

Abstract
The quaternary stability of purified, detergent-solubilized, cytochrome c oxidase (CcO) was probed using two chemical denaturants, urea, and guanidinium chloride. Each chaotrope induces dissociation of five subunits in a concentration dependent manner. These five subunits are not scattered over the surface of CcO, but are clustered together in close contact at the dimer interface. Increasing the concentration of urea selectively dissociates subunits from CcO in the following order: VIa & VIb, followed by III & VIIa, and finally Vb. After incubation in urea for 10 min at room temperature, the sigmoidal dissociation transitions were centered at 3.7 M, 4.6 M, and 7.0 M urea, respectively. The secondary structure of CcO was only minimally perturbed, indicating that urea causes disruption of subunit interactions without urea-induced conformational changes. Incubation of CcO in urea for 120 min produced similar results, but shifted the sigmoidal dissociation curves to lower urea concentrations. Incubation of CcO with increasing concentrations of guanidinium chloride (GdmCl) produces an analogous effect; however, the GdmCl-induced dissociation of subunits occurs at lower concentrations and with a narrower concentration range. Thermodynamic parameters for each subunit dissociation were evaluated from the sigmoidal dissociation data by assuming a single transition from bound to dissociated subunit. The free energy change accompanying urea-induced dissociation of each subunit ranged from 18.0 to 29.7 kJ/mol, which corresponds to 0.32 to 0.59 kJ/mol per 100 Å2 of newly exposed solvent accessible surface area. These values are 30-50-fold smaller than previously reported for the unfolding of soluble or membrane proteins.
Bovine cytochrome c oxidase (CcO) (ferrocytochrome c: O2 oxidoreductase; EC 1.9.3.1) is the terminal enzyme (complex IV) of the inner mitochondrial electron transport chain that catalyzes electron transfer from reduced cytochrome c to molecular oxygen. The mitochondrial complex is composed of 13 non-identical protein subunits, three of which are products of the mitochondrial genome, 10 of which are nuclear encoded. The three mitochondrially encoded subunits (I, II and III2) form the core of the enzyme and contain 2 heme a moieties and 2 copper centers as prosthetic groups (1,2). The three core subunits are also homologous with the three major subunits of bacterial terminal heme oxidases (the latter contain only three to four subunits, 3,4,5). The ten nuclear-encoded subunits surround the core and are unique to mitochondrial terminal oxidases. Because bacterial a3 terminal oxidases function perfectly well without counterparts to the nuclear encoded subunits, it raises a question regarding the structural and/or functional importance of these subunits.
Several attempts have been made to clarify the role of the nuclear encoded subunits within mitochondrial CcO. Two of these subunits, VIa and VIb2, are located at the dimer interface of CcO (2) and their dissociation during cardiolipin removal irreversibly produces monomeric enzyme (6). Subunits VIa, VIIa, VIIc, and possibly VIII map near cardiolipin binding sites since they are photo-labeled by arylazido-cardiolipin (7). Subunits IV, Va, Vb, and VIa may serve regulatory roles since ATP, ADP, and diiodothyronine allosterically affect enzymatic activity when they bind to one or more of these subunits (8). Attempts have also been made to clarify the functional role of the nuclear-encoded subunits by their selective proteolytic enzyme cleavage (9,10,11), or subunit-specific monoclonal antibody binding to these subunits (12). However, these types of experiments are complex and relatively non-specific, making their interpretation often ambiguous.
An alternative approach has been to use denaturants or chaotropes to perturb the quaternary structure of the enzyme. Multi-subunit water-soluble proteins, when exposed to structural perturbants such as guanidinium chloride (GdmCl) or urea, often exhibit a concentration-dependent transition from the native state to an assembly of partially folded, and then completely unfolded polypeptides (13,14,15,16). Exposure of CcO to increasing concentrations of GdmCl, however, suggests the presence of two structural domains with differing denaturant sensitivity, i.e., a more stable, membrane-embedded, hydrophobic domain, and a second, less stable hydrophilic domain that protrudes into the intra-membrane space (17). Some subunit interactions within CcO must be relatively weak since rather minor structural perturbations, such as removal of cardiolipin, are directly linked to the dissociation of specific subunits (i.e., VIa and VIb CcO; 18). Based on these findings, together with the observation that the secondary structure of integral proteins is very resistant to denaturation (19,20,21,22), we predict that dissociation of certain subunits from CcO will precede their denaturant-induced unfolding. If true, selective dissociation of subunits may be possible without disruption of the enzymatic core of CcO. In this work, we confirm the validity of this hypothesis and present a straightforward method for the sequential and selective removal of several subunits from CcO using either urea, or guanidinium chloride as a structural perturbant.
EXPERIMENTAL PROCEDURES
Materials
Bovine cytochrome c oxidase was prepared from Keilin-Hartree heart particles by the method of Fowler et al. (23) with modifications described by Mahapatro and Robinson (24). Two different preparations of the enzyme, from two different sources of bovine hearts, were similar in terms of heme content (9.4-9.9 nmol/mg) and phosphorus content (15-30 mol/mol oxidase), and electron transfer activity (350 - 400 sec-1, determined spectrophotometrically using ferrocytochrome c as a substrate).
Individual drops of purified enzyme (20-25 mg/mL protein, 10 mg/mL sodium cholate, 100 mM phosphate buffer, pH 7.4) were quickly frozen by slowly dripping the protein solution into liquid nitrogen. Individual frozen aliquots of enzyme, ∼ 25 μL, were stored at -80 °C. Before each experiment, dodecylmaltoside was substituted for the sodium cholate in the enzyme preparation by diluting the enzyme to 1 mg/mL with 1 mg/mL dodecylmaltoside followed by removal of sodium cholate by extensive dialysis against buffer containing greater than CMC concentrations of the new detergent. The resulting dodecylmaltoside-solubilized enzyme was monomeric as judged by sedimentation velocity analysis (25,26,27). CcO concentrations were calculated based on ε422 = 1.54 × 105 M-1cm-1 (28). Reduced horse heart ferrocytochrome c (ε550 = 29.5 mM-1cm-1) for activity measurements was freshly prepared by dithionite reduction followed by Sephadex G-25 gel filtration for removal of excess dithionite.
The C18 reversed phase column for subunit analysis (10 μ, 4.6 mm × 250 mm, Cat. No. 218TP104) was purchased from Vydac; the HiTrap Q HP anion exchange column (1-mL disposable column) for removal of dissociated subunits was purchased from Amersham Biosciences. Horse heart cytochrome c (Type III) was obtained from Sigma Chemical Co. Acetonitrile and phosphoric acid were of HPLC grade and were obtained from Fisher Scientific. HPLC grade chloroform and methanol were from EM Science. Ultrapure dodecylmaltoside was obtained from either Boehringer Mannheim or Anatrace, Inc., respectively. All other chemicals were reagent grade.
Urea-Induced Dissociation of CcO Subunits
Dodecylmaltoside-solubilized CcO (0.5-1.0 mg of the protein in 200 μL of 40 mM MOPS, pH 7.2) was mixed with the appropriate amount of 9 M urea in 200 mM MOPS, pH 7.2, and an appropriate amount of the same buffer without urea to achieve the desired urea concentration (between 0.0 and 7.0 M in the final volume 1 mL). After incubation for either 10, or 120 min at room temperature, the solution was diluted 5 fold with 20 mM MOPS, pH 7.2, and stored at 4 °C until dissociated subunits were removed by HiTrap Q anion exchange chromatography. To remove dissociated subunits, the 5-mL sample was applied to a 1-mL HiTrap Q column at 0.5 mL/min and the column washed with buffer A for 15 min. Subunit depleted CcO was subsequently eluted with a linear 5 min. gradient from 100% buffer A (20 mM MOPS, pH 7.2, containing 0.5 mg/mL dodecylmaltoside) to 100% buffer B (the same as buffer A, but containing 0.4 M Na2SO4). The detergent concentration in the sample was adjusted to produce a protein to detergent ratio of 1:1 (w/w). Sample injection and control of HPLC elution from the HiTrap Q anion-exchange column were similar to that previously described (18). Digital absorbance data from the Gilson variable wavelength detector was collected using a Waters SATIN A/D interface module connected to a 50 MHz 486 PC Computer running Waters Millennium 2010 software, Ver. 2.0.
Quantitative Determination of CcO Subunit Content
The subunit content after exposure to urea was determined by a combination of RP-HPLC and SDS-PAGE subunit analyses. The dissociation of nuclear-encoded subunits, i.e., IV-VIII2, was quantified using C18 reversed phase HPLC (29). The content of subunits IV and Va were assumed to be unperturbed by exposure to urea and were used as internal standards. The normalized area under each of the other elution peaks for urea-treated CcO was compared with that obtained for untreated CcO to quantify subunit loss. The content of the three core subunits, (I - III), was determined after resolution of subunits by SDS-polyacrylamide gel electrophoresis on 15% acrylamide gels that contained 2 M urea in addition to 0.1% SDS (30). The gel was fixed, stained with Coomassie blue, scanned and quantified using a Molecular Dynamics gel scanner and Ver. 4.0 software. The amount of subunits II and III that remained associated with the core of CcO after its exposure to urea was determined relative to that of untreated CcO, assuming the amount of subunit I remained constant.
Fitting of Sigmoidal Subunit Dissociation Data
Denaturant-induced dissociation of a subunit was found to be a two stage process: 1) the transition from subunit “x”, Sx, bound to the enzyme core, CcO:Sx, to the fully dissociated subunit, CcO + Sx; and 2) the unfolding of dissociated subunit, Sx, to produce denatured subunit, Dx. This process can be described by the following equation:
| (1) |
Step (2) is much slower than step (1); therefore, exposure of CcO to urea for short periods of time, i.e., 10 min, primarily involves step (1) and does not involve subunit unfolding. Exposure of CcO to urea for longer periods of time, i.e., 2 h, at least partially involves step (2). Although only step (1) appears to be an equilibrium process with CcO subunits, urea-induced dependences of subunit dissociation were analyzed after either 10 min, or 2 hr of urea exposure by the linear extrapolation model formally equal to the denaturant-induced protein unfolding model (31), according to the following equations:
| (2) |
| (3) |
| (4) |
| (5) |
where, fD is the fraction of the dissociated subunit, K and ΔG are the equilibrium constant and associated Gibbs energy change for subunit dissociation, respectively, and m is the slope of the linear correlation between ΔG and urea concentration. ΔGH2O is the Gibbs energy change for subunit dissociation in the absence of urea, i.e., in water. Non-linear least-squares fitting algorithms within SigmaPlot©, ver. 1.02, were used to fit equation 5 to the experimental data, (fD as a function of [urea]), to evaluate best fit values for the parameters ΔGH2O and m. The value of [urea]1/2, which represents the urea concentration when K is unity, was evaluated from the equality, [urea]1/2 = ΔGH2O /m.
Evaluation of Increased Solvent Accessible Surface Contact Area upon Dissociation of CcO Subunits
The increase in solvent accessible surface area was assumed to be the difference between total surface area before and after dissociation of a particular subunit. For this purpose, the CcO coordinates from 1v54.pdb and PyMol® software (DeLano Scientific) were used to generate separate pdf files for CcO comprised of 13, 12, 11, 10, and 9 subunits corresponding to the sequential removal of subunits VIb, VIa, VIIa and III, together with separate pdf files for the four subunits. The software program PISA (Protein Interfaces Surfaces and Assemblies), which uses a 1.4 Å “rolling ball” to evaluate the solvent exposed surface area, was then utilized to measure the surface area for each subunit and subunit-depleted form of CcO. From these values, increased solvent exposure corresponding to the dissociation of each subunit was evaluated. For example, the increase in the solvent accessible surface area upon dissociation of subunit III, which dissociates after dissociation of subunits VIa, VIb and VIIa, was calculated by the difference in total surface area of: 1) 9-subunit CcO (missing VIa, VIb, VIIa & III) plus the surface area of subunit III alone; and 2) 10-subunit CcO (missing VIa, VIb, & VIIa).
RESULTS
Urea-Induced Dissociation of Subunits from CcO
The content of subunits remaining associated with the core of CcO was quantified after exposure to a series of different urea concentrations. The procedure was to remove dissociated subunits using HiTrap Q anion exchange chromatography followed by subunit analysis of residual CcO subunits using a combination of reversed phase C18 HPLC (all subunits except for I, II & III; Figure 1) and SDS polyacrylamide gel electrophoresis (subunits I, II & III; data not shown). Resolution of subunits was similar after exposure to 0 - 7 M urea, but certain subunits were progressively absent as the urea concentration was increased, particularly subunits III, VIa, VIb, VIIa, and Vb.
Figure 1.

Subunit composition of CcO before and after exposure to various concentrations of urea. In each sample, dodecylmaltoside solubilized CcO was exposed to 0 - 7 urea for 2 hr at room temperature, the dissociated subunits and urea were removed by HiTrap Q anion exchange chromatography, and the subunit composition determined by either SDS-PAGE or reversed phase HPLC. Top Panel: SDS-PAGE analysis of the mitochondrial-encoded subunit composition CcO before and after exposure to 0, 3.0, 5.0 and 7.0 M urea. Bottom Panel: Reversed-phase C18-HPLC subunit analysis of the nuclear-encoded subunit composition determined by analysis of 100μg (0.5 nmol) of the purified CcO using reversed-phase C18 HPLC (refer to Methods for details). Elution peaks are labeled by roman numerals to indicate the elution peak corresponding to each of the nuclear-encoded subunits. The four elution profiles from the top down correspond to results obtained after exposure to 0, 4.0, 5.5, and 7.0 M urea, and are labeled accordingly. Prolonged incubation at high urea concentrations did not affect either the elution position or the shape of the elution peaks suggesting that covalent modification by urea break down products, e.g., cyanate, did not occur.
Analysis of two different preparations of CcO, characterized by identical subunit composition, similar phospholipid content and comparable electron-transfer activity, produced qualitatively similar results when exposed to urea at room temperature for either 10 min. (Figure 2a), or 120 min (Figure 2b). Exposure of CcO to urea for 10 min induced the selective dissociation of subunits in the order VIb > VIa >> VIIa > III >>> Vb, with sigmoidal dissociation transitions centered at 3.7, 3.8, 4.4, 4.8, and ∼7.0 M urea, respectively. The almost coincident dissociation of VIa + VIb and then of III + VIIa (Figure 2) are each consistent with the crystal structure of CcO which shows these pairs of subunits are in close contact with each other, Figure 5 (2). Analysis of CcO after a 2-hr exposure to urea at room temperature produced similar results, i.e., urea-induced dissociation of subunits from CcO took place in the same relative order as after exposure to urea for 10 min. However, prolonged exposure to urea shifted the subunit dissociation transitions to lower urea concentrations and the dissociation curves for each subunit were less well separated (Figure 2, & Table 1).
Figure 2.

Concentration dependence of urea-induced dissociation of subunits from CcO. CcO was exposed to urea (0 - 7 M), and the subunit content determined after removal of urea and dissociated subunits by HiTrap Q anion exchange column chromatography (refer to Methods for details). Data were collected after exposure of CcO to urea for either 10 min (top panel), or 2 h (bottom panel) at room temperature. Five subunits dissociated with a sigmoidal dependence upon the urea concentration: III (grey-filled circles); Vb (black-filled circles); VIa black-filled triangles); VIb (unfilled triangles); and VIIa (unfilled circles). A full complement of the other eight subunits remained associated with the core of CcO, even at the highest urea concentration. Dissociation of each subunit could be fitted by non-linear regression analysis according to equation (5). For clarity, only the best-fit line through dissociation data collected for subunit VIIa are shown. Molar ellipticity data, θ222nm (unfilled diamonds), is also included in each panel, which indicates that only minimal perturbation of the secondary structure occurs after exposure of CcO to 7 M urea for 10 min, or 4 M urea for 2 h at room temperature.
Figure 5.

Location of subunits within the three-dimensional structure of CcO that are either susceptible, or resistant to urea, or GdmCl-induced dissociation. The five subunits that dissociate (colored in this figure) are located near the dimer interface, and are in close contact with each other. This group of subunits is also the group that is sensitive to dissociation by elevated hydrostatic pressure [31]. Subunits that are resistant to dissociation, i.e., I, II, IV, Vb, VIc, VIIb, VIIc and VIII, are also clustered to form the catalytic core of CcO, (colored grey in this figure). Dimeric CcO can be considered to consist of three domains, two core domains that are urea and GdmCl resistant, and a third fragile, domain that bridges the other two (artificial separation of the CcO into these three domains is shown at the bottom of the figure). The third, fragile domain is roughly donut shaped with a hole in the center when viewed down the y-axis (yellow line). Dimeric CcO contains two copies of each subunit; therefore, the fragile domain is comprised of 10 subunits including two copies of subunits III (bright and lemon yellow), Vb (dark and light blue), VIa (bright and pale orange), VIb (bright and light pink), and VIIa (bright and dark cyan). All of the bright-colored subunits are associated with one CcO monomer, while all of the light, pale colored subunits are associated with the second monomer.
Table 1.
Apparent thermodynamic parameters for dissociation of subunits from CcO derived from urea, or GdmCl-induced, isothermal dissociation data acquired at room temperature
| Subunit | 10 min Exposure to Urea |
120 min Exposure to Urea |
10 min GdmCl Exposure |
|||
|---|---|---|---|---|---|---|
| ΔGH2O (kJ/mol) | [urea]1/2a (M) | ΔGH2O (kJ/mol) | [urea]1/2a (M) | ΔGH2O (kJ/mol) | [GdmCl]1/2a (M) | |
| VIb | 18.8±1.1 | 3.7 | 14.4±1.4 | 3.5 | 18.3±3.1 | 1.03 |
| VIa | 18.0±2.6 | 3.8 | 14.2±1.4 | 3.6 | 19.3±3.2 | 1.04 |
| VIIa | 24.7±3.0 | 4.4 | 13.7±1.2 | 3.9 | 14.6±2.6 | 1.12 |
| III | 29.7±3.1 | 4.8 | 12.3±1.4 | 3.9 | 14.9±1.5 | 1.14 |
| Vb | --- | --- | 19.9±1.9 | 4.9 | --- | --- |
[urea]1/2 = ΔGH2O/m; standard error in m = 5-10%; standard error in [urea]1/2 = 15-20%.
[GdmCl]1/2 = ΔGH2O/m; standard error in m = 5-10%; standard error in [GdmCl]1/2 = 15-20%.
The sigmoidal dependence of each subunit dissociation upon the urea concentration suggests that each can be described by a pseudo-equilibrium transition (First step of equation 1). Pseudo-equilibrium also indicates that dissociated subunits reassociate with the remainder of the enzyme, but this was not investigated. Consistent with a pseudo-equilibrium model, the experimental subunit dissociation data for each subunit were adequately fitted to equation (5) using non-linear regression analysis to evaluate the parameters ΔGH2O and [urea]1/2. A typical solution is illustrated in Figure 2 as a “best-fit” line superimposed upon subunit VIIa dissociation data. Similar analysis of the other dissociation data yielded ΔGH2O and [urea]1/2 values for each subunit that undergoes urea-induced dissociation (Table 1).
The above analysis is purely empirical and does not prove, or require reversibility. In fact, it is likely that reassociation of dissociated subunits is slowed, or prevented by two factors: 1) the solubilizing detergent, dodecylmaltoside; and 2) urea-induced unfolding of the dissociated subunit (second step of equation 1). Detergent would be expected to affect reversibility since it would coat the newly exposed hydrophobic surfaces on both the released subunit and the remainder of the protein, thereby preventing reassociation. If this occurs, equilibrium would be shifted to the “right”, i.e., towards subunit dissociation, and would result in an underestimation of ΔGH2O. This “detergent-effect” cannot be avoided and influences data collected exposure to urea for either 10, or 120 min. Exposure to elevated urea concentration may also produce coincident unfolding of the dissociated subunit, or induce structural perturbations within the CcO core. If either occurs, it would alter the interpretation of ΔGH2O and [urea]1/2 since any conformational changes would shift the equilibrium toward subunit dissociation. Such urea-induced structural changes are strongly time-dependent. Exposure of CcO to urea for short time periods, i.e., 10 min, causes the dissociation of subunits III, VIa, VIb and VIIa before structural alterations are detected by circular dichroism, i.e., before any significant change occurs in the molar ellipticity at 222 nm (Figure 2). Dissociation of Vb, however, occurs coincident with a significant decrease in the molar ellipticity, suggesting that dissociation is coupled to significant structural changes in either Vb, or the core subunits of CcO, even after 10 min exposure to high concentrations of urea. Subunit dissociation data acquired after 120 min exposure to urea are also not independent of subunit unfolding or structural perturbations. Although the subunit dissociation curves remain sigmoidal shaped, significant changes begin to occur in the molar ellipticity at 222 nm with relatively low urea concentration, i.e., 3 M. It is, therefore, not surprising that ΔGH2O values evaluated from the 120 min data are significantly smaller than values evaluated from 10 min data (Table 1).
Confirmation that subunit dissociation precedes significant structural changes in either the dissociated subunit or the CcO core was obtained by monitoring urea-induced changes in the visible spectrum, which is sensitive to the conformational state surrounding heme a and a3. The visible spectrum, difference spectrum and second derivative spectrum were each recorded before and after exposure of CcO to 6 M urea (Figure 3). After 10 min exposure, none of these was significantly perturbed as compared with spectra recorded prior to exposure of CcO to 6 M urea suggesting the absence of structural changes near the catalytic core. For example, the two minima at 416 nm and 431 nm in the second derivative spectrum, corresponding to signals from high spin heme a3 and low spin heme a (32), remain unchanged. Prolonged exposure to 6 M urea, i.e., for 120 min, did produce changes that are consistent with partial reduction of heme a3, but do not suggest major changes in the heme environments. The difference spectrum exhibits a minimum at 416 nm with a maximum at 451 nm, both of which are consistent with a small amount of heme a3 reduction. The changes in the second derivative spectrum are consistent with this interpretation: a decrease in the oxidized high spin heme a3 signal (negative trough at 416 nm), together with a slight red shift so that it no longer is resolved from the oxidized low spin heme a signal (negative trough at 431 nm).
Figure 3.

Urea-induced changes in the visible spectrum of CcO. Top Panel, absorbance spectrum of CcO after exposure to 6 M urea for 0 min (solid line), 10 min (dashed line), or 120 min (dotted-dashed line). Inset, corresponding difference spectrum relative to CcO exposed to urea for 0 min. Bottom Panel, second derivative spectrum of CcO after exposure to 6 M urea at room temperature for 0 min (solid line), 10 min (dashed line), or 120 min (dotted-dashed line). In each case spectra were acquired at 25 ° after CcO was incubated in 6M for the given length of time, and subsequently diluted 5-fold with 20 mM MOPS buffer, pH 7.2, to quench the dissociation reaction.
GdmCl-induced dissociation of subunits in CcO
The content of subunits remaining associated with the core of CcO was also quantified after exposure to a series of different GdmCl concentrations using an experimental approach identical to that used with urea. Exposure of CcO to a series of GdmCl concentrations for 10 min at room temperature leads to the sequential dissociation of the same subunits in the same order as does urea, i.e., VIa + VIb > III + VIIa >> Vb. However, in accordance with stronger denaturant properties of GdmCl, all of these subunits exhibit sigmoidal dissociation within a narrow concentration range at a significantly lower denaturant concentration, i.e., between 1.0-1.4 M GdmCl (Figure 4, Table 1). GdmCl is generally considered to be at least 2-fold more efficient as a structural perturbant than is urea, at least as has been demonstrated for water-soluble proteins (33). However, dissociation of subunits from CcO yields values for [GdmCl]1/2 that are 4-5-fold lower then for [urea]1/2, indicating that GdmCl is much more efficient at dissociating subunits from CcO than is urea. GdmCl is also a stronger perturbant of CcO secondary structure since significant changes in the 222 nm molar ellipticity occur at quite low GdmCl concentration, i.e., <1 M, making it impossible to separate secondary structural changes from subunit dissociation. Analysis of GdmCl-induced subunit dissociation after prolonged incubation was not possible due to CcO precipitation.
Figure 4.
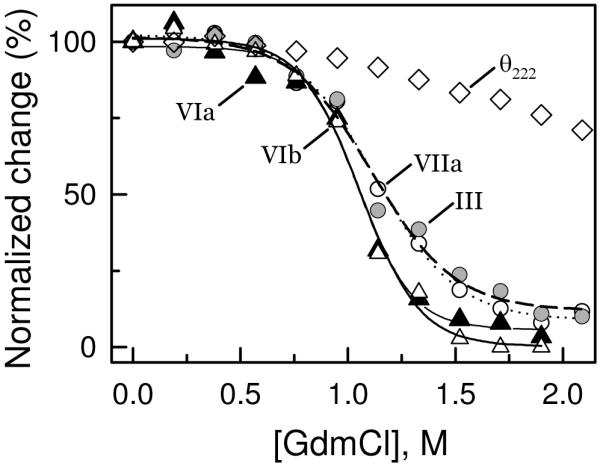
Concentration dependence of GdmCl-induced dissociation of subunits from CcO. CcO was exposed to GdmCl (0 - 2.2 M), and the subunit content determined after removal of GdmCl and dissociated subunits by HiTrap Q anion exchange column chromatography (refer to Methods for details). Data were collected and analyzed as described in Figure 2 for urea-induced dissociation of subunits from CcO. Data are shown for dissociation of subunits III (grey-filled circles), VIa (solid-filled triangles), VIb (unfilled triangles), and VIIa (unfilled circles), together with accompanying changes in the molar ellipticity, θ222nm (unfilled diamonds).
DISCUSSION
Mitochondrial cytochrome c oxidase consists of an enzymatic core of three mitochondrially-encoded subunits surrounded by a number of much smaller, nuclear-encoded, subunits that contain one or two trans-membrane α-helices. The structural stability of the individual subunits is much stronger than the interactions between subunits. Therefore, exposure of CcO to low or moderate concentrations of urea or GdmCl for relatively short times effectively disrupts specific subunit interactions without significant perturbation of either the protein secondary structure, or the environment surrounding the hemes. As a consequence, we have utilized these chaotropes to dissociate five subunits from CcO. The five subunits dissociate sequentially, not coincidently, as the urea, or GdmCl concentration is increased. The associations of subunits VIa and VIb are the weakest since they dissociate together at a relatively low concentration of urea or GdmCl. The associations of subunits III and VIIa with CcO are significantly stronger and they dissociate from CcO at a higher concentration of urea or GdmCl. Subunit Vb is bound the most tightly of the five subunits since it only begins to dissociate from CcO at the highest chaotrope concentrations when the protein secondary structure is significantly perturbed, i.e., as θ222 becomes less negative. No other subunits dissociate from CcO, even at the very highest concentrations of urea, or GdmCl suggesting that their association with the core of CcO is significantly stronger than the five other subunits. The relative association of the five subunits is best summarized as: VIb<VIa << VIIa <III <<< Vb.
The five subunits that can be removed from the core of CcO by exposure to urea, or GdmCl are not scattered over the surface of the enzyme. Rather, they are clustered together at the dimer interface, forming a fragile domain that connects the two resistant core domains of the CcO dimer (Figure 5). Dissociation of these five subunits is not limited to chaotrope-induced dissociation. The same subunits are also sensitive to pressure-induced dissociation from CcO. In fact, the extent and order of subunit loss when the detergent-solubilized enzyme is exposed to 2 - 3 kbar hydrostatic pressure is nearly identical to that produced by exposure of enzyme to either urea or GdmCl (34). One structural consequence of CcO dimerization is that it should stabilize this group of intrinsically unstable subunits, which was confirmed by the hydrostatic pressure experiments (34). We conclude that monomerization almost certainly precedes the initial dissociation of subunits VIa and VIb. Once they dissociate, reassociation or retention of the native dimeric structure is highly improbable.
Further evidence to the fragility of this interfacial domain is the observation that these subunits are also often lost during enzyme purification, or delipidation in the presence of relatively high concentrations of dodecylmaltoside, or Triton X-100 (29). In fact, such inadvertent subunit loss during enzyme isolation is often responsible for the polydispersity present with purified CcO. Hence, exposure of purified CcO to chemical, or physical perturbants only increases the probability of a “natural” disintegration of CcO (35). The order of denaturant-induced subunit dissociation also mimics the reverse of CcO assembly. For example, the last steps in human CcO assembly involve the incorporation of subunits III, Vb and VIb, followed by the association of subunits VIa and VIIa (36,37). Therefore, we predict that association energies derived from perturbant-induced subunit dissociation are directly applicable to CcO assembly.
One direct consequence of the structural perturbant approach is that it allows evaluation of subunit association/dissociation free energies. The approach is similar to that used to quantify protein protein-unfolding/folding energy. By assuming that forces stabilizing the CcO quaternary structure are similar to those known to stabilize protein secondary and tertiary structure, we could utilize equations normally associated with the evaluation of protein-unfolding/folding to evaluate ΔGdissociation as a function of chaotrope concentration. By analogy with protein unfolding, ΔGdissociation for each subunit is expected to be linearly dependent upon the chaotrope concentration (equation 4). Therefore, the Gibbs energy of subunit dissociation in the absence of chaotrope, , is obtained by extrapolation to zero concentration (Table 1). Such an analysis presumes independent subunit binding and an equilibrium dissociation/association process. With relatively short exposure-times to urea, i.e., 10 min, these assumptions appear to be valid and result in Gibbs energy values for dissociation of the four subunits from CcO in the range of 18 - 30 kJ/mol. However, with longer exposure-times, urea begins to induce significant changes in the secondary structure of the dissociated subunit or in the CcO core, which shifts the equilibrium towards dissociation and results in smaller values for (refer to Table 1). Such time-dependent changes in the apparent are more pronounced for subunits that dissociate at higher urea concentration; therefore, the values obtained for subunits III, VIIa and Vb are affected more than values obtained for subunits VIa and VIb. This probably explains why the values for the five subunits are all very similar when they are based upon data collected after 120 min exposure to urea, but the four values vary from 18 to 30 kJ/mol when evaluated from data collected after only a 10 min exposure (the ΔG for the fifth subunit, Vb, could not be evaluated from the 10 min exposure data). We believe that the latter values, which are the least perturbed by urea-induced structural changes, more closely correspond to the true thermodynamic parameters for subunit dissociation/reassociation.
We were unable to compare our values for subunit dissociation from CcO with values obtained with other multi-subunit, intrinsic membrane proteins because our study is the first attempt to quantify the energetics of such interactions. Therefore, we could only compare our values with those previously obtained for dissociation of subunits from soluble proteins, a very different process since it does not involve transfer of the dissociated monomer into a detergent micelle. Surprisingly, despite the large structural difference between these two systems, for dimer dissociation is quite comparable, i.e., they are in the range of 17 - 24 kJ/mol (38,39). However, as discussed earlier our values for subunit dissociation could easily be underestimated. The presence of solubilizing detergent certainly complicates the comparison and may greatly perturb the subunit dissociation/reassociation equilibrium.
The Gibbs free energy for subunit dissociation from homo-multimeric proteins, however, is known to be directly proportional to the change in solvent accessible surface area. Few attempts have been made to quantify the kJ/mol of free energy associated with exposure of a defined amount of buried surface area. The single value reported for soluble proteins is 0.9 kJ/mol per 100 Å2 of newly exposed solvent accessible surface [40], but a value of 10.5 kJ/mol per 100 Å2 has been suggested based upon theoretical arguments [41]. The higher value is consistent with the free energy change associated with the unfolding of either soluble or membrane proteins, i.e., 11.0 - 15.5 kJ/mol per 100 Å2 of newly exposed surface area [42]. We applied a similar approach to quantify the free energy change per 100 Å2 of newly exposed surface for dissociation of each of the four subunits from CcO. The increase in solvent accessible surface accompanying the dissociation of each subunit was evaluated in silico using: 1) PyMol® to sequentially remove the four subunits; and 2) the software program PISA to evaluate the surface accessible area before and after subunit removal (refer to Experimental Procedures for details). The resulting values fall into two groups, i.e., 0.59 kJ/mol per 100 Å2 for the more peripheral subunits VIb and VIIa, and 0.32 kJ/mol per 100 Å2 for the more hydrophobic, buried subunits VIa and III (Table 2). Both values are considerably smaller than for those previously reported. However, the free energy values associated with the dissociation of subunits from soluble homo-multimeric proteins, or the unfolding of membrane proteins is very different from the dissociation of subunits from CcO. The previous studies concern free energy changes accompanying the transfer of amino acids buried within the protein interior into solution, i.e., all of the proteins before and after dissociation are completely hydrated and exposed to solvent. Our experimentally derived values, however, concern the free energy associated with the transfer of a subunit from a detergent-solubilized multi-subunit complex to the interior of a detergent micelle, i.e., the buried amino acid is transferred from one apolar environment to another apolar environment. A more accurate description for this process would be rather than . Such a free energy change should be smaller by the decrease in free energy accompanying the transfer of each subunit from solvent to the interior of a detergent micelle. Free energy of transfer of a hydrophobic membrane protein from water is predicted to be energetically very favorable, especially for those containing multiple trans-membrane helices. This probably accounts for our values being 1.5 - 3.0 times smaller than for proteins that become solvated by water upon dissociation.
Table 2.
Increases in solvent accessible area accompanying dissociation of a CcO subunits compared with experimental values for the change in free energy, ΔGexp
| Dissociated subunit | Increase in Solvent Accessible Surface Area (Å2) | ΔGexp (kJ/mol) | ΔGdissociation (kJ/mol per 100 Å2) |
|---|---|---|---|
| VIb | 3178 | 18.8 | 0.592 |
| VIa | 5712 | 18.0 | 0.315 |
| VIIa | 4148 | 24.7 | 0.595 |
| III | 9056 | 29.7 | 0.328 |
Lastly, a major advantage of the structural perturbant approach is that different chemical stabilizing factors, e.g., sulfate anion, trehalose, or altered pH, may preferentially stabilize one subunit association more than another. If this occurs, it would shift or modify the order of subunit dissociation from the complex. The simplicity, variability and relative specificity of this approach makes it more advantageous than methods using either selective proteolytic cleavage (9,10,11), or monoclonal antibodies (12) to remove subunits. Our approach is also quite different from genetic removal of a subunit (43,44). With chaotrope-induced subunit depletion, subunits are removed from a pre-formed and isolated complex; with the gene-depletion approach, CcO assembly is never completed. If the energetics of subunit assembly are in fact the reverse of CcO assembly as discussed earlier, then the two experimental approaches would be expected to directly complement each other. However, if subunit removal from a pre-formed complex is significantly different from CcO assembly, then the two approaches provide different information and the simultaneous use of both methods may be needed to completely define subunit function within a particular enzyme complex.
ACKNOWLEDGEMENT
We wish to thank Dr. Alex Taylor for his assistance in surface area calculations for CcO and its individual subunits. We also thank Drs. Andrej Musatov and LeAnn K. Robinson for their helpful comments and assistance in preparing this manuscript.
Abbreviations
- CcO
cytochrome c oxidase
- GdmCl
guanidinium chloride
- RP-HPLC
reversed-phase high performance liquid chromatography
Footnotes
This work was supported by research grants from National Institute of General Medical Sciences (R01 GM024795) and The Robert A. Welch Foundation (AQ1481).
Nomenclature for CcO subunits is according to Kadenbach et al., 1983 (1).
REFERENCES
- 1.Kadenbach B, Jarausch J, Hartmann R, Merle P. Separation of mammalian cytochrome c oxidase into 13 polypeptides by sodium dodecyl sulfate-gel electrophoresis procedure. Anal.Biochem. 1983;129:517–521. doi: 10.1016/0003-2697(83)90586-9. [DOI] [PubMed] [Google Scholar]
- 2.Tsukihara T, Aoyama H, Yamashita E, Tomizaki T, Yamaguchi H, Shinzawa-Itoh K, Nakashima R, Yaono R, Yoshikawa S. The whole structure of the 13-subunit oxidized cytochrome c oxidase at 2.8 Å. Science. 1996;272:1136–1144. doi: 10.1126/science.272.5265.1136. [DOI] [PubMed] [Google Scholar]
- 3.Hosler JP, Fetter J, Tecklenburg MM, Espe M, Lerma C, Ferguson-Miller S. Cytochrome aa3 of Rhodobacter sphaeroides as a model for mitochondrial cytochrome c oxidase. Purification, kinetics, proton pumping, and spectral analysis. J. Biol. Chem. 1992;267:24264–24272. [PubMed] [Google Scholar]
- 4.Abramson J, Svensson-Ek M, Byrne B, Iwata S. Structure of cytochrome c oxidase: a comparison of the bacterial and mitochondrial enzymes. Biochim. Biophys. Acta. 2001;1544:1–9. doi: 10.1016/s0167-4838(00)00241-7. [DOI] [PubMed] [Google Scholar]
- 5.Svensson-Ek M, Abramson J, Larsson G, Törnroth S, Brzezinski P, Iwata S. The X-ray crystal structures of wild-type and EQ(I-286) mutant cytochrome c oxidases from Rhodobacter sphaeroides. J. Mol. Biol. 2002;321:329–339. doi: 10.1016/s0022-2836(02)00619-8. [DOI] [PubMed] [Google Scholar]
- 6.Musatov A, Robinson NC. Cholate-Induced Dimerization of Detergent- or Phospholipid-Solubilized Bovine Cytochrome c oxidase. Biochemistry. 2002;41:4371–4376. doi: 10.1021/bi016080g. [DOI] [PubMed] [Google Scholar]
- 7.Sedlák E, Panda M, Dale MP, Weintraub ST, Robinson NC. Photolabeling of cardiolipin binding subunits within bovine heart cytochrome c oxidase. Biochemistry. 2006;45:746–754. doi: 10.1021/bi050870z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kadenbach B, Hüttemann M, Arnold S, Lee I, Bender E. Mitochondrial energy metabolism is regulated via nuclear-coded subunits of cytochrome c oxidase. Free Radic. Biol. Med. 2000;29:211–221. doi: 10.1016/s0891-5849(00)00305-1. [DOI] [PubMed] [Google Scholar]
- 9.Büge V, Kadenbach B. Effect of trypsin on the kinetic properties of reconstituted beef heart cytochrome c oxidase. J.Bioenerg.Biomembr. 1985;17:375–384. doi: 10.1007/BF00743110. [DOI] [PubMed] [Google Scholar]
- 10.Planques Y, Capitanio N, Capitanio G, De Nitto E, Villani G, Papa S. Role of supernumerary subunits in mitochondrial cytochrome c oxidase. FEBS Lett. 1989;258:285–288. doi: 10.1016/0014-5793(89)81675-8. [DOI] [PubMed] [Google Scholar]
- 11.Capitanio N, Peccarisi R, Capitanio G, Villani G, De Nitto E, Scacco S, Papa S. Role of nuclear-encoded subunits of mitochondrial cytochrome c oxidase in proton pumping revealed by limited enzymatic proteolysis. Biochemistry. 1994;33:12521–12526. doi: 10.1021/bi00207a020. [DOI] [PubMed] [Google Scholar]
- 12.Lincoln JA, Donat N, Palmer G, Prochaska LJ. Site-specific antibodies against hydrophilic domains of subunit III of bovine heart cytochrome c oxidase affect enzyme function. Arch.Biochem.Biophys. 2003;416:81–91. doi: 10.1016/s0003-9861(03)00202-9. [DOI] [PubMed] [Google Scholar]
- 13.Parr GR, Hammes GG. Subunit dissociation and unfolding of rabbit muscle phosphofructokinase by guanidine by hydrochloride. Biochemistry. 1975;14:1600–1605. doi: 10.1021/bi00679a009. [DOI] [PubMed] [Google Scholar]
- 14.Blackburn MN, Noltmann EA. Evidence for an intermediate in the denaturation and assembly of phosphoglucose isomerase. Arch.Biochem.Biophys. 1981;212:161–169. doi: 10.1016/0003-9861(81)90355-6. [DOI] [PubMed] [Google Scholar]
- 15.Jaenicke R, Vogel W, Rudolph R. Dimeric intermediates in the dissociation of lactic dehydrogenase. Eur.J.Biochem. 1981;114:525–531. doi: 10.1111/j.1432-1033.1981.tb05176.x. [DOI] [PubMed] [Google Scholar]
- 16.Abu-Soud HM, Loftus M, Stuehr DJ. Subunit dissociation and unfolding of macrophage NO synthase: relationship between enzyme structure, prosthetic group binding, and catalytic function. Biochemistry. 1995;34:11167–11175. doi: 10.1021/bi00035a023. [DOI] [PubMed] [Google Scholar]
- 17.Hill BC, Cook K, Robinson NC. Subunit Dissociation and Protein Unfolding in the Bovine Heart Cytochrome Oxidase Complex Induced by Guanidine Hydrochloride. Biochemistry. 1988;27:4741–4747. doi: 10.1021/bi00413a024. [DOI] [PubMed] [Google Scholar]
- 18.Sedlák E, Robinson NC. Phospholipase A2 Digestion of Cardiolipin Bound to Bovine Cytochrome c oxidase Alters both Activity and Quaternary Structure. Biochemistry. 1999;38:14966–14972. doi: 10.1021/bi9914053. [DOI] [PubMed] [Google Scholar]
- 19.Nozaki Y, Reynolds JA, Tanford C. Conformational states of a hydrophobic protein. The coat protein of fd bacteriophage. Biochemistry. 1978;17:1239–1246. doi: 10.1021/bi00600a017. [DOI] [PubMed] [Google Scholar]
- 20.Haltia T, Freire E. Forces and factors that contribute to the structural stability of membrane proteins. Biochim.Biophys.Acta. 1995;1228:1–27. doi: 10.1016/0005-2728(94)00161-w. [DOI] [PubMed] [Google Scholar]
- 21.Booth PJ, Templer RH, Meijberg W, Allen SJ, Curran AR, Lorch M. In vitro studies of membrane protein folding. Crit. Rev. Biochem. Mol. Biol. 2001;36:501–603. doi: 10.1080/20014091074246. [DOI] [PubMed] [Google Scholar]
- 22.Minetti CA, Remeta DP. Energetics of membrane protein folding and stability. Arch. Biochem. Biophys. 2006;453:32–53. doi: 10.1016/j.abb.2006.03.023. [DOI] [PubMed] [Google Scholar]
- 23.Fowler LR, Richardson SH, Hatefi Y. A Rapid Method for the Preparation of Highly Purified Cytochrome Oxidase. Biochim.Biophys.Acta. 1962;64:170–173. doi: 10.1016/0006-3002(62)90770-9. [DOI] [PubMed] [Google Scholar]
- 24.Mahapatro SN, Robinson NC. Effect of Changing the Detergent Bound to Bovine Cytochrome c oxidase upon Its Individual Electron Transfer Steps. Biochemistry. 1990;29:764–770. doi: 10.1021/bi00455a025. [DOI] [PubMed] [Google Scholar]
- 25.Robinson NC, Talbert L. Triton X-100 Induced Dissociation of Beef Heart Cytochrome c oxidase into Monomers. Biochemistry. 1986;25:2328–2335. doi: 10.1021/bi00357a005. [DOI] [PubMed] [Google Scholar]
- 26.Robinson NC, Gomez B, Musatov A, Ortega-Lopez J. Analysis of Detergent Solubilized Membrane Proteins in the Analytical Ultracentrifuge. ChemTracts: Biochem. Molec. Biol. 1998;11:960–968. [Google Scholar]
- 27.Musatov A, Ortega-Lopez J, Robinson NC. Detergent-Solubilized Bovine Cytochrome c oxidase: Dimerization Depends upon the Amphiphilic Environment. Biochemistry. 2000;39:12996–13004. doi: 10.1021/bi000884z. [DOI] [PubMed] [Google Scholar]
- 28.van Gelder BF. Optical Properties of Cytochromes from Beef Heart Mitochondria, Submitochondrial Vesicles, and Derived Preparations. Methods Enzymol. 1978;53:125–128. doi: 10.1016/s0076-6879(78)53020-6. [DOI] [PubMed] [Google Scholar]
- 29.Liu Y-Ch., Sowdal LH, Robinson NC. Separation and Quantitation of Cytochrome c oxidase Subunits by Mono Q Fast Liquid Chromatography and C18 Reverse Phase HPLC. Arch.Biochem.Biophys. 1995;324:135–142. doi: 10.1006/abbi.1995.9917. [DOI] [PubMed] [Google Scholar]
- 30.Robinson NC, Strey F, Talbert L. Investigation of the Essential Boundary Layer Phospholipids of Cytochrome c oxidase Using Triton X-100 Delipidation. Biochemistry. 1980;19:3656–3661. doi: 10.1021/bi00557a003. [DOI] [PubMed] [Google Scholar]
- 31.Pace CN. Determination and analysis of urea and guanidine hydrochloride denaturation curves. Methods Enzymol. 1986;131:266–280. doi: 10.1016/0076-6879(86)31045-0. [DOI] [PubMed] [Google Scholar]
- 32.Copeland RA. Long-distance cofactor interactions in terminal oxidases studied by second-derivative absorption spectroscopy. J.Bioenerg.Biomembr. 1993;25:93–102. doi: 10.1007/BF00762851. [DOI] [PubMed] [Google Scholar]
- 33.Akhtar MS, Ahmad A, Bhakuni V. Guanidinium chloride- and urea-induced unfolding of the dimeric enzyme glucose oxidase. Biochemistry. 2002;41:3819–3827. doi: 10.1021/bi0116700. [DOI] [PubMed] [Google Scholar]
- 34.Stanicová J, Sedlák E, Musatov A, Robinson NC. Differential stability of dimeric and monomeric cytochrome c oxidase exposed to elevated hydrostatic pressure. Biochemistry. 2007;46:7146–7152. doi: 10.1021/bi700548a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Heinrichs M, Schönert H. Identification of different quaternary structures of beef heart cytochrome-c oxidase by two-dimensional polyacrylamide gel electrophoresis. FEBS Lett. 1987;223:255–261. doi: 10.1016/0014-5793(87)80300-9. [DOI] [PubMed] [Google Scholar]
- 36.Nijtmans LGJ, Taanman J-W, Muijsers AO, Speijer D, Van den Bogert C. Assembly of cytochrome-c oxidase in cultured human cells. Eur.J.Biochem. 1998;254:389–394. doi: 10.1046/j.1432-1327.1998.2540389.x. [DOI] [PubMed] [Google Scholar]
- 37.Taanman J-W, Williams SL. Assembly of cytochrome c oxidase: what can we learn from patients with cytochrome c oxidase deficiency? Biochem. Soc. Transac. 2001;29:446–451. doi: 10.1042/bst0290446. [DOI] [PubMed] [Google Scholar]
- 38.Moore JMR, Patapoff TW, Cromwell MEM. Kinetics and thermodynamics of dimer formation and dissociation for a recombinant humanized monoclonal antibody to vascular endothelial growth factor. Biochemistry. 1999;38:13960–13967. doi: 10.1021/bi9905516. [DOI] [PubMed] [Google Scholar]
- 39.Apiyo D, Jones K, Guidry J, Wittung-Stafshede P. Equilibrium unfolding of dimeric desulfoferrodoxin involves a monomeric intermediate: iron cofactors dissociate after polypeptide unfolding. Biochemistry. 2001;40:4940–4948. doi: 10.1021/bi002653y. [DOI] [PubMed] [Google Scholar]
- 40.Wójciak P, Mazurkiewicz A, Bakalova A, Kuciel R. Equilibrium unfolding of dimeric human prostatic acid phosphatase involves an inactive monomeric intermediate. Int. J. Biol. Macromolec. 2003;32:43–54. doi: 10.1016/s0141-8130(03)00024-2. [DOI] [PubMed] [Google Scholar]
- 41.Janin J, Miller S, Chothia C. Surface, subunit interfaces and interior of oligomeric proteins. J. Mol. Biol. 1988;204:155–164. doi: 10.1016/0022-2836(88)90606-7. [DOI] [PubMed] [Google Scholar]
- 42.Faham S, Yang D, Bare E, Yohannan S, Whitelegge JP, Bowie JU. Side-chain contributions to membrane protein structure and stability. J. Mol. Biol. 2004;335:297–305. doi: 10.1016/j.jmb.2003.10.041. [DOI] [PubMed] [Google Scholar]
- 43.Aggeler R, Capaldi RA. Yeast cytochrome c oxidase subunit VII is essential for assembly of an active enzyme. Cloning, sequencing, and characterization of the nuclear-encoded gene. J. Biol. Chem. 1990;265:16389–93. [PubMed] [Google Scholar]
- 44.Poyton RO, McEwen JE. Crosstalk between nuclear and mitochondrial genomes. Annu. Rev. Biochem. 1996;65:563–607. doi: 10.1146/annurev.bi.65.070196.003023. [DOI] [PubMed] [Google Scholar]