

Abstract
The rates of RNA decay and transcription determine the steady state levels of all mRNAs and both can be subject to regulation. While the details of transcriptional regulation are becoming increasingly understood, the mechanism(s) controlling mRNA decay remain unclear. In yeast, a major pathway of mRNA decay begins with deadenylation followed by decapping and 5’-3’ exonuclease digestion. Importantly, it is hypothesized that ribosomes must be removed from mRNA before transcripts are destroyed. Contrary to this prediction, here we show that decay takes place while mRNAs are associated with actively translating ribosomes. The data indicate that dissociation of ribosomes from mRNA is not a prerequisite for decay and we suggest that the 5’-3’ polarity of mRNA degradation has evolved to ensure that the last translocating ribosome can complete translation.
In eukaryotic cells, mRNA is predominately degraded by two alternative pathways that are both initiated by shortening of the 3’ polyadenosine tail (deadenylation). Following deadenylation, either the 5’ 7mGpppN cap is removed (decapping) and the message is digested exonucleolytically 5’ to 3’ or alternatively the transcript is destroyed 3’ to 5’ by the cytoplasmic exosome1. The two mechanisms of mRNA decay together determine basal mRNA levels thereby significantly contributing to overall gene expression.
Translation is postulated to be a key determinant in controlling mRNA decapping1. The translational initiation complex eIF-4F occupies the cap during translation implying that its binding must be antagonized and translational repression must ensue before decapping can occur1-4. This hypothesis is supported by several observations. First, translational initiation rate is inversely proportional to decapping rate3. Second, the decapping regulators Dhh1p and Pat1p are translational repressors and their role in promoting mRNA decapping is partially a function of this activity8,19. Third, mRNA decapping can occur at an unquantified level in ribosome-free cellular foci, termed P-bodies2. Collectively, a two-step model for mRNA decay has been proposed where ribosome dissociation is a necessary first step prior to mRNA decapping1-4.
Deadenylated mRNA remains on polyribosomes
The aforementioned model for mRNA decay predicts that following deadenylation but before decapping a ribosome-free state exists1-4. We reasoned that in a decapping defective cell (dcp2Δ), deadenylated RNA would accumulate in this ribosome-free state. We used sucrose-density gradients to survey mRNA ribosome association in wild-type (WT) and decapping defective cells (dcp2Δ). Greater than 90% of total cellular mRNA is analyzed by this method (data not shown) and ribosome-free ribonucleoprotein (RNP) structures can be clearly separated from polyribosomes (Fig. S2c). As predicted, inhibition of decapping did result in accumulation of deadenylated mRNA (Fig. S2a, b&f); however, the mRNAs continued to sediment deep into a sucrose gradient even when deadenylated (Fig. S2d, g&h). In fact, the sedimentation profiles of several mRNAs in dcp2Δ cells were indistinguishable from those in wild-type cells (Fig. S2d, g&h). The rapid sedimentation of these RNAs could occur either because they were sequestered in heavy particles (perhaps P-bodies)1,2 or because they were associated with ribosomes. The fact that sedimentation correlated with the length of the open reading frame (Fig. S2d, g&h) strongly suggested that the mRNAs were ribosome associated (see below).
Decapped mRNAs are found on polyribosomes
Because deadenylated mRNAs are the substrates for decapping3 we also assessed the sedimentation profiles of decapped RNAs. This was done in cells defective for the 5’-3’ exonuclease (xrn1Δ). In these cells a stable decapped decay intermediate shortened by 2 nucleotides accumulates (indicated by - cap; Fig. 1a) and can be detected using quantitative primer extension analysis (Fig. S10)5-7. Interestingly, the decapped intermediate showed the same sedimentation profile as the deadenylated RNA (Fig. 1a); the vast majority (83-95%) of decapped mRNA being present in polyribosomes (Fig. 1a & d). To determine if the decay intermediate was associated with ribosomes, we took four approaches. First, introduction of a premature termination codon that shortened the ORF of PGK1 by 393 codons resulted in a dramatic shift to significantly lighter fractions (Fig. 1b&c). Second, introduction of a stem-loop to limit translation8 caused a shift towards the top of the gradient for both capped and uncapped mRNAs (Fig. 1c). Third, treatment with EDTA (known to dissociate ribosomes) shifted the sedimentation to the top of the gradient (Fig. 1c). Finally, we showed that decapped mRNAs were associated with ribosomes by ribosome immunoprecipitation9 (Fig. S3).
Figure 1. Decapped mRNA is associated with polyribosomes.
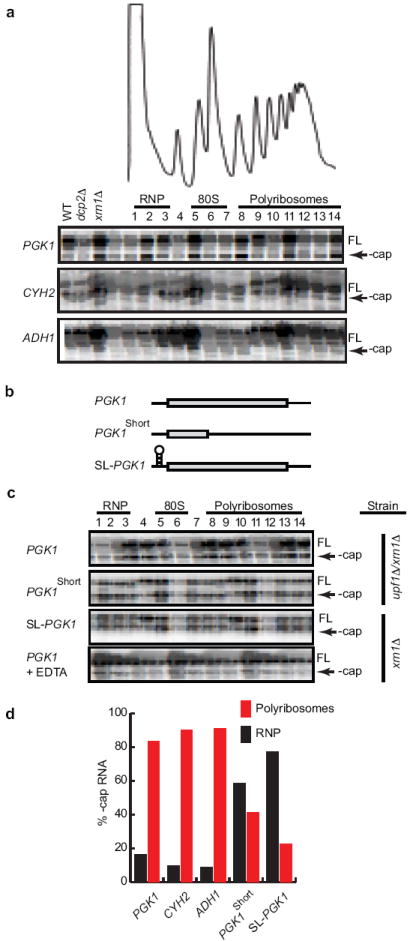
(a) Primer extension analysis on endogenous PGK1, CYH2, and ADH1 mRNA was performed on RNA isolated from sucrose gradient fractions of an xrn1Δ cell lysate. RNP, 80S, and polyribosomes are indicated above fraction numbers. FL, full length mRNA; - cap, decapped mRNA. Primer extension analyses on total RNA (15 μg) from WT, dcp2Δ, and xrn1Δ cells are shown on left side of each panel to indicate −cap mRNA is observed only in xrn1Δ cells. (b) Representation of PGK1 reporter, PGK1 reporter with a PTC (PGK1short), and PGK1 reporter with a stem-loop in its 5’ UTR (SL-PGK1). (c) Primer extension on RNA from sucrose gradient fractions from lysates of upf1Δ/xrn1Δ cells expressing PGK1 reporter or PGK1short reporter, and from xrn1Δ cells expressing SL-PGK1 or PGK1 reporter. In the bottom panel, lysates from xrn1Δ cells expressing the PGK1 reporter were incubated in presence of 50 mM EDTA prior to loading on sucrose gradients. (d) Quantification of – cap mRNAs as a percentage of total reverse transcription product in RNP and polyribosome fractions.
To investigate ribosome-associated decapping further and to exclude the possibility that decapping had occurred prior to initiation of protein synthesis, we took a transcriptional-pulse chase approach using the PGK1 mRNA reporter7. Using a circularization-based RT-PCR (cRT-PCR)10 analysis we noted that decapped RNA started to appear around 60 min following initiation of transcription (Fig. 2a-c). Separation of cell lysate into non-translating and polyribosome-associated fractions indicated that when decapping is initiated at 60 min, the vast majority of decapped mRNA was polyribosome associated (Fig. 2d). To further exclude the possibility that association of uncapped mRNA with polyribosomes is a consequence of reloading ribosomes, we used a transcriptional shut-off approach3 with the PGK1 reporter and monitored decapping using primer extension analysis. Transcription was arrested and further translation was blocked by addition of cycloheximide. Because cycloheximide inhibits ribosome elongation, newly initiated strands would be arrested at 80S11. Strikingly, mRNAs trapped on ribosomes continued to be decapped until greater than 50% was achieved after 120 min (Fig. 2e & f). In the absence of cycloheximide, the bolus of newly decapped mRNA sediments to the top of the gradient by 120 min (Fig. 2g), indicating that ribosomal run-off ensued. These results clearly show that decapping can occur when mRNAs are associated with actively translocating ribosomes.
Figure 2. mRNA decapping is initiated on polyribosomes.

All experiments in Fig. 2 were performed using cells expressing PGK1 reporter under control of the GAL1 promoter. (a) Total RNA from WT, dcp2Δ, and xrn1Δ cells was treated with (+) or without (-) tobacco acid pyrophosphatase (TAP) and circularization RT-PCR (cRT-PCR) was performed to detect decapped PGK1 reporter. (b-d) Transcriptional pulse-chase of PGK1 performed in xrn1Δ cells. (b) Poly(A) tail status of PGK1 was analyzed by oligonucleotide-directed RNase H cleavage, PAGE and Northern analysis. (c) Decapping of PGK1 mRNA monitored by cRT-PCR. (d) Cell lysates from the pulse-chase were separated on sucrose gradients. RNA from gradient fractions was pooled into non-translating (RNP) and polysome pools and decapped PGK1 was detected by cRT-PCR. (e-g) Transcriptional shut-off of PGK1 was performed in xrn1Δ cells. Lysates from cells at 0 min after shut-off (e), 120 min after shut-off in the presence of 25 μg/mL cycloheximide (f), and 120 min after shut-off without cycloheximide (g) were separated ny sucrose gradients. RNA from gradient fractions was analyzed by primer extension for PGK1 reporter. The quantifications of full length (FL) and decapped (-cap) mRNA as a percentage of total extension product are shown for each time point.
Decapping occurs on polyribosomes when translation is slowed in cis
The foregoing studies were all conducted in xrn1Δ cells to allow for the enrichment of decay intermediates. To detect decay intermediates in wild-type cells, we designed a reporter with 10 consecutive rare codons (PGK1RC; Fig. 3a). We reasoned that the presence of rare codons might slow ribosome transit12 and result in accumulation of decapped, ribosome-associated decay intermediates13. Importantly, the PGK1RC reporter’s decay is dependent on decapping and is not a major substrate for No-Go mRNA decay (Fig. S4)14. We analyzed the PGK1RC reporter on sucrose gradients and detected decay intermediates using high resolution PAGE followed by Northern blot. Strikingly, using a 3’ end-specific probe, decay intermediates of ~500 nt were detected in the region of the gradient associated with a single ribosome (i.e. 80S; Fig. 3b). In addition, mRNA intermediates of increasing length were also detected in polyribosome fractions and their size correlated well with possible ribosome occupancy (Fig. 3b). Addition of formaldehyde prior to cell lysis was used to ensure that the decay intermediates were generated in vivo (Fig. 3b), however, similar fragments were seen without formaldehyde treatment (Fig. S6a). A probe complementary to the 5’ end of the mRNA failed to detect decay intermediates confirming that the truncated mRNA was trimmed from the 5’ end (Fig. 3b & Fig. S5). Most importantly, polyribosome-associated decay intermediates were lost in dcp2Δ and xrn1Δ mutants (Fig. 3c & Fig. S6b), indicating their formation requires mRNA decapping and 5’-3’ exonucleolytic digestion. Moreover, the PGK1RC mRNA decay fragments were not a result of No-Go decay14 (Fig. S6c).
Figure 3. mRNA decapping occurs on polyribosomes in wild-type cells.

The PGK1RC reporter is depicted (a). (b) Northern blot analysis of PGK1RC mRNA after sucrose gradient fractionation. RNA detected using a 5’ or 3’ probe as depicted in (a). The same analysis as in (b) performed in dcp2Δ cells (c). (d) Splinted-ligation RT-PCR assay to detect endogenous decapped mRNA in wild-type cells. An RNA adaptor is ligated specifically to decapped mRNA via a DNA splint by T4 DNA ligase. The DNA splint is removed by DNase I treatment and ligation product is detected by RT-PCR using a gene and adaptor specific primers. The PCR product is indicative of decapped mRNA. (e) Splinted-ligation RT-PCR analysis for endogenous PGK1 and RPL41A mRNAs on total RNA from WT and dcp2Δ cells. + TAP: total RNA treated with tobacco acid pyrophosphatase to remove the 5’ cap in vitro; −ligase: no T4 ligase; -RT: no reverse transcriptase; -cDNA: no cDNA template added to PCR. (f) RNA recovered from sucrose gradient fractions of wild-type cell lysate was analyzed by splinted-ligation RT-PCR or Northern blot using gene specific probe.
We used four experiments to demonstrate that the sedimentation pattern of the PGK1RC mRNA decay intermediates is a result of polyribosome association. First, we inhibited translation of the mRNA. Insertion of a stem-loop structure into the 5’ UTR (SL-PGK1RC; Fig. S7a) shifted the full-length mRNA to the top of the gradient, and no decay intermediates were detectable deep in the gradient (Fig. S7b). Second, we terminated ribosome elongation before rare-codon recognition by introduction of a stop codon upstream of the rare codon stretch (PGK1PTC-RC; Fig. S7a). This experiment was performed in upf1Δ cells to prevent NMD15. Terminating ribosome translocation prior to the rare codons completely inhibited the formation of polyribosome-associated decay intermediates (Fig. S7c vs. S7d). Further demonstrating that ribosome recognition of the rare-codon stretch is required, repositioning the rare codon stretch within the PGK1 ORF resulted in a predictable size shift in polyribosome-associated decay fragments (Fig. S8). Finally, we performed affinity purification of polyribosomes9 and demonstrated that the decay fragments are ribosome bound (Fig. S9). In sum, these data strongly demonstrate that decapping can be detected on polyribosomes in wild-type cells if translational elongation is slowed in cis.
Endogenous mRNAs are decapped on polyribosomes in wild-type cells
The foregoing experiment utilzed a reporter harboring rare codons. To determine if endogenous mRNA in wild-type cells were also decapped when associated with ribosomes we developed a splinted ligation assay followed by RT-PCR (Fig. 3d). The RNA ligation mediated by the DNA splint is sequence specific16, thereby allowing us to directly detect the transient product generated by the decapping reaction (i.e. an RNA with 5’ phosphate). Using this assay, decapped products from endogenous PGK1 and RPL41A mRNA were detected in wild-type cells (Fig. 3e). A product was not detected in dcp2Δ cells (Fig. 3e), indicating formation requires decapping in vivo. Consistent with this, in vitro removal of the 5’ cap by tobacco acid pyrophosphate (TAP) resulted in detection of RT-PCR products in both wild-type and dcp2Δ cells (Fig. 3e). Together, these data indicate that the splinted ligation/RT-PCR assay monitors 5’ decapping. We performed this assay on RNA recovered from sucrose gradient fractions of wild-type cell lysate, and found that the decapped mRNAs from endogenous PGK1 and RPL41A were predominately detected on polyribosomes (Fig. 3f). Notably the sedimentation pattern of the decapped mRNA correlates with the total mRNA detected by Northern blot (Fig. 3f) and mRNA ORF length (Fig. 3f). Consistent with our earlier findings (Fig. 2) the sedimentation of decapped mRNA on polyribosomes is unlikely a result of ribosome reloading since the decapped intermediate is exceptionally transient in a wild-type cell. Collectively, these data indicate that in wild-type cells, endogenous mRNA are decapped on polyribosomes.
Conclusions and perspective
In sum, we have shown that decapping and 5’-3’ degradation of mRNA can occur when the transcripts are associated with actively translating ribosomes (Fig. S1). Co-translational degradation of mRNA has been previously hypothesized17,18. Here we experimentally demonstrate this hypothesis and show mRNA remains associated with active ribosomes during the process of mRNA decapping and exonucleolytic degradation. The data clearly indicate that sequestration into a ribosome-free state (e.g. P-bodies) is not a prerequisite for initiation of mRNA decay. These findings are consistent with the demonstration in yeast, Drosophila, and humans that mRNA metabolism can be uncoupled from P-body formation19-22. Moreover, they also help to explain why decay factors (e.g. hDCP2 and Xrn1p) have been found to co-sediment with polyribosomes18,23. Our findings raise several interesting mechanistic questions, for instance how mRNA half-lives are determined in the context of on-going translation. Moreover, it is unclear how the decapping machinery associates and functions on an actively translating mRNA. Interestingly, it has previously been proposed that decapping regulators promoted a ribosome-free state1,2,8,19; it now seems likely that they function in response to yet unknown cues to render the cap more accessible to the decapping enzyme during translation (Fig. S1).
Finally, we note that co-translational mRNA degradation makes sense from an evolutionary point of view. Specifically, the three steps of decay each serve to systematically limit translational events without interfering with them. Deadenylation may reduce translational efficiency perhaps through loss of the poly(A) binding protein, Pab1p15 or association of decapping regulators8. mRNA decapping inhibits further translation initiation events. Finally, degradation from the 5’ end while the mRNA is ribosome associated ensures decay does not impede residual ribosomes undergoing translocation. In this way, the final polypeptide expressed prior to the mRNA being destroyed is full length and functional.
Methods Summary
All experiments were performed using early log phase cells grown at 24 °C in synthetic medium containing appropriate sugars. RNA and polysome analyses were performed as described previously8. The cRT-PCR assay was performed as described previously10 with oJC620 for reverse transcription and oJC620/oJC635 for PCR amplification. The PGK1RC reporter was generated from fragments amplified from a previously described PGK1 reporter7 using oJC558/oJC556 and oJC557/oJC559; fragments were combined to produce a template for amplification of full length PGK1RC using oJC558/oJC559, followed by cloning onto the PGK1 reporter backbone at the BamHI and HindIII sites. Affinity purification of polyribosomes was performed as described previously9. Detection of endogenous decapped mRNA was achieved by ligating an RNA adaptor (oJC706) to the 5’ end of decapped mRNA via splinted ligation, removal of the DNA splint by DNase I, cDNA synthesis by Superscript II reverse transcriptase using a gene specific primer, and DNA amplified by PCR using a primer complementary to the RNA adaptor and a gene specific primer.
Supplementary Material
Acknowledgments
The authors would like to thank Pat Maroney and Tim Nilsen for sharing unpublished data regarding the poly(A) tailing assay. The authors also thank Dr. Tim Nilsen for his insight, suggestions, and critical evaluation of the manuscript. We also thank Drs. Roy Parker, Ambro van Hoof, and Marv Wickens for reagents and advice. Funding was provided by the American Heart Association and National Institute of Health.
Footnotes
Author Contributions W.H., K.B., and J.C. wrote the manuscript. W.H. and T.S. performed the experiments. S.C. provided technical experitice with poly(A) tailing assays. All of the authors contributed to discussion and the design of the research. All authors commented on the manuscript.
Full Methods and any associated references are available in the online version of the paper at www.nature.com/nature.
Supplementary Information is linked to the online version of the paper at www.nature.com/nature.
References
- 1.Franks TM, Lykke-Andersen J. The control of mRNA decapping and P-body formation. Mol Cell. 2008;32(5):605–615. doi: 10.1016/j.molcel.2008.11.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Parker R, Sheth U. P bodies and the control of mRNA translation and degradation. Mol Cell. 2007;25(5):635–646. doi: 10.1016/j.molcel.2007.02.011. [DOI] [PubMed] [Google Scholar]
- 3.Coller J, Parker R. Eukaryotic mRNA decapping. Annu Rev Biochem. 2004;73:861–890. doi: 10.1146/annurev.biochem.73.011303.074032. [DOI] [PubMed] [Google Scholar]
- 4.Eulalio A, Behm-Ansmant I, Izaurralde E. P bodies: at the crossroads of post-transcriptional pathways. Nat Rev Mol Cell Biol. 2007;8(1):9–22. doi: 10.1038/nrm2080. [DOI] [PubMed] [Google Scholar]
- 5.He F, Jacobson A. Upf1p, Nmd2p, and Upf3p regulate the decapping and exonucleolytic degradation of both nonsense-containing mRNAs and wild-type mRNAs. Mol Cell Biol. 2001;21(5):1515–1530. doi: 10.1128/MCB.21.5.1515-1530.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hsu CL, Stevens A. Yeast cells lacking 5’-->3’ exoribonuclease 1 contain mRNA species that are poly(A) deficient and partially lack the 5’ cap structure. Mol Cell Biol. 1993;13(8):4826–4835. doi: 10.1128/mcb.13.8.4826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Muhlrad D, Decker CJ, Parker R. Turnover mechanisms of the stable yeast PGK1 mRNA. Mol Cell Biol. 1995;15(4):2145–2156. doi: 10.1128/mcb.15.4.2145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Coller J, Parker R. General translational repression by activators of mRNA decapping. Cell. 2005;122(6):875–886. doi: 10.1016/j.cell.2005.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Inada T, et al. One-step affinity purification of the yeast ribosome and its associated proteins and mRNAs. Rna. 2002;8(7):948–958. doi: 10.1017/s1355838202026018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Couttet P, Fromont-Racine M, Steel D, Pictet R, Grange T. Messenger RNA deadenylylation precedes decapping in mammalian cells. Proc Natl Acad Sci U S A. 1997;94(11):5628–5633. doi: 10.1073/pnas.94.11.5628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Amrani N, Ghosh S, Mangus DA, Jacobson A. Translation factors promote the formation of two states of the closed-loop mRNP. Nature. 2008;453(7199):1276–1280. doi: 10.1038/nature06974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhang S, et al. Polysome-associated mRNAs are substrates for the nonsense-mediated mRNA decay pathway in Saccharomyces cerevisiae. Rna. 1997;3(3):234–244. [PMC free article] [PubMed] [Google Scholar]
- 13.Stevens A. 5’-exoribonuclease 1: Xrn1. Methods Enzymol. 2001;342:251–259. doi: 10.1016/s0076-6879(01)42549-3. [DOI] [PubMed] [Google Scholar]
- 14.Doma MK, Parker R. Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature. 2006;440(7083):561–564. doi: 10.1038/nature04530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Jacobson A, Peltz SW. Interrelationships of the pathways of mRNA decay and translation in eukaryotic cells. Annu Rev Biochem. 1996;65:693–739. doi: 10.1146/annurev.bi.65.070196.003401. [DOI] [PubMed] [Google Scholar]
- 16.Moore MJ, Query CC. Joining of RNAs by splinted ligation. Methods Enzymol. 2000;317:109–123. doi: 10.1016/s0076-6879(00)17009-0. [DOI] [PubMed] [Google Scholar]
- 17.Beelman CA, Parker R. Differential effects of translational inhibition in cis and in trans on the decay of the unstable yeast MFA2 mRNA. J Biol Chem. 1994;269(13):9687–9692. [PubMed] [Google Scholar]
- 18.Mangus DA, Jacobson A. Linking mRNA turnover and translation: assessing the polyribosomal association of mRNA decay factors and degradative intermediates. Methods. 1999;17(1):28–37. doi: 10.1006/meth.1998.0704. [DOI] [PubMed] [Google Scholar]
- 19.Chu CY, Rana TM. Translation Repression in Human Cells by MicroRNA-Induced Gene Silencing Requires RCK/p54. PLoS Biol. 2006;4(7):e210. doi: 10.1371/journal.pbio.0040210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Decker CJ, Teixeira D, Parker R. Edc3p and a glutamine/asparagine-rich domain of Lsm4p function in processing body assembly in Saccharomyces cerevisiae. J Cell Biol. 2007;179(3):437–449. doi: 10.1083/jcb.200704147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sweet TJ, Boyer B, Hu W, Baker KE, Coller J. Microtubule disruption stimulates P-body formation. Rna. 2007;13(4):493–502. doi: 10.1261/rna.355807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Eulalio A, Behm-Ansmant I, Schweizer D, Izaurralde E. P-body formation is a consequence, not the cause, of RNA-mediated gene silencing. Mol Cell Biol. 2007;27(11):3970–3981. doi: 10.1128/MCB.00128-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang Z, Jiao X, Carr-Schmid A, Kiledjian M. The hDcp2 protein is a mammalian mRNA decapping enzyme. Proc Natl Acad Sci U S A. 2002;99(20):12663–12668. doi: 10.1073/pnas.192445599. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.