

SYNOPSIS
β-Arrestins are known to regulate G protein signaling through interactions with their downstream effectors. Here, we report that β-arrestin1 associates with the G protein β1γ2 subunits in transfected cells, and purified β-arrestin1 interacts with Gβ1γ2 derived from in vitro translation. Deletion mutagenesis of β-arrestin1 led to the identification of a region, comprising amino acids 181-280, as being responsible for its interaction with Gβ1γ2. Overexpression of β-arrestin1 facilitates Gβ1γ2-mediated Akt phosphorylation, and inhibition of endogenous β-arrestin1 expression by small interfering RNA (siRNA) diminishes this effect. Through investigation of nuclear factor κB (NF-κB), a transcription factor regulated by Akt signaling, we have found that overexpression of β-arrestin1 significantly enhances Gβ1γ2-mediated nuclear translocation of NF-κB proteins and expression of a NF-κB-directed luciferase reporter. Overexpression of β-arrestin1 also promotes bradykinin-induced, Gβγ-mediated NF-κB luciferase reporter expression, which is reverted by silencing the endogenous β-arrestin1 with a specific siRNA. These results identify novel functions of β-arrestin1 in binding to the β1γ2 subunits of heterotrimeric G proteins and promoting Gβγ-mediated Akt signaling for NF-κB activation.
Keywords: GTP-binding protein βγ subunits (Gβγ), nuclear factor κB (NF-κB), Akt, β-arrestins
INTRODUCTION
Receptors bearing seven transmembrane structures that are functionally coupled to heterotrimeric guanine nucleotide-regulatory proteins (G proteins)1 respond to a wide range of stimuli, including light, hormones, odorants, chemoattractants, and neurotransmitters. Agonist stimulation of these G protein-coupled receptors (GPCRs) triggers the activation of heterotrimeric G proteins by catalyzing the exchange of GDP for GTP on G protein α subunits. The resulting changes in the G protein βγ subunits conformation, and their dissociation from Gα, lead to activation of numerous downstream effectors including phospholipase Cβ, adenylyl cyclase, ion channels, mitogen-activated protein kinase (MAPK), and phosphatidylinositol 3-Kinase (PI3K) [1]. Agonist occupancy of GPCRs also initiates desensitization and internalization of the receptors, leading to termination of GPCR-mediated signaling. Homologous desensitization of GPCRs requires G protein-coupled receptor kinases and a group of adapter proteins termed β-arrestins [2-4]. Recent findings demonstrate that, in addition to their roles in GPCR desensitization and internalization, β-arrestins can function as GPCR signal transducers by forming protein complexes with signaling molecules downstream of G proteins [5]. β-arrestins interact with a large number of cellular proteins, many of them signaling molecules, and together form the β-arrestin interactome [6]. Among the signaling molecules that interact with β-arrestins are the Src family tyrosine kinases [7], components of the MAPK signaling cascade [8, 9] and the regulatory subunit of Type IA PI3Ks [10]. Others have reported that β-arrestins interact with several components of the classical nuclear factor κB (NF-κB) signaling pathway, including IκBα, IκB kinase (IKK) α and β, and NF-κB-inducing kinase, resulting in the stabilization of IκBα and reduction in NF-κB activation [11, 12]. These findings indicate that the effects of β-arrestins are pleiotropic, and whether it positively or negatively regulates NF-κB and other cellular functions may be dependent on the cell type, the activation states, and the stimuli used for cell activation.
One of the major signaling pathways downstream of Gβγ is the PI3K-Akt pathway, which plays a critical role in cell survival and inhibition of apoptosis [13, 14]. Activation of the transcription factor NF-κB contributes to the anti-apoptotic effect of Akt [14, 15]. The transcription factor NF-κB, which can be homo- or heterodimer consisting of different subunits, is critical to the inducible expression of many genes involved in immunity, inflammation, and cell survival [16, 17]. The most abundant form of NF-κB is a heterodimer consisting of the p65 (RelA) and p50 subunits, in which p65/RelA contains the transcriptional activation domain. Agonist stimulation of many GPCRs leads to the activation of NF-κB via Gαq, Gα13, and/or Gβγ [18-21]. We have previously reported that stimulation of the B2 bradykinin receptor (B2BKR) and D2 (long) dopamine receptor causes Gβγ-dependent NF-κB activation [22, 23]. B2BKR-induced NF-κB activation involves Gαq, Gβγ, PI3K, Akt, and the activation of IKKβ for NF-κB signaling. In comparison, the D2 dopamine receptor-induced NF-κB activation uses a mechanism that involves Gβγ-dependent recruitment of the c-Src tyrosine kinase and does not seem to require IκBα degradation [23]. Interestingly, the D2 dopamine receptor agonist-induced NF-κB activation is significantly enhanced by overexpression of β-arrestin1 (also termed arrestin-2) [23]. These results suggest that, in the activation state, the ability of β-arresin1 to promote GPCR signaling may surpass its inhibitory effect through binding of IκBα, resulting in a net increase in NF-κB activation.
Studies conducted by others have shown that the β-arrestin2 interaction with Akt and the phosphatase PP2A is believed to be important for dopaminergic neurotransmission and behavior [24]. To further understand the mechanism of Gβγ-mediated NF-κB activation, we investigated the potential interaction between these G protein subunits and β-arrestin1 and the biological function of β-arrestin1 in NF-κB activation. We report here that β-arrestin1 interacts with Gβ1γ2. Moreover, β-arrestin1 facilitates the activation of the serine/threonine kinase Akt that is a downstream effector of Gβγ. The increased Akt activity in turn contributes to the Gβ1γ2-mediated NF-κB activation.
EXPERIMENTAL
Reagents
Bradykinin was purchased from Sigma-Aldrich. Pertussis toxin was obtained from Calbiochem. Bovine arrestin-2 (β-arresin1) was purified using a previously described procedure [25]. Protein A/G-Sepharose beads and antibodies against Gβ1, Gγ2, or actin were purchased from Santa Cruz Biotechnology. An antibody against AU5 was purchased from Covance. Antibodies against Akt and phospho-Akt (serine 473) were obtained from Cell Signaling Technology. An anti-human β-arrestin1 antibody was obtained from BD Biosciences. A Gβ1 subunit expression vector (FLAG-tagged at its N-terminus) and Gγ2 expression vector (HA-tagged at its N-terminus) were obtained from the University of Missouri–Rolla cDNA Research Center. The expression vectors for AU5-tagged β-arrestin1 and NF-κB luciferase reporter were described previously [23]. The expression vectors of B2BKR and the Gβγ scavenger T8βARK-myc with a βARK (β-adrenergic receptor kinase or GRK2) carboxyl-terminal fragment were previously described [22, 26]. The siRNA oligonucleotides for control (Cat No: 4611) or β-arrestin1 (siRNA ID: 5026) were purchased from Ambion.
Cell culture, transfection, and luciferase reporter assay
All cells were derived from American Type Culture Collection (ATCC). HEK293 and HeLa cells were maintained in DMEM supplemented with 10% heat-inactivated fetal bovine serum, 2 mM L-glutamine, 100 IU/ml penicillin, and 50 μg/ml streptomycin. HeLa cells (∼ 70% confluence) in 6-well plates were transfected with empty vectors or plasmid expression vectors coding for a 3 × κB-directed luciferase reporter, B2BKR, and β-galactosidase (β-gal), as indicated in the associated figure legends. The total cDNA concentration in each sample was adjusted to 1 μg by addition of the empty vector pCI (Promega) to minimize variations in transfection efficiency. Transient transfection was performed as described [27] using the Lipofectamine Plus reagent or Lipofectamine 2000 (Invitrogen), according to the manufacturer's instructions. Twenty-four hours after transfection, cells were serum-starved for 16-18 h, washed twice with PBS, and assayed with or without agonist stimulation. Reporter lysis buffer (Promega) was then added to the cells. The expressed luciferase activity was measured in a Femtomaster FB12 luminometer (Zylux) or Wallac Victor 1420 Microplate Reader (PerkinElmer). Unless otherwise indicated, all luciferase assays were performed with duplicate or triplicate samples, and 2-4 independent experiments usually were conducted. Data were plotted using the GraphPad Prism 4 software (GraphPad Software).
In vitro translation
In vitro translation was conducted with a TNT system (Promega) in the presence of [35S]-methionine, 0.5 μg each of DNA for the Gβ1 and Gγ2 expression vector (in pcDNA3.1), for 90 min at 30°C. As a negative control, 1 μg of the vector DNA was added to the translation reaction. Approximately half of the resulting translation product was incubated with 200 ng of purified bovine β-arrestin1 for 1 h at 4°C. Immunoprecipitation was carried out with an anti-βarr1 antibody (1:100 dilution) immobilized on protein-A/G beads. Both the supernatant and immunoprecipitates were collected. The immunoprecipitated proteins were eluted, boiled, and analyzed by SDS-PAGE and autoradiography.
Deletion mutagenesis
β-arrestin1 deletion mutants were generated using PCR, with oligonucleotide primers based on the desired sequences. A carboxyl terminal AU5 tag was encoded with the 3′ primers. After 25 cycles of amplification, the PCR products were subcloned into the pCI expression vector. The sequences of the cDNA inserts were verified by automated DNA sequencing.
Immunoprecipitation
This experiment followed a previously proven procedure [28]. Cells in 100 mm dishes were harvested using 1 ml ice-cold lysis buffer (50 mM Tris-HCl, pH 7.6, 150 mM NaCl, 1 % igepal ca-630, 0.5 % Sodium Deoxycholate, 0.1% SDS, 2 mM EDTA) containing a cocktail of protease and phosphatase inhibitors (Sigma-Aldrich). Cell samples in centrifuge tubes were sonicated for 10 second at 4°C and centrifuged at 14,000 × g to remove insoluble material. The supernatant in each tube was incubated with 20 μl protein A/G-Sepharose beads for 2 h at 4°C and centrifuged at 14,000 × g for 1 min. The supernatant then was transferred into fresh tubes and incubated with 10 μl of the selected antibody overnight at 4°C. Immuno-complexes were isolated the next morning by the addition of 20 μl protein A/G-Sepharose beads followed by incubation at 4°C for 4 h. Immunoprecipitates were then washed five times with modified lysis buffer (containing 1 mM Sodium orthovanadate), the last wash used lysis buffer without detergent. Washed immunoprecipitate pellets were dissolved in 50 μl 2 × Laemmli sample buffer. Proteins were denatured by heating to 95°C for 5 min, and the protein A/G-Sepharose were removed by centrifugation at 14,000 × g for 1 min at room temperature (23°C) before electrophoresis.
Western blot analysis
Proteins from whole cell extracts were separated on 8%, 10%, or 12% acrylamide SDS-PAGE gels by electrophoresis at 50 mA. Proteins were then electrotransfered to nitrocellulose membranes at 100 V for 1 h at 4°C. The membrane was pretreated with 5% non-fat milk in TTBS (20 mM Tris-HCl, pH 7.5, 120 mM NaCl, 0.05% Tween-20) for 1-2 h at room temperature. Incubation with primary antibody was done at 4°C in TTBS with 5% BSA, for 16 h. The membrane was then washed for 10 min, 3 times, with TTBS and incubated with horseradish peroxidase-conjugated secondary antibody for 1 h at room temperature. After 3 washes with TTBS, the bound antibody was detected by enhanced chemiluminescence (Pierce Biotechnology).
EMSA (electrophoretic mobility shift assay)
Nuclear protein extracts were prepared as described [18]. A double-stranded NF-κB oligonucleotide, containing the forward strand sequence 5′-AGTTGAGGGGACTTTCCCAGGC–3′ (Promega), was end-labeled using [γ-32P]-ATP and T4 polynucleotide kinase. EMSA was performed according to a previously described procedure [18] using 6% acrylamide gels and 0.5 × TBE buffer (1 × TBE = 89 mM Tris/borate and 2 mM EDTA). The gels were dried, and an autoradiograph was taken using a PhosphoImager cassette (Molecular Dynamics). Results were analyzed using software from Molecular Dynamics.
RESULTS
β-Arrestin1 interacts with Gβ1γ2
It is well documented that β-arrestin1 not only interacts with phosphorylated GPCRs, but also binds to a variety of signaling molecules downstream of these receptors [5]. However, interactions between β-arrestins and heterotrimeric G protein subunits have not been well characterized. In this study, we examined the association between G protein β1γ2 subunits with β-arrestin1 in co-immunoprecipitation assays. In β-arrestin1-transfected human embryonic kidney 293 (HEK293) cells, an antibody against the endogenous Gβ1 subunit co-immunoprecipitated β-arrestin1, which was subsequently detected by Western blotting (Figure 1A, top panel, Lane 3). This result was confirmed by overexpression of Gβ1 together with β-arrestin1, which further enhanced the amount of β-arrestin1 precipitated by the anti-Gβ1 (Figure 1A, top panel, Lane 4). However, overexpression of Gγ2 together with β-arrestin1 did not lead to additional increase in the amount of the associated β-arrestin1 (Figure 1A, top panel, Lanes 5 and 6, compared to Lanes 3 and 4). In a parallel experiment, an anti-Gγ2 was able to co-immunoprecipitate a small amount of β-arrestin1 (Figure 1B, top panel, Lane 3). However, the amount of β-arrestin1 that was co-precipitated by the anti-Gγ2 did not increase by overexpression of Gγ2 (Lane 4), unless Gβ1 was also overexpressed in the same cells (Lane 5). Therefore, it is likely that β-arrestin1 co-immunoprecipitation with Gγ2 using the anti-Gγ2 antibody (Figure 1B, Lane 3) reflects an indirect interaction due to Gγ2 association with Gβ1. Reciprocal experiments employing an anti-AU5 antibody to immunoprecipitate the AU5-tagged β-arrestin1 showed a similar pattern of interaction, i.e., an increased association between β-arrestin1 and Gβ1γ2 was detectable only when Gβ1 was overexpressed (data not shown). These results suggest an interaction between Gβ1 and β-arrestin1.
Figure 1. Interaction between β-arrestin 1 and Gβ1γ2 in transfected cells.
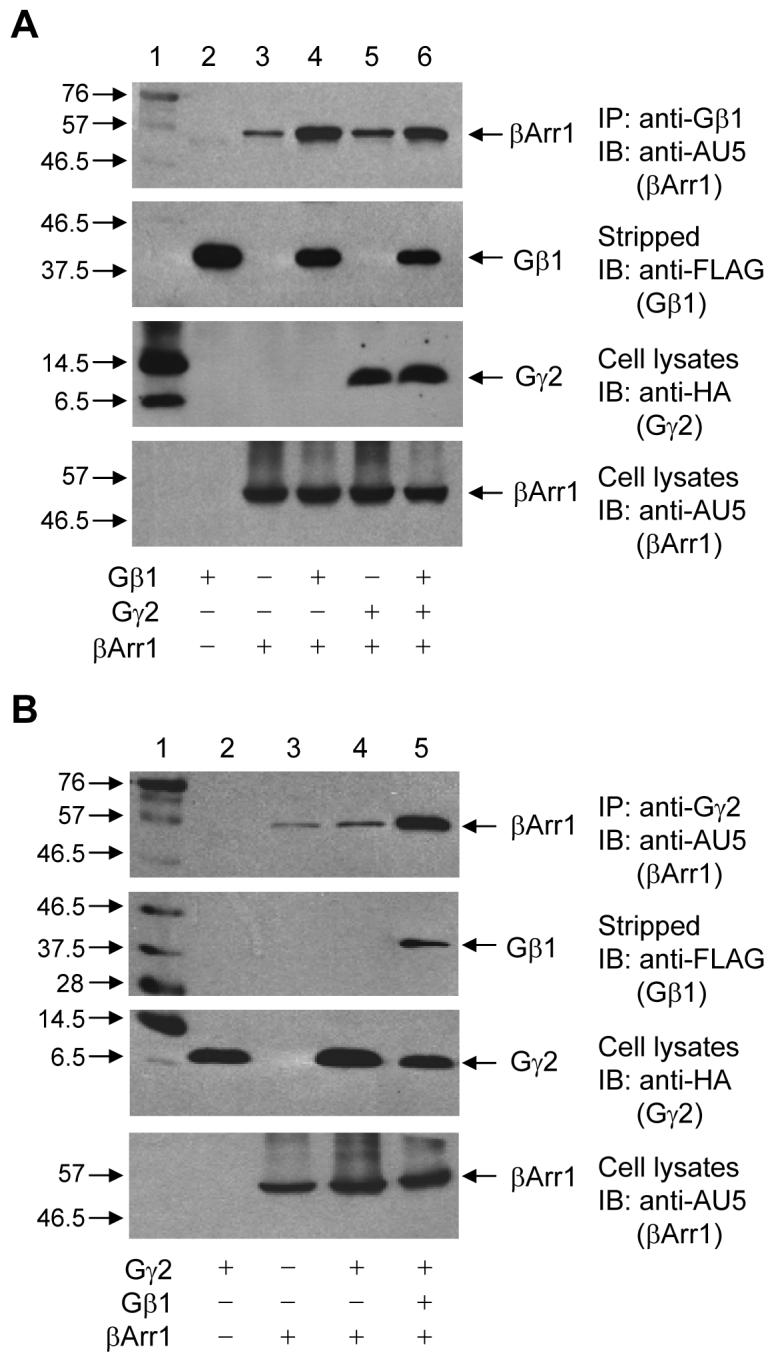
(A) HEK 293 cells were transiently transfected with the expression vectors for AU5-tagged β-arrestin1 (βArr1), FLAG-tagged Gβ1, and HA-tagged Gγ2 as indicated, and the control vector pCI when needed for normalization of the total cDNA input. After 48 h, cell lysate was prepared for immunoprecipitation (IP) with an anti-Gβ1 antibody. Top panel: A representative Western blot (IB) showing detection of the AU5-tagged β-arrestin1 in the anti-Gβ1 immunoprecipitates. Lane 1, molecular markers with the sizes in kilodalton indicated on the left. The contents in Lane 2 to 6 are indicated at the bottom of the figure. The same nitrocellulose membrane was stripped and blotted again with anti-FLAG to detect the precipitated Gβ1, present in Lanes 2, 4, and 6 (second panel). Equal amounts of cell lysate before immunoprecipitation were used for Western blot to determine the expression level of Gγ2 by anti-HA (third panel) and β-arrestin1 by anti-AU5 (bottom panel). (B) HEK 293 cells were transfected similarly as above. Top panel: An anti-Gγ2 was used for IP, and the associated βArr1 was detected by anti-AU5 in a representative Western blot. The same nitrocellulose membrane was stripped and the associated Gβ1 was detected by anti-FLAG (second panel). Cell lysate before immunoprecipitation was collected for Western blots to detect the expression level of Gγ2 by anti-HA (third panel) and β-arrestin1 by anti-AU5 (bottom panel). The blots shown are representative of at least 3 experiments with similar results.
To further determine whether β-arrestin1 directly interacts with Gβ1γ2, a radiolabeled preparation of Gβ1 was made from in vitro translation in the presence of [35S]-methionine. A Gγ2 expression vector was included to facilitate synthesis and formation of the Gβ1γ2 heterodimer, which increases its expression [29]. The radiolabeled Gβ1γ2 was incubated with purified bovine β-arrestin1 at 4°C for 1 h. Immunoprecipitation was carried out using an anti-β-arrestin1 antibody. As shown in Figure 2, a radiolabeled Gβ1 was co-immmunoprecipitated from samples containing the Gβ1 and Gγ2 expression vectors, but not in samples with the control vector (pcDNA3.1 without insert). The anti-β-arrestin1 antibody precipitated significantly more (∼3-fold) radiolabeled Gβ1 in the presence of purified β-arrestin1 than in its absence, suggesting a direct interaction between β-arrestin1 and Gβ1. Based on radioactivity of the sample, the β-arrestin1-associated Gβ1 is approximately 1.63% of the total in vitro translation products.
Figure 2. Interaction between β-arrestin 1 and radiolabeled, in vitro translated Gβ1.

In vitro translation (IVT) was conducted with rabbit reticular lysate using a coupled transcription-translation system, in the presence of [35S]-methionine as described in Materials and Methods. (A) Four microliters of the IVT product (in 50 μl total volume) was loaded to SDS-PAGE and autoradiograph was conducted to show major IVT products. The radiolabeled Gγ2, which is separate from the Gβ1 under denaturing condition, is not visible on the gel due to its small size (69 amino acids). (B) The radiolabeled IVT products, from reactions containing Gβ1 and Gγ2 DNA or the empty vector (pcDNA3.1), were incubated with 200 ng of purified bovine β-arrestin1 for 1 h. β-arrestin1 (βArr1) interaction with the radiolabeled Gβ1 was determined after immunoprecipitation (IP) with an anti-β-arrestin1 polyclonal antibody immobilized on protein A/G Sepharose beads. Both the beads and the supernatant were collected. One-third of the immunoprecipitate was applied to the SDS gel (top panel), showing in autoradiograph an enrichment of the radiolabeled Gβ1 (arrow) only when the IVT products were incubated with the purified bovine β-arrestin1. The radiolabeled Gβ1 that remains in the supernatant is shown in the bottom panel (with one-sixth of the supernatant loaded to SDS gel). Based on radioactivity, approximately 1.63% of the total IVT products were complexed with the bovine β-arrestin1 and immunoprecipitated. A representative autoradiograph from two repeating experiments is shown.
Amino acids 181-280 of β-arrestin1 constitute a critical determinant for β-arrestin1 interaction with Gβ1γ2
Four β-arrestin1 deletion mutants were prepared, tagged with an AU5 epitope at their carboxyl termini, and expressed in the HEK293 cells (Figure 3). All four truncated β-arrestin1 constructs were able to express in the transfected cells, albeit at different levels (Figure 3B, bottom panel). Immunoprecipitation was carried out using an anti-β1 antibody, which recognizes both the endogenous and the recombinant Gβ1 (Figure 3B, top panel, Lanes 2 and 3, respectively). An anti-AU5 monoclonal antibody was used to detect the full-length and mutant β-arrestin1 constructs. An anti-FLAG monoclonal antibody, which detects the FLAG-tagged recombinant Gβ1, was used for evaluation of equal loading of samples (Figure 3B, middle panel). Of the four β-arrestin1 mutants, three were found to associate with Gβ1 (Figure 3B, top panel). The βarr1-180S mutant, however, was not co-immunoprecipitated with the anti-Gβ1 antibody, suggesting that this truncated β-arrestin1 construct was unable to interact with Gβ1. Because both βarr1-280S and βarr1-180N were found to associate with Gβ1, the result suggests that a structural determinant for Gβ1 binding is located in a region consisting of amino acids 181-280 of β-arrestin1.
Figure 3. Identification of a structural determinant of β-arrestin1 in its interaction with Gβ1γ2.
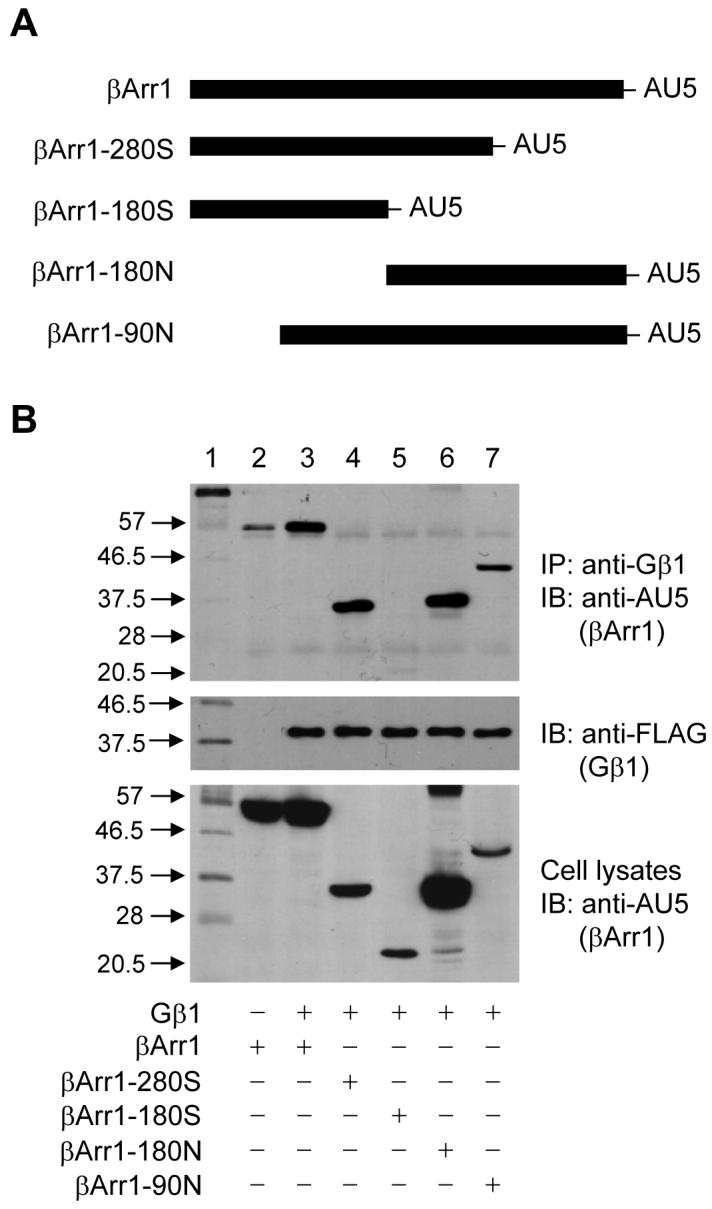
Deletion mutants of β-arrestin1 were prepared as described in Materials and Methods and schematically shown in (A). Expression vectors coding for the 4 deletion mutants and the full-length β-arrestin1 were individually expressed in HEK 293 cells by means of transient transfection (bottom panel in B). A FLAG-tagged Gβ1 construct was co-transfected with each β-arrestin1 construct, and expression level of the tagged Gβ1 was determined by Western blotting using an anti-FLAG monoclonal antibody (middle panel in B). Interaction between Gβ1 and the AU5-tagged β-arrestin1 constructs was determined with Western blotting (upper panel) using an anti-AU5, after immunoprecipitation of Gβ1 from cell lysates with an anti-β1 antibody. The size of the molecular markers (Lane 1) is shown on the left side. The experiment was repeated 2 more times and results similar to the ones shown were obtained.
β-Arrestin1 enhances Gβ1γ2-mediated Akt phosphorylation
The finding that β-arrestin1 interacts with Gβ1γ2 prompted us to investigate the function of β-arrestin1 in Gβγ downstream signaling. One of the major downstream signaling molecules of Gβγ is Akt which, when activated, undergoes auto-phosphorylation. As shown in Figure 4A, exogenous expression of Gβ1γ2 resulted in Akt phosphorylation at Ser473. Whereas expression of β-arrestin1 alone did not increase Akt phosphorylation (Figure 4A, upper panel, Lanes 3 and 4), expression of β-arrestin1 together with Gβ1γ2 significantly enhanced the level of Akt phosphorylation (Lanes 5 and 6). The relative levels of Akt phosphorylation were quantified and shown in a bar graph.
Figure 4. Role of β-arrestin1 in Gβ1γ2-mediated Akt phosphorylation.
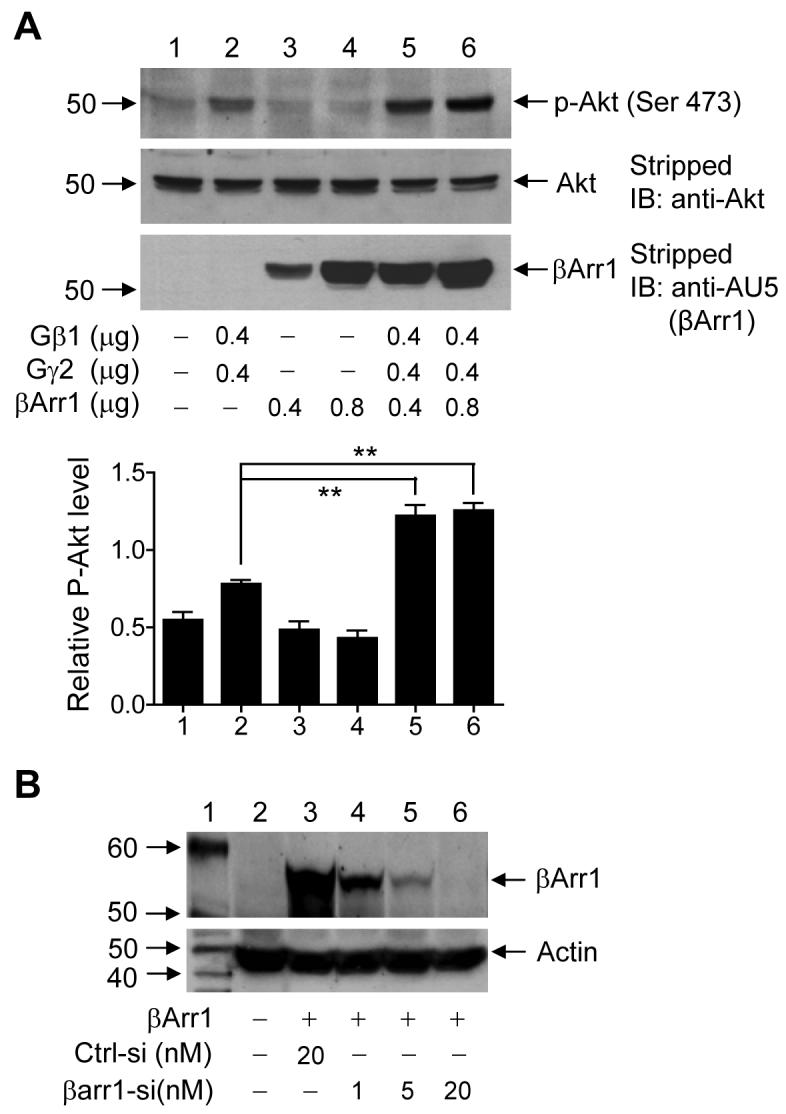
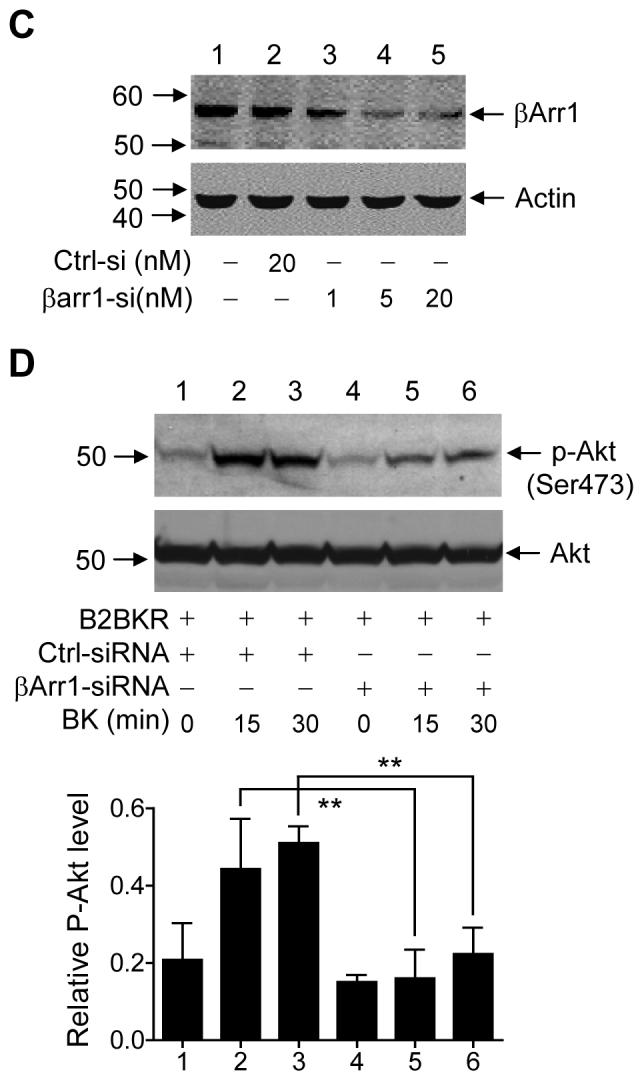
(A) Akt phosphorylation by Gβ1γ2, in the presence or absence of exogenous β-arrestin1. HeLa cells were transiently transfected with different amounts of expression vectors (μg DNA) as indicated under the bottom of the blots. After 24 h, cells were serum-starved overnight, and cell lysate was prepared for Western blotting. Representative results of three experiments are shown in three panels: Levels of phosphorylation of Akt at Ser473 (Upper panel); total Akt levels (Middle panel, using the same nitrocellulose membrane); and levels of transfected β-arrestin1 (Lower panel, with the stripped nitrocellulose membrane probed using an anti-AU5 for the AU5-tagged β-arrestin1). The relative Akt phosphorylation levels are shown in the bar graph after normalization against total Akt. ** : p<0.01. (B) Inhibition of β-arrestin1 expression by siRNA. HeLa cells were transfected with the same amount of expression constructs for AU5-tagged β-arrestin1, together with a control siRNA (Ctrl-si, 20 nM) or a siRNA for β-arrestin1 (βarr1-siRNA) at 3 different concentrations. After 24 h, cell lysate was prepared for Western blotting using anti-AU5 to detect the AU5-tagged β-arrestin1. (C) HeLa cells in 6-well plates were transfected with 20 nM of control siRNA (Ctrl-si) and 3 different concentrations of β-arrestin1 siRNA (βarr1-si) as shown. After 24 h, cell lysate was prepared and endogenous β-arrestin1 levels were detected by anti-human β-arrestin1. The same nitrocellulose membrane was stripped, and the levels of actin were shown as loading controls. (D) Effect of the siRNAs on bradykinin (BK)-induced Akt phosphorylation. HeLa cells were transiently transfected with expression vectors for B2BKR (0.2 μg), the control siRNA (Ctrl-siRNA, 20 nM) or β-arrestin1 siRNA (βArr1-siRNA, 20 nM). After 24 h, cells were serum-starved for 16 h and then stimulated with 100 nM BK for 0, 15, and 30 min. Cell lysate was prepared for Western blotting. Upper panel: Antibody detection of Akt phosphorylation at serine 473. Lower panel: Antibody detection of total Akt that serves as a loading control. The bar graph shows relative phospho-Akt levels after normalized against total Akt (n=3). ** indicates p< 0.05.
To further investigate the role of β-arrestin1 in Akt activation, we examined the effect of small interfering RNA (siRNA)-mediated knockdown of β-arrestin1 on Gβγ-mediated Akt phosphorylation in response to GPCR activation. The effectiveness of siRNA-mediated inhibition of β-arrestin1 expression was first determined. HeLa cells, which were also used in reporter assays described below, were transiently transfected with an AU5-tagged β-arrestin1 expression vector, together with either a control siRNA or a β-arrestin1-specific siRNA. After 24 h, cell lysate was prepared for Western blotting. As shown in Figure 4B, introduction of the β-arrestin1-specific siRNA into the cells reduced the expression level of the AU5-tagged β-arrestin1 in a dose-dependent manner. Maximum inhibition was observed with 20 nM of the siRNA oligonucleotide, a concentration that was sufficiently low to avoid off-target effects [30]. The β-arrestin1-specific siRNA also reduced the level of endogenous β-arrestin1 when applied at concentrations of 1, 5 and 20 nM (Figure 4C).
We next investigated the effect of β-arrestin1 knockdown on Gβγ-mediated Akt activation. In experiments described above, cotransfection of Gβγ does not permit a time-dependent measurement of agonist-induced Akt activation. Therefore, we expressed the B2 bradykinin receptor (B2BKR), which mediates Akt activation in part through Gβγ [22], for time-dependent stimulation of Akt phosphorylation. HeLa cells were transiently transfected with a B2BKR expression vector, with either the control siRNA oligonucleotide or β-arrestin1-specific siRNA oligonucleotide. Twenty-four hours after transfection, the cells were serum starved for 16 h and then stimulated with bradykinin for 0, 15, and 30 min. As shown in Figure 4D, bradykinin increased Akt phosphorylation after 15-30 min of stimulation in the control group (Lanes 2-3). In the presence of the β-arrestin1-specific siRNA, the bradykinin-induced Akt phosphorylation was markedly reduced (Lanes 5 and 6, compared to Lanes 2 and 3). This result suggests that β-arrestin1 facilitates bradykinin-induced, Gβγ-mediated Akt activation.
β-Arrestin1 enhances Gβ1γ2-mediated NF-κB activation
Previous reports suggested that the PI3K–Akt pathway plays an important role in NF-κB activation [15, 22, 31-34]. Our observations that β-arrestin1 interacts with Gβ1 and also promotes Gβγ-mediated Akt phosphorylation prompted us to investigate its role in Gβ1γ2-mediated NF-κB activation. Accordingly, we performed an NF-κB-driven luciferase reporter assay in HeLa cells. The cells were transiently transfected with expression vectors coding for an NF-κB luciferase reporter, Gβ1, and/or Gγ2, with or without the β-arrestin1 expression vector. Coexpression of Gβ1γ2 induced a tenfold increase in NF-κB luciferase reporter activity (Figure 5A). This activity was further enhanced by overexpression of β-arrestin1 (p < 0.01). Likewise, overexpression of β-arrestin1 enhanced Gβ1γ2-mediated NF-κB nuclear translocation and DNA binding in a gel mobility shift assay (Figure 5B, Lanes 5 and 6 compared to Lane 2). We have shown in a previous study that overexpression of β-arrestin1 in the absence of agonist stimulation or Gβγ coexpression does not significantly alter NF-κB luciferase reporter expression [23].
Figure 5. β-Arrestin1 enhances Gβ1γ2-mediated NF-κB activation.
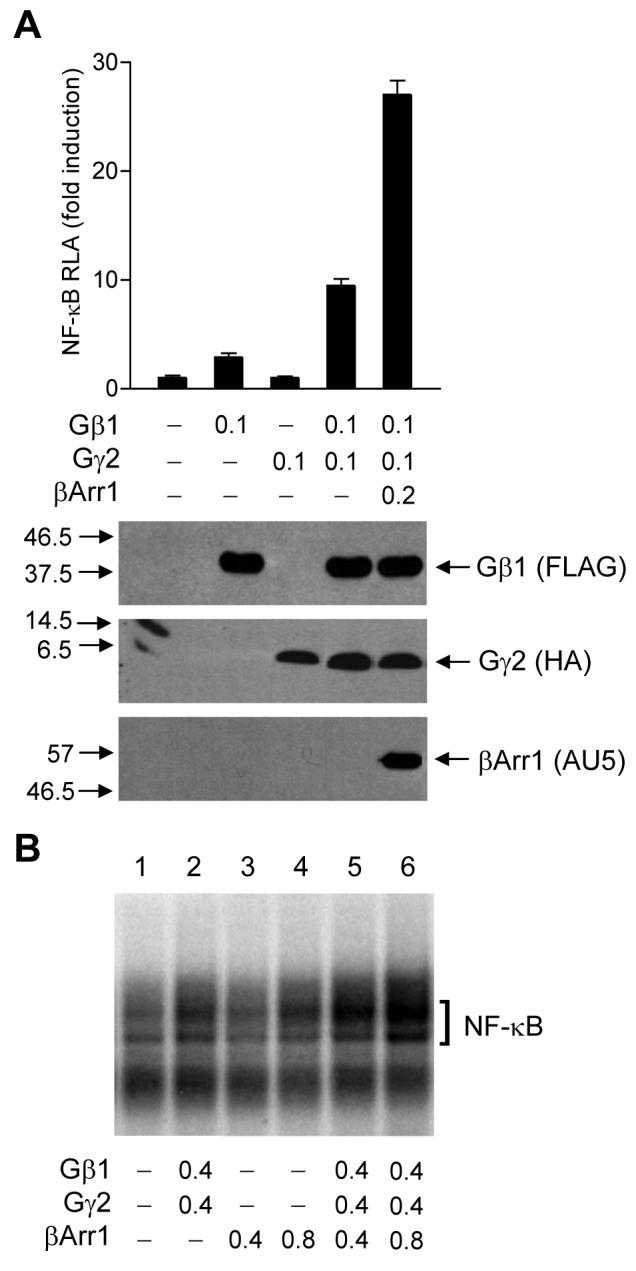
(A) NF-κB luciferase reporter assay. HeLa cells in 6-well plates were transiently transfected with the expression vectors (in μg) for the NF-κB luciferase reporter, Gβ1, and Gγ2, with or without a βArr1 expression vector as indicated. After 48 h, serum-starved cells were harvested, and luciferase reporter gene activities were measured in a Femtomaster FB12 luminometer. Lower panel: Lysate was prepared from HeLa cells transfected as above for Western blotting to detect the expression of FLAG-tagged Gβ1, HA-tagged Gγ2, and AU5-tagged β-arrestin1 using antibodies against the respective tags. (B) Electrophoretic mobility shift assay. HeLa cells in 60 mm dishes were transfected with expression vectors (in μg) as shown. After 24 h, cells were serum-starved for 16 h, and nuclear proteins were harvested for DNA-protein binding assay with [γ-32P]ATP-labeled double-stranded NF-κB oligonucleotides. The DNA-protein complexes were separated on a 6% acrylamide gel. NF-κB is marked with a bracket.
To investigate the role of β-arrestin1 in GPCR-induced and Gβγ-mediated NF-κB activation, we used B2BKR as a model because activation of NF-κB through B2BKR is known to involve Gβγ, PI3K and Akt [22]. In HeLa cells transiently transfected with expression vectors coding for B2BKR and the NF-κB luciferase reporter, stimulation with bradykinin produced more than 40-fold induction of the NF-κB luciferase reporter activity (Figure 6A). The dependence of this activation on Gβγ was shown by coexpression of a Gβγ scavenger, the β-adrenergic receptor kinase carboxyl terminal fragment (βARK-c), which abolished this effect (Figure 6A). Coexpression of β-arrestin1 significantly enhanced the bradykinin-induced NF-κB activity (p < 0.01; Figure 6A), and the increased luciferase reporter activity was also blocked by βARK-c. β-arrestin1 potentiated bradykinin-induced NF-κB activation in a dose-dependent manner (Figure 6B) when its expression vector was used in the range of 0.1 to 0.4 μg DNA per sample. The expression levels of β-arrestin1 was determined by Western blotting and shown in Figure 6C.
Figure 6. Role of β-arrestin1 in B2BKR-induced NF-κB activation.

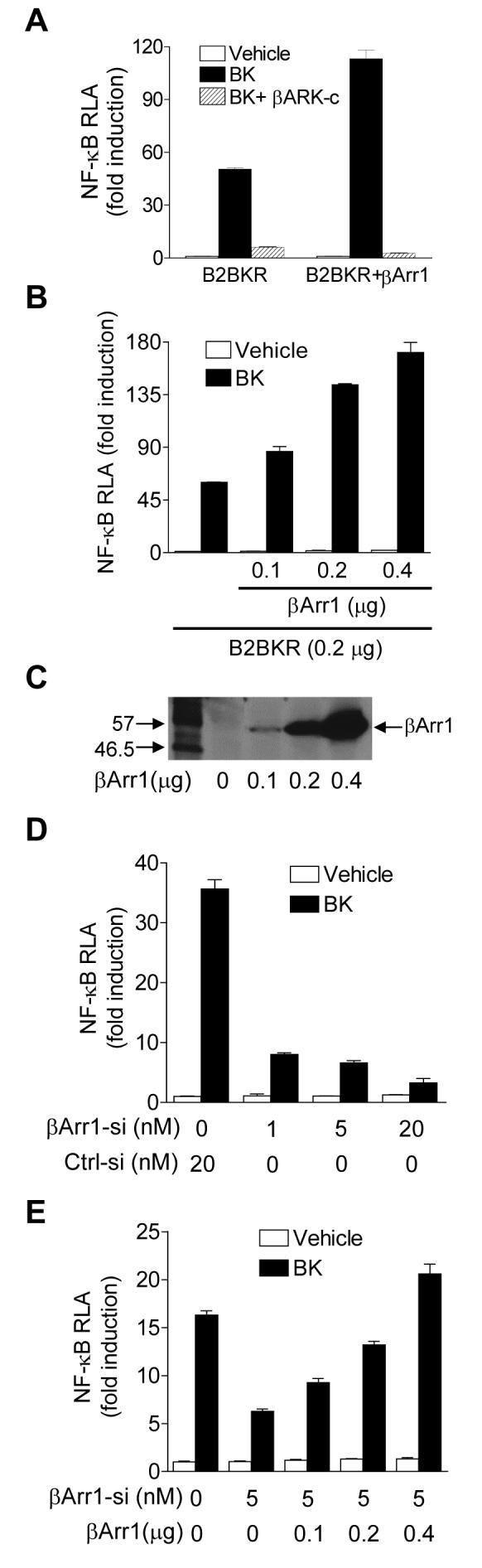
(A) B2BKR-mediated NF-κB luciferase reporter expression. HeLa cells were transfected with expression vectors for B2BKR, βARK-c, β-galactosidase with or without β-arrestin1 at a total cDNA concentration of 1.0 μg. Twenty-four hours after transfection, cells were serum-starved for 16 h, and then treated with either a vehicle (assay buffer) or BK (100 nM) for 5 h. Relative luciferase activities were measured and normalized against activities of β-galactosidase and shown as fold induction over control. (B) Dose response based on the amount of the β-arrestin1 expression vector used for transfection of HeLa cells. Total DNA concentration was adjusted to 1.0 μg with the pCI vector. Luciferase reporter assay was conducted similarly as described in (A). (C) Cell lysate was prepared from HeLa cells transfected as above for Western blotting, showing DNA dose-dependent expression of β-arrestin1 as detected by anti-AU5. (D) Increased inhibition of BK-induced NF-κB luciferase reporter by the β-arrestin1 siRNA, used at 3 different concentrations. HeLa cells were transfected with constructs for B2BKR, β-galactosidase and the control siRNA (Ctrl-siRNA, 20 nM) or different concentrations of the β-arrestin1 siRNA (βArr1-siRNA), for 24 h. Serum-starved cells were treated with a vehicle or 100 nM BK for 5 h. Relative luciferase activities were measured and normalized against β-galactosidase activities and shown as a fold induction over the control. (E) β-arrestin1 dose-dependently reversed the inhibitory effect of β-arrestin1 siRNA. The luciferase assay was done similarly as in (D) above. For all luciferase assays, duplicate samples were taken, and at least 3 independent experiments were conducted to produce mean ± SEM.
To further examine the role of β-arrestin1 in Gβγ-mediated NF-κB activation, we conducted a reciprocal experiment in which the expression of β-arrestin1 was inhibited by a specific siRNA used in Figure 4. HeLa cells were transiently transfected with expression vectors coding for the NF-κB luciferase reporter and B2BKR and either the control siRNA or the β-arrestin1-specific siRNA. Our results demonstrate that the β-arrestin1-specific siRNA dose-dependently inhibited bradykinin-induced NF-κB luciferase reporter activity (Figure 6D). When the β-arrestin1 siRNA was used at fixed concentration (5 nM), increasing the concentrations of the β-arrestin1 cDNA expression vector reversed the inhibitory effect of the siRNA in a dose-dependent manner (Figure 6E).
DISCUSSION
Previous studies have demonstrated that β-arrestins play a critical role in GPCR desensitization [2-4]. More recent evidence suggests that, in addition to their negative regulation of GPCR activation, β-arrestins also enhance GPCR signaling through interaction with effectors downstream of G proteins [5]. Examples for this function include β-arrestin1 interaction with c-Src and components of the MAPK cascade [7-9]. In this study, we found that β-arrestin1 can also associate with Gβ1γ2 and promote the activation of Akt, a downstream effector of the Gβγ heterodimer. This association most likely is mediated through an interaction between β-arrestin1 and Gβ1 because it is not affected significantly by overexpression of Gγ2. Also, results from in vitro translation experiment demonstrate an association of β-arrestin1 with Gβ1, indicating a direct interaction between these proteins. Our findings provide a previously unrecognized interaction between β-arrestin1 and a β subunit of heterotrimeric G proteins, and support a role of β-arrestin1 in regulating GPCR functions through its interaction with components of the GPCR signaling pathways.
In HeLa cells, overexpression of β-arrestin1 facilitates Gβ1γ2 downstream signaling through enhancement of Akt phosphorylation. Since the potentiation of Akt phosphorylation can be reverted by siRNA-mediated silencing of endogenous β-arrestin1, there is a correlation between β-arrestin1 expression level and Gβγ-mediated Akt activation. The mechanism underlying the observed potentiation effect by β-arrestin1 may involve its direct interaction with Gβ1γ2, although other signal-promoting functions of β-arrestin1 cannot be excluded due to its scaffolding functions as discussed below. Our results provide a possible mechanism for a recently reported function of β-arrestin1 in α-thrombin-induced rapid phosphorylation of Akt [35, 36]. Activation of Akt by α-thrombin receptors predominantly depends on the function of Gβγ released from Gαi2 and Gαq [10]. Therefore, β-arrestin1 interaction with signaling components, such as Gβ1γ2, can possibly contribute to Gβγ-mediated Akt phosphorylation. β-arrestin1 also contributes to insulin-like growth factor 1-mediated Akt activity as reported [37], suggesting that the enhancement effect of β-arrestin1 on Akt activation is not confined to one type of stimulus. Because β-arrestins are scaffolds for a large number of signaling molecules [5], they can facilitate the formation of multiple signaling complexes and contribute to the increased Akt activity. β-arrestin2 interaction with Akt and the phosphatase PP2A is believed to be important for dopaminergic neurotransmission and behavior [24]. Additionally, immunoprecipitation experiments have identified β-arrestin1 interaction with a catalytic subunit of PI3K, p110γ, which is a class 1B enzyme activated by the Gβγ heterodimers [38]. Therefore, β-arrestin1 may enhance Gβγ-mediated Akt activity through its interaction with Gβ1γ2 and other signaling molecules.
Our findings that overexpression of β-arrestin1 enhances Gβ1γ2-mediated NF-κB activity, and that siRNA-mediated silencing of endogenous β-arrestin1 significantly reverses this effect, suggest that β-arrestin1 plays a role in the regulation of NF-κB transcriptional activity through Gβγ signaling. Bradykinin, acting through B2BKR, induces potent NF-κB activation in a large part through Gβγ-mediated signaling. As shown in Figure 6, scavenging Gβγ effectively blocks the bradykinin-induced NF-κB activation and its potentiation by β-arrestin1, supporting a critical role of Gβγ in NF-κB activation downstream of B2BKR. NF-κB is an important antiapoptotic factor [39-41], and the potentiation effect of β-arrestin1 on Gβγ-mediated Akt activation may promote cell survival through NF-κB activation. Akt, known for its suppression of apoptosis [13], stimulates the transcriptional potential of NF-κB partially through phosphorylation and transactivation of p65/RelA [15, 42]. Thus, β-arrestin1 association with Gβγ may be one of the mechanisms for optimal Akt signaling through GPCRs. This notion is supported by published results demonstrating that expression of β-arrestins is responsible for the protection from GPCR-mediated apoptosis [43] and enhancement of GPCR-mediated antiapoptotic effect [44].
Two recent reports showed that β-arrestins directly interact with IκBα through its carboxyl terminal domain, and, when overexpressed, inhibit TNFα-induced IκBα degradation and NF-κB activation [11, 12]. Although these results differ from the potentiation effect we observed in the current study, there are several possible explanations for the discrepancy. First, β-arrestin1 can have opposite roles in different systems. β-arrestin1 promotes signaling downstream of a large number of GPCRs. Therefore, it may enhance NF-κB activation through interactions with signaling molecules such as Gβ1γ2 and protein kinases. β-arrestins may also limit the basal activity of NF-κB through binding and stabilizing IκBα. Therefore, the outcome of NF-κB activation depends on the balance of the two signaling events. In the case of certain GPCRs that rely primarily on Gβγ for downstream signaling, the balance may be in favor of NF-κB activation as demonstrated in this study and in a previously published report [23]. Conversely, for GPCRs that primarily use Gq for signaling, the NF-κB activation pathway is mediated mainly by the Gαq-Ca2+-PKC pathway [21, 45], and the net outcome may be different. The M1 muscarinic receptor that was used in one of the published studies [11] is strongly coupled to Gαq and can be susceptible to β-arrestin1-mediated stabilization of IκBα. β-arrestin-mediated signaling through GPCRs can lead to biased agonism [46], indicating the diversity of its role in biological functions. Secondly, the current model entails that β-arrestins inhibit NF-κB activation through reduction of IκBα degradation following serine phosphorylation at positions 32 and 36 [12], which is induced by many physiological stimuli including TNFα. NF-κB activation mechanisms that do not involve IκBα degradation [47] are less likely to be subjective to this inhibitory mechanism. The D2 dopamine receptor-mediated NF-κB activation requires Gβγ and involves tyrosine phosphorylation of IκBα, but not its degradation [23] 2. Likewise, Akt stimulates NF-κB activation primarily through p65/RelA phosphorylation that enhances the transactivation potential of this NF-κB component [15, 42]. This activation process is in parallel with, but not subsequent to, IκBα degradation. Finally, although β-arrestins can regulate steady-state level of NF-κB activation, and may have an important role in preventing excessive activation of this transcriptional factor, numerous cellular activation mechanisms can overcome this inhibition. Therefore it is not surprising that cells respond to physiological stimuli such as TNFα and IL-1β with a potent NF-κB activation despite the presence of endogenous β-arrestins. A growing number of GPCRs have been shown to mediate NF-κB activation in cells that express β-arrestins [21], indicating that the signaling capability of β-arrestin1 may overwhelm its inhibitory effect upon agonist stimulation and, thereby, lead to increased NF-κB activation. β-arrestin1 is known to translocate to nuclei upon delta opioid receptor activation and can regulate the recruitment of the histone acetyltransferase p300 [48]. P300 is a transcription cofactor, and its recruitment is expected to increase transcriptional activity. However, published information only indicates upregulation of a very limited number of genes such as p27 and c-fos [48], and it remains unclear whether the nuclear localized β-arrestin1 positively regulates NF-κB activation.
β-arrestin1 may regulate the transcriptional activity of multiple transcription factors through its interaction with Gβγ. As reported previously, β-arrestin1 enhances the transcriptional activation of lymphoid enhancer factor, mediated by the non-conventional GPCR frizzled receptors, via a direct interaction with phosphorylated disheveled proteins [49]. Therefore, different mechanisms may be involved in β-arrestin1-mediated regulation of GPCR signaling and gene transcription. The finding that β-arrestin1 enhancement of Akt activity and NF-κB activation depends on Gβ1γ2 indicates that β-arrestin1 can potentially play an important role in regulating Gβγ-mediated cell survival, cell proliferation, and differentiation. For instance, in β-arrestin1 transgenic mice, rapid xenograft tumor progression is observed [50]. The accelerated growth of tumors in these mice is PI3K-dependent, and relies on the increased expression of matrix metalloproteinase 9. These findings are consistent with our observation that β-arrestin1 promotes Akt activation. More recent studies also demonstrate interactions between Gβγ subunits and other proteins that result in either positive or negative regulation of cellular functions [51-53]. An interaction between β-arrestin1 and Gβ1γ2 provides a possible mechanism for scaffolding that may facilitate signal complex formation by bringing the relevant proteins to proximity. Further investigation of the detailed molecular interactions between β-arrestin1 and different Gβγ dimers will be necessary to understand its function in cell signaling.
ACKNOWLEDGEMENT
This work was supported in part by National Institutes of Health Grants GM066182 and AI040176 (to R.D.Y.) and GM47417 (to J.L.B).
Footnotes
Abbreviations used: G proteins, guanine nucleotide-binding regulatory proteins; GPCR, G protein-coupled receptor; MAPK, mitogen-activated protein kinase; PI3K, phosphatidylinositol 3-kinase; NF-κB, nuclear factor κB; IKK, IκB kinase; B2BKR, type 2 bradykinin receptor; HEK293, human embryonic kidney cell line 293; siRNA, small interfering RNA; EMSA, electrophoretic mobility shift assay.
M. Yang and R.D. Ye, unpublished data.
REFERENCES
- 1.Clapham DE, Neer EJ. G protein βγ subunits. Annu. Rev. Pharmacol. Toxicol. 1997;37:167–203. doi: 10.1146/annurev.pharmtox.37.1.167. [DOI] [PubMed] [Google Scholar]
- 2.Lohse MJ, Benovic JL, Codina J, Caron MG, Lefkowitz RJ. β-Arrestin: a protein that regulates β-adrenergic receptor function. Science. 1990;248:1547–1550. doi: 10.1126/science.2163110. [DOI] [PubMed] [Google Scholar]
- 3.Goodman OB, Jr., Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, Keen JH, Benovic JL. β-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature. 1996;383:447–450. doi: 10.1038/383447a0. [DOI] [PubMed] [Google Scholar]
- 4.Freedman NJ, Liggett SB, Drachman DE, Pei G, Caron MG, Lefkowitz RJ. Phosphorylation and desensitization of the human β1-adrenergic receptor. Involvement of G protein-coupled receptor kinases and cAMP-dependent protein kinase. J. Biol. Chem. 1995;270:17953–17961. doi: 10.1074/jbc.270.30.17953. [DOI] [PubMed] [Google Scholar]
- 5.Lefkowitz RJ, Shenoy SK. Transduction of receptor signals by β-arrestins. Science. 2005;308:512–517. doi: 10.1126/science.1109237. [DOI] [PubMed] [Google Scholar]
- 6.Xiao K, McClatchy DB, Shukla AK, Zhao Y, Chen M, Shenoy SK, Yates JR, 3rd, Lefkowitz RJ. Functional specialization of β-arrestin interactions revealed by proteomic analysis. Proc. Natl. Acad. Sci. U. S. A. 2007;104:12011–12016. doi: 10.1073/pnas.0704849104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, Lin F, Kawakatsu H, Owada K, Luttrell DK, Caron MG, Lefkowitz RJ. β-arrestin-dependent formation of β2 adrenergic receptor-Src protein kinase complexes. Science. 1999;283:655–661. doi: 10.1126/science.283.5402.655. [DOI] [PubMed] [Google Scholar]
- 8.McDonald PH, Chow CW, Miller WE, Laporte SA, Field ME, Lin FT, Davis RJ, Lefkowitz RJ. β-arrestin 2: a receptor-regulated MAPK scaffold for the activation of JNK3. Science. 2000;290:1574–1577. doi: 10.1126/science.290.5496.1574. [DOI] [PubMed] [Google Scholar]
- 9.DeFea KA, Zalevsky J, Thoma MS, Dery O, Mullins RD, Bunnett NW. β-arrestin-dependent endocytosis of proteinase-activated receptor 2 is required for intracellular targeting of activated ERK1/2. J. Cell Biol. 2000;148:1267–1281. doi: 10.1083/jcb.148.6.1267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Goel R, Phillips-Mason PJ, Gardner A, Raben DM, Baldassare JJ. α-thrombin-mediated phosphatidylinositol 3-kinase activation through release of Gβγ dimers from Gαq and Gαi2. J. Biol. Chem. 2004;279:6701–6710. doi: 10.1074/jbc.M308753200. [DOI] [PubMed] [Google Scholar]
- 11.Witherow DS, Garrison TR, Miller WE, Lefkowitz RJ. β-Arrestin inhibits NF-κB activity by means of its interaction with the NF-κB inhibitor IκBα. Proc. Natl. Acad. Sci. U. S. A. 2004;101:8603–8607. doi: 10.1073/pnas.0402851101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Gao H, Sun Y, Wu Y, Luan B, Wang Y, Qu B, Pei G. Identification of β-arrestin2 as a G protein-coupled receptor-stimulated regulator of NF-κB pathways. Mol. Cell. 2004;14:303–317. doi: 10.1016/s1097-2765(04)00216-3. [DOI] [PubMed] [Google Scholar]
- 13.Kennedy SG, Wagner AJ, Conzen SD, Jordan J, Bellacosa A, Tsichlis PN, Hay N. The PI 3-kinase/Akt signaling pathway delivers an anti-apoptotic signal. Genes Dev. 1997;11:701–713. doi: 10.1101/gad.11.6.701. [DOI] [PubMed] [Google Scholar]
- 14.Mitsiades CS, Mitsiades N, Koutsilieris M. The Akt pathway: molecular targets for anti-cancer drug development. Curr. Cancer Drug Targets. 2004;4:235–256. doi: 10.2174/1568009043333032. [DOI] [PubMed] [Google Scholar]
- 15.Madrid LV, Wang CY, Guttridge DC, Schottelius AJ, Baldwin AS, Jr., Mayo MW. Akt suppresses apoptosis by stimulating the transactivation potential of the RelA/p65 subunit of NF-κB. Mol. Cell. Biol. 2000;20:1626–1638. doi: 10.1128/mcb.20.5.1626-1638.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kopp EB, Ghosh S. NF-κB and rel proteins in innate immunity. Adv. Immunol. 1995;58:1–27. doi: 10.1016/s0065-2776(08)60618-5. [DOI] [PubMed] [Google Scholar]
- 17.Bonizzi G, Karin M. The two NF-κB activation pathways and their role in innate and adaptive immunity. Trends Immunol. 2004;25:280–288. doi: 10.1016/j.it.2004.03.008. [DOI] [PubMed] [Google Scholar]
- 18.Kravchenko VV, Pan Z, Han J, Herbert JM, Ulevitch RJ, Ye RD. Platelet-activating factor induces NF-κB activation through a G protein-coupled pathway. J. Biol. Chem. 1995;270:14928–14934. doi: 10.1074/jbc.270.25.14928. [DOI] [PubMed] [Google Scholar]
- 19.Shahrestanifar M, Fan X, Manning DR. Lysophosphatidic acid activates NF-κB in fibroblasts. A requirement for multiple inputs. J. Biol. Chem. 1999;274:3828–3833. doi: 10.1074/jbc.274.6.3828. [DOI] [PubMed] [Google Scholar]
- 20.Shi CS, Kehrl JH. PYK2 links Gqα and G13α signaling to NF-κB activation. J. Biol. Chem. 2001;276:31845–31850. doi: 10.1074/jbc.M101043200. [DOI] [PubMed] [Google Scholar]
- 21.Ye RD. Regulation of nuclear factor κB activation by G-protein-coupled receptors. J. Leukoc. Biol. 2001;70:839–848. [PubMed] [Google Scholar]
- 22.Xie P, Browning DD, Hay N, Mackman N, Ye RD. Activation of NF-κB by bradykinin through a Gαq- and Gβγ-dependent pathway that involves phosphoinositide 3-kinase and Akt. J. Biol. Chem. 2000;275:24907–24914. doi: 10.1074/jbc.M001051200. [DOI] [PubMed] [Google Scholar]
- 23.Yang M, Zhang H, Voyno-Yasenetskaya T, Ye RD. Requirement of Gβγ and c-Src in D2 dopamine receptor-mediated nuclear factor-κB activation. Mol. Pharmacol. 2003;64:447–455. doi: 10.1124/mol.64.2.447. [DOI] [PubMed] [Google Scholar]
- 24.Beaulieu JM, Sotnikova TD, Marion S, Lefkowitz RJ, Gainetdinov RR, Caron MG. An Akt/beta-arrestin 2/PP2A signaling complex mediates dopaminergic neurotransmission and behavior. Cell. 2005;122:261–273. doi: 10.1016/j.cell.2005.05.012. [DOI] [PubMed] [Google Scholar]
- 25.Milano SK, Pace HC, Kim YM, Brenner C, Benovic JL. Scaffolding functions of arrestin-2 revealed by crystal structure and mutagenesis. Biochemistry. 2002;41:3321–3328. doi: 10.1021/bi015905j. [DOI] [PubMed] [Google Scholar]
- 26.Coso OA, Teramoto H, Simonds WF, Gutkind JS. Signaling from G protein-coupled receptors to c-Jun kinase involves βγ subunits of heterotrimeric G proteins acting on a Ras and Rac1-dependent pathway. J. Biol. Chem. 1996;271:3963–3966. doi: 10.1074/jbc.271.8.3963. [DOI] [PubMed] [Google Scholar]
- 27.Yang M, Sang H, Rahman A, Wu D, Malik AB, Ye RD. Gα16 couples chemoattractant receptors to NF-κB activation. J. Immunol. 2001;166:6885–6892. doi: 10.4049/jimmunol.166.11.6885. [DOI] [PubMed] [Google Scholar]
- 28.Yang M, Leonard JP. Identification of mouse NMDA receptor subunit NR2A C-terminal tyrosine sites phosphorylated by coexpression with v-Src. J. Neurochem. 2001;77:580–588. doi: 10.1046/j.1471-4159.2001.00255.x. [DOI] [PubMed] [Google Scholar]
- 29.Wang Q, Mullah B, Hansen C, Asundi J, Robishaw JD. Ribozyme-mediated suppression of the G protein γ7 subunit suggests a role in hormone regulation of adenylylcyclase activity. J. Biol. Chem. 1997;272:26040–26048. doi: 10.1074/jbc.272.41.26040. [DOI] [PubMed] [Google Scholar]
- 30.Persengiev SP, Zhu X, Green MR. Nonspecific, concentration-dependent stimulation and repression of mammalian gene expression by small interfering RNAs (siRNAs) RNA. 2004;10:12–18. doi: 10.1261/rna5160904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ozes ON, Mayo LD, Gustin JA, Pfeffer SR, Pfeffer LM, Donner DB. NF-κB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature. 1999;401:82–85. doi: 10.1038/43466. [DOI] [PubMed] [Google Scholar]
- 32.Romashkova JA, Makarov SS. NF-κB is a target of AKT in anti-apoptotic PDGF signalling. Nature. 1999;401:86–90. doi: 10.1038/43474. [DOI] [PubMed] [Google Scholar]
- 33.Yang CH, Murti A, Pfeffer SR, Kim JG, Donner DB, Pfeffer LM. Interferon α/β promotes cell survival by activating nuclear factor κB through phosphatidylinositol 3-kinase and Akt. J. Biol. Chem. 2001;276:13756–13761. doi: 10.1074/jbc.M011006200. [DOI] [PubMed] [Google Scholar]
- 34.Pan ZK, Christiansen SC, Ptasznik A, Zuraw BL. Requirement of phosphatidylinositol 3-kinase activity for bradykinin stimulation of NF-κB activation in cultured human epithelial cells. J. Biol. Chem. 1999;274:9918–9922. doi: 10.1074/jbc.274.15.9918. [DOI] [PubMed] [Google Scholar]
- 35.Goel R, Baldassare JJ. β-Arrestin 1 couples thrombin to the rapid activation of the Akt pathway. Ann. N. Y. Acad. Sci. 2002;973:138–141. doi: 10.1111/j.1749-6632.2002.tb04622.x. [DOI] [PubMed] [Google Scholar]
- 36.Goel R, Phillips-Mason PJ, Raben DM, Baldassare JJ. α-thrombin induces rapid and sustained Akt phosphorylation by β-arrestin1-dependent and -independent mechanisms, and only the sustained Akt phosphorylation is essential for G1 phase progression. J. Biol. Chem. 2002;277:18640–18648. doi: 10.1074/jbc.M108995200. [DOI] [PubMed] [Google Scholar]
- 37.Povsic TJ, Kohout TA, Lefkowitz RJ. β-arrestin1 mediates insulin-like growth factor 1 (IGF-1) activation of phosphatidylinositol 3-kinase (PI3K) and anti-apoptosis. J. Biol. Chem. 2003;278:51334–51339. doi: 10.1074/jbc.M309968200. [DOI] [PubMed] [Google Scholar]
- 38.Stephens L, Smrcka A, Cooke FT, Jackson TR, Sternweis PC, Hawkins PT. A novel phosphoinositide 3 kinase activity in myeloid-derived cells is activated by G protein βγ subunits. Cell. 1994;77:83–93. doi: 10.1016/0092-8674(94)90237-2. [DOI] [PubMed] [Google Scholar]
- 39.Beg AA, Sha WC, Bronson RT, Ghosh S, Baltimore D. Embryonic lethality and liver degeneration in mice lacking the RelA component of NF-κB. Nature. 1995;376:167–170. doi: 10.1038/376167a0. [DOI] [PubMed] [Google Scholar]
- 40.Wang CY, Mayo MW, Baldwin AS., Jr. TNF- and cancer therapy-induced apoptosis: potentiation by inhibition of NF-κB. Science. 1996;274:784–787. doi: 10.1126/science.274.5288.784. [DOI] [PubMed] [Google Scholar]
- 41.Van Antwerp DJ, Martin SJ, Kafri T, Green DR, Verma IM. Suppression of TNF-α-induced apoptosis by NF-κB. Science. 1996;274:787–789. doi: 10.1126/science.274.5288.787. [DOI] [PubMed] [Google Scholar]
- 42.Madrid LV, Mayo MW, Reuther JY, Baldwin AS., Jr. Akt stimulates the transactivation potential of the RelA/p65 Subunit of NF-κB through utilization of the IκB kinase and activation of the mitogen-activated protein kinase p38. J. Biol. Chem. 2001;276:18934–18940. doi: 10.1074/jbc.M101103200. [DOI] [PubMed] [Google Scholar]
- 43.Revankar CM, Vines CM, Cimino DF, Prossnitz ER. Arrestins block G protein-coupled receptor-mediated apoptosis. J. Biol. Chem. 2004;279:24578–24584. doi: 10.1074/jbc.M402121200. [DOI] [PubMed] [Google Scholar]
- 44.DeFea KA, Vaughn ZD, O'Bryan EM, Nishijima D, Dery O, Bunnett NW. The proliferative and antiapoptotic effects of substance P are facilitated by formation of a β-arrestin-dependent scaffolding complex. Proc. Natl. Acad. Sci. U. S. A. 2000;97:11086–11091. doi: 10.1073/pnas.190276697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Siehler S, Wang Y, Fan X, Windh RT, Manning DR. Sphingosine 1-phosphate activates nuclear factor-κB through Edg receptors. Activation through Edg-3 and Edg-5, but not Edg-1, in human embryonic kidney 293 cells. J. Biol. Chem. 2001;276:48733–48739. doi: 10.1074/jbc.M011072200. [DOI] [PubMed] [Google Scholar]
- 46.Shukla AK, Violin JD, Whalen EJ, Gesty-Palmer D, Shenoy SK, Lefkowitz RJ. Distinct conformational changes in β-arrestin report biased agonism at seven-transmembrane receptors. Proc. Natl. Acad. Sci. U. S. A. 2008;105:9988–9993. doi: 10.1073/pnas.0804246105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Imbert V, Rupec RA, Livolsi A, Pahl HL, Traenckner EB, Mueller-Dieckmann C, Farahifar D, Rossi B, Auberger P, Baeuerle PA, Peyron JF. Tyrosine phosphorylation of IκBα activates NF-κB without proteolytic degradation of IκBα. Cell. 1996;86:787–798. doi: 10.1016/s0092-8674(00)80153-1. [DOI] [PubMed] [Google Scholar]
- 48.Kang J, Shi Y, Xiang B, Qu B, Su W, Zhu M, Zhang M, Bao G, Wang F, Zhang X, Yang R, Fan F, Chen X, Pei G, Ma L. A nuclear function of β-arrestin1 in GPCR signaling: regulation of histone acetylation and gene transcription. Cell. 2005;123:833–847. doi: 10.1016/j.cell.2005.09.011. [DOI] [PubMed] [Google Scholar]
- 49.Chen W, Hu LA, Semenov MV, Yanagawa S, Kikuchi A, Lefkowitz RJ, Miller WE. β-Arrestin1 modulates lymphoid enhancer factor transcriptional activity through interaction with phosphorylated dishevelled proteins. Proc. Natl. Acad. Sci. U. S. A. 2001;98:14889–14894. doi: 10.1073/pnas.211572798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Zou L, Yang R, Chai J, Pei G. Rapid xenograft tumor progression in beta-arrestin1 transgenic mice due to enhanced tumor angiogenesis. FASEB J. 2008;22:355–364. doi: 10.1096/fj.07-9046com. [DOI] [PubMed] [Google Scholar]
- 51.Chen S, Lin F, Hamm HE. RACK1 binds to a signal transfer region of G betagamma and inhibits phospholipase C β2 activation. J. Biol. Chem. 2005;280:33445–33452. doi: 10.1074/jbc.M505422200. [DOI] [PubMed] [Google Scholar]
- 52.Blackmer T, Larsen EC, Bartleson C, Kowalchyk JA, Yoon EJ, Preininger AM, Alford S, Hamm HE, Martin TF. G protein βγ directly regulates SNARE protein fusion machinery for secretory granule exocytosis. Nat. Neurosci. 2005;8:421–425. doi: 10.1038/nn1423. [DOI] [PubMed] [Google Scholar]
- 53.Spiegelberg BD, Hamm HE. G βγ binds histone deacetylase 5 (HDAC5) and inhibits its transcriptional co-repression activity. J. Biol. Chem. 2005;280:41769–41776. doi: 10.1074/jbc.M504066200. [DOI] [PubMed] [Google Scholar]