Z.-H., Fang L., Zhang X., Li H., Yan X.-G., Lu J.-H., Hu Z.-H., Huang J.-C., Wan Z.-Y., Hou J.-L., Lin J.-Y., Song H.-D., Wang S.-Y., Zhou X.-J., Zhang G.-W., Gu B.-W., Zheng H.-J., Zhang X.-L., He M., Zheng K., Wang B.-F., Fu G., Wang X.-N., Chen S.-J., Chen Z., Hao P., Tang H., Ren S.-X., Zhong Y., Guo Z.-M., Liu Q., Miao Y.-G., Kong X.-Y., He W.-Z., Li Y.-X., Wu C.-I., Zhao G.-P., Chiu R.W.K., Chim S.S.C., Tong Y.-K., Chan P.K.S., Tam J.S., Lo Y.M.D. Science. 2004;303:1666. [Google Scholar]
- 3.Drosten C., Gunther S., Preiser W., van der Werf S., Brodt H.R., Becker S., Rabanau H., Panning M., Kolesnikova L., Fouchier R.A., Berger A., Burguiere A.M., Cinatl J., Eickmann M., Escriou N., Grywna K., Kramme S., Manuguerra J.C., Muller S., Rickerts V., Sturmer M., Vieth S., Klenk H.D., Osterhaus A.D., Schmitz H., Doerr H.W. N. Engl. J. Med. 2003;348:1967. doi: 10.1056/NEJMoa030747. [DOI] [PubMed] [Google Scholar]
- 4.Ksiazek T.G., Erdman D., Goldsmith C.S., Zaki S.R., Peret T., Emery S., Tong S., Urbani C., Comer J.A., Lim W., Rollin P.E., Dowell S.F., Ling A.E., Humphrey C.D., Shieh W.J., Guarner J., Paddock C.D., Rota P., Fields B., DeRisi J., Yang J.Y., Cox N., Hughes J.M., LeDuc J.W., Bellini W.J., Anderson L.J. N. Engl. J. Med. 2003;348:1953. doi: 10.1056/NEJMoa030781. [DOI] [PubMed] [Google Scholar]
- 5.(a) Bartlam M., Yang H., Rao Z. Curr. Opin. Struct. Biol. 2005;15:664. doi: 10.1016/j.sbi.2005.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Thiel V., Ivanov K.A., Putics A., Hertzig T., Schelle B., Bayer S., Weibrich B., Snijder E.J., Rabenau H., Doerr H.W., Gorbalenya A.E., Ziebuhr J. J. Gen. Virol. 2003;84:2305. doi: 10.1099/vir.0.19424-0. [DOI] [PubMed] [Google Scholar]
- 6.Ziebuhr J. Curr. Top. Microbiol. Immunol. 2005;287:57. doi: 10.1007/3-540-26765-4_3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.(a) Chou K., Wei D., Zhong W. Biochem. Biophys. Res. Commun. 2003;308:148. doi: 10.1016/S0006-291X(03)01342-1. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Ghosh A.K., Xi K., Johnson M.E., Baker S.C., Mesecar A.D. Ann. Rep. Med. Chem. 2006;41:183. doi: 10.1016/S0065-7743(06)41011-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.(a) Ghosh A.K., Xi K., Grum-Tokars V., Xu X., Ratia K., Fu W., Houser K., Baker S.C., Johnson M.E., Mesecar A.D. J. Med. Chem. 2005;48:6767. doi: 10.1021/jm050548m. [DOI] [PubMed] [Google Scholar]; (b) Ghosh A.K., Xi K., Ratia K., Santarsiero B.D., Fu W., Harcourt B.H., Rota P.A., Baker S.C., Johnson M.E., Mesecar A.D. Bioorg. Med. Chem. Lett. 2007;17:5876. doi: 10.1016/j.bmcl.2007.08.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Anand K., Ziebuhr J., Wadhwani P., Mesters J.R., Hilgenfeld R. Science. 2003;300:1763. doi: 10.1126/science.1085658. [DOI] [PubMed] [Google Scholar]
- 10.Wu C.-Y., King K.-Y., Kuo C.-J., Fang J.-M., Wu Y.-T., Ho M.-Y., Liao C.-L., Shie J.-J., Liang P.-H., Wong C.-H. Chem. Biol. 2006;13:261. doi: 10.1016/j.chembiol.2005.12.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Thus far, no antiviral activity has been reported for these active ester-derived inhibitors. Inhibitor 3 did not exhibit any antiviral activity in our assay.
- 12.Blanchard J.E., Elowe N.H., Huitema C., Fortin P.D., Cechetto J.D., Eltis L.D., Brown E.D. Chem. Biol. 2004;11:1445. doi: 10.1016/j.chembiol.2004.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.(a) Zhang J., Pettersson H.I., Huitema C., Niu C., Yin J., James M.N., Eltis L.D., Vederas J.C. J. Med. Chem. 2007;50:1850. doi: 10.1021/jm061425k. [DOI] [PubMed] [Google Scholar]; (b) Niu C., Yin J., Zhang J., Vederas J.C., James M.N. Bioorg. Med. Chem. 2008;16:293. doi: 10.1016/j.bmc.2007.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grunewald G.L., Romero F.A., Criscione K.R. J. Med. Chem. 2005;48:134. doi: 10.1021/jm049368n. The indolecarboxylic acids are either from a commercial source or made from hydrolysis of corresponding esters or oxidation of corresponding aldehydes. The starting material for 13 is from Boc protection of 6-aminopenicillanic acid. For preparation of the starting material of 14, 2-acetyl-1,2,3,4-tetrahydroisoquinoline-3-carboxylic acid, please see: [DOI] [PubMed] [Google Scholar]
- 15.Gribble G.W., Hoffman J.H. Synthesis. 1977:859. [Google Scholar]
- 16.Chandra T., Zou S., Brown K.L. Tetrahedron Lett. 2004;45:7783. [Google Scholar]
- 17.Grum-Tokars V., Ratia K., Begaye A., Baker S.C., Mesecar A.D. Virus Res. 2008;133:63. doi: 10.1016/j.virusres.2007.02.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.SARS-CoV antiviral activity assays.Vero E6 cells were maintained in Minimal Essential Media (MEM) (Gibco) supplemented with 100 U/mL penicillin, 100 μg/mL streptomycin (Gibco) and 10% fetal calf serum (FCS) (Gemini Bio-Products). The SARS-CoV Urbani strain used in this study was provided by the Centers for Disease Control and Prevention.4 All experiments using SARS-CoV were carried out in a Biosafety Level 3 facility using approved biosafety protocols.Vero E6 cells were seeded onto flat-bottom, 96-well plates at a density of 9 × 103 cells/well. Cells were either mock infected with serum-free MEM or infected with 300 TCID50/well of SARS-CoV Urbani in 100 μL of serum-free MEM and incubated for 1 h at 37 °C with 5% CO2. Following the 1 h incubation period, the viral inoculum was removed and, 100 μL of MEM supplemented with 2% FCS and containing the inhibitor compound of interest at the desired concentration (serial twofold dilutions from 50 to 0.1 μM) was added. Cells were incubated for a period of 48 h at 37 °C with 5% CO2. Each condition was set up in triplicate and antiviral assays were performed independently on at least two separate occasions. Cell viability was measured 48 h post infection using the CellTiter-Glo Luminescent Cell Viability Assay (Promega), according to manufacturer’s recommendations. Cell viability for the CellTiter-Glo Luminescent Cell Viability Assay was measured as luminescence and output expressed as relative luciferase units (RLU).
- 19.Authentic SARS-CoV 3CLpro was diluted to 1 μM concentration using cold 20 mM HEPES pH 7.5. Five micrometers of compound 10 was added using a 1:400 dilution into the enzyme solution. The final volume was 200 μL and contained a final DMSO concentration of 0.25%. Control reactions were performed in the same buffer in the presence of 0.25% DMSO in the absence of inhibitor. Incubation was performed at room temperature for 20 min. Sinapinic acid was prepared as a saturating solution in 50% acetonitrile and 1% TFA. The sandwich method of preparation was used for MALDI. One microliters of the matrix was spotted onto a plate followed by 1 μL of the enzyme inhibitor complex followed by a top layering of 1 μL of matrix. A Voyager DE Pro Time of Flight (TOF) mass spectrometer was used for determining the molecular weights.
- 20.Verschueren K.H.G., Pumpor K., Anemuller S., Chen S., Mesters J.R., Hilgenfeld R. Chem. Biol. 2008;15:597. doi: 10.1016/j.chembiol.2008.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Verdonk M.L., Cole J.C., Hartshorn M.J., Murray C.W., Taylor R.D. Proteins. 2003;52:609. doi: 10.1002/prot.10465. and references within. [DOI] [PubMed] [Google Scholar]
- 22.Xue X., Yang H., Shen W., Zhao Q., Li J., Rao Z. J. Mol. Biol. 2007;366:965. doi: 10.1016/j.jmb.2006.11.073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Huitema C., Zhang J., Yin J., James M.N.G., Vederas J.C., Eltis L.D. Bioorg. Med. Chem. 2008;16:5761. doi: 10.1016/j.bmc.2008.03.059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Niu C., Yin J., Zhang J., Verderas J.C., James M.N. Bioorg. Med. Chem. 2008;16:293. doi: 10.1016/j.bmc.2007.09.034. [DOI] [PMC free article] [PubMed] [Google Scholar]