

Abstract
Previous clinical and animal studies suggest that selective activators of M1 and/or M4 muscarinic acetylcholine receptors (mAChRs) have potential as novel therapeutic agents for treatment of schizophrenia and Alzheimer’s disease. However, highly selective centrally penetrant activators of either M1 or M4 have not been available, making it impossible to determine the in vivo effects of selective activation of these receptors. We previously identified VU10010 [3-amino-N-(4-chlorobenzyl)-4, 6-dimethylthieno[2,3-b]pyridine-2-carboxamide] as a potent and selective allosteric potentiator of M4 mAChRs. However, unfavorable physiochemical properties prevented use of this compound for in vivo studies. We now report that chemical optimization of VU10010 has afforded two centrally penetrant analogs, VU0152099 [3-amino-N-(benzo[d][1,3]dioxol-5-ylmethyl)-4,6-dimethylthieno[2,3-b]pyridine carboxamide] and VU0152100 [3-amino-N-(4-methoxybenzyl)-4,6-dimethylthieno[2,3-b]pyridine carboxamide], that are potent and selective positive allosteric modulators of M4. VU0152099 and VU0152100 had no agonist activity but potentiated responses of M4 to acetylcholine. Both compounds were devoid of activity at other mAChR subtypes or at a panel of other GPCRs. The improved physiochemical properties of VU0152099 and VU0152100 allowed in vivo dosing and evaluation of behavioral effects in rats. Interestingly, these selective allosteric potentiators of M4 reverse amphetamine-induced hyperlocomotion in rats, a model that is sensitive to known antipsychotic agents and to nonselective mAChR agonists. This is consistent with the hypothesis that M4 plays an important role in regulating midbrain dopaminergic activity and raises the possibility that positive allosteric modulation of M4 may mimic some of the antipsychotic-like effects of less selective mAChR agonists.
To date, five muscarinic acetylcholine receptor (mAChR) subtypes have been identified (M1–M5) and play important roles in mediating the actions of ACh in the peripheral and central nervous systems (Wess, 1996). Of these, M1 and M4 are the most heavily expressed in the CNS and represent attractive therapeutic targets for cognition, Alzheimer’s disease, and schizophrenia (Bymaster et al., 2002; Messer, 2002; Raedler et al., 2007). In contrast, the adverse effects of cholinergic agents are thought to be primarily due to activation of peripheral M2 and M3 mAChRs (Bymaster et al., 2003a,b). Due to the high sequence homology and conservation of the orthosteric ACh binding site among the mAChR subtypes, development of chemical agents that are selective for a single subtype has been largely unsuccessful, and in the absence of highly selective activators of M4, it has been impossible to test the role of selective M4 activation. Clinical trials with xanomeline (1) (Fig. 1), a M1/M4-preferring orthosteric agonist, demonstrated efficacy as both a cognition-enhancing agent and an antipsychotic agent (Bodick et al., 1997; Shekhar et al., 2001, 2008). In follow-up studies in rats, xanomeline displayed an antipsychotic-like profile comparable to clozapine (Stanhope et al., 2001). However, a long standing question concerned whether or not the antipsychotic efficacy or antipsychotic-like activity in animal models is mediated by activation of M1, M4, or a combination of both receptors. Data from mAChR knockout mice led to the suggestion that a selective M1 agonist would be beneficial for cognition, whereas an M4 agonist would provide antipsychotic activity for the treatment of schizophrenia (Felder et al., 2001; Bymaster et al., 2003a,b). This proposal is further supported by recent studies demonstrating that M4 receptors modulate the dynamics of cholinergic and dopaminergic neurotransmission and that loss of M4 function results in a state of dopamine hyperfunction (Tzavara et al., 2004). These data, coupled with findings that schizophrenic patients have altered hippocampal M4 but not M1 receptor expression (Scarr et al., 2007), suggest that selective activators of M4 may provide a novel treatment strategy for schizophrenia patients. However, multiple studies suggest that M1 may also play an important role in the antipsychotic effects of mAChR agonists and that the relative contributions of M1 and M4 to the antipsychotic efficacy of xanomeline or antipsychotic-like effects of this compound in animal models are not known. Unfortunately, the lack of highly selective, systemically active activators of M1 and M4 has made it difficult to fully evaluate the effects of activation of these mAChR subtypes in animal models.
Fig. 1.

Chemical structures of xanomeline [3-[3-hexyloxy-1,2,5-thiadiazo-4-yl]-1,2,5,6-tetrahydro-1-methylpyridine] (1) and VU10010 (2).
Recently, we reported discovery of a number of positive allosteric modulators for class C GPCRs that bind to allosteric sites, provide high levels of subtype selectivity, and display behavioral effects in vivo comparable to direct acting agonists (O’Brien et al., 2003, 2004; Lindsley et al., 2004; Kinney et al., 2005; Galici et al., 2006; Hemstapat et al., 2006; Marino and Conn, 2006; Zhao et al., 2007). In addition, we identified a highly selective positive allosteric modulator of M4 termed VU10010 (2) (Fig. 1) (Shirey et al., 2008). This compound induces a 47-fold potentiation of the M4 ACh concentration response curve (CRC), possesses an EC50 value in the 400 nM range, and causes no activation of the other mAChR subtypes. Additional in vitro pharmacological characterization studies suggested that VU10010 binds to an allosteric site on the M4 receptor to increase affinity for ACh and coupling to G proteins (Shirey et al., 2008). Subsequent studies with VU10010 revealed that selective potentiation of M4 increased carbachol-induced depression at excitatory but not inhibitory synapses and that the effect on excitatory currents was not mimicked by an inactive analog of VU10010 or in M4 knockout mice (Shirey et al., 2008).
Despite this notable advance, VU10010 suffered from poor physiochemical properties (log P ~4.5), and in vivo studies proved unfeasible because we were unable to formulate VU10010 into a homogeneous solution in any acceptable vehicle, regardless of salt form or particle size. Several suspensions were prepared and dosed intraperitoneally; however, VU10010 was not found to be centrally active. To evaluate the role of selective M4 activation in vivo, VU10010 would require further chemical optimization. Here we report the development and characterization of two novel analogs of VU10010 that are CNS penetrant following systemic administration.
Materials and Methods
Materials
All tissue culture reagents, as well as Fluo-4 acetoxymethyl ester and benzothiazole coumarin-acetoxymethyl ester, were obtained from Invitrogen (Carlsbad, CA). ACh, probenecid, Pluronic F-127, and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (St. Louis, MO). Costar 96-well cell culture plates and V-bottom compound plates were purchased from Corning Inc. (Corning, NY). Poly-d-lysine-coated 96-well assay plates were purchased from BD Biosciences (Franklin Lakes, NJ). l-[N-methyl-3H]Scopolamine methyl chloride was purchased from GE Healthcare (Chalfont St. Giles, UK).
General Medicinal Chemistry Methods
All NMR spectra were recorded on a 400-MHz Bruker NMR. 1H chemical shifts were reported in δ values in parts per million downfield from tetramethylsilane as the internal standard in DMSO. Data are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, br = broad, and m = multiplet), integration, and coupling constant (in Hertz). 13C chemical shifts are reported in δ values in parts per million with the DMSO carbon peak set to 39.5 ppm. Low resolution mass spectra were obtained on an Agilent 1200 LC/MS with electrospray ionization (Agilent Technologies, Santa Clara, CA). High-resolution mass spectra were recorded on a Quadrupole Time of Flight (Q-ToF)-API-US plus Acquity system (Waters, Milford, MA). Analytical thin-layer chromatography was performed on 250-µm Silica Gel 60 F254 plates. Analytical HPLC was performed on an Agilent 1200 analytical LC/MS with UV detection at 214 and 254 nm along with ELSD detection. Preparative purification was performed on a custom Agilent 1200 preparative LC/MS with collection triggered by mass detection. Solvents for extraction, washing, and chromatography were of HPLC grade. All reagents were purchased from Aldrich Chemical Co. (Milwaukee, WI), Maybridge Chemicals (Trevillet, UK), ChemBridge Corporation (San Diego, CA), and SPECS Technologies Corporation (Sarasota, FL) and were used without purification. All polymer-supported reagents were purchased from Biotage AB (Uppsala, Sweden).
General Procedure for Library Synthesis of Analogs 7
Each of the 31 glass vials containing 3 ml of CH2Cl2 was loaded with N,N-diisopropylethylamine (0.3 ml, 1.70 mmol), AM-807/25050004 (50 mg, 0.225 mmol; SPECS Technologies Corporation), 1-hydroxybenzotriazole hydrate (30.4 mg, 0.225 mmol, 1.0 equivalents), polystyrene-bound N-cyclohexylcarbodiimide (317 mg, 0.450 mmol, 1.42 mmol/g, 2.0 equivalents), and one of 31 amines (0.225 mmol, 1.0 equivalents). The reactions were stirred for 48 h at room temperature. Macroporous triethylammonium methylpolystyrene carbonate (145 mg, 0.225 mmol, 3.11 mmol/g, 2.0 equivalents) was added, and the reactions were stirred for an additional 3 h at room temperature. Then, the reactions were filtered and concentrated on a heat-air block to afford 84 to 99% pure products. Those <99% pure were purified by mass-directed HPLC.
VU0152099
The following components were added to a stirred solution of 3-amino-4,6-dimethylthioenol[2,3-b]-pyridine-2-carboxylic acid (3.0 g, 13.51 mmol; ChemBridge Corporation) in CH2Cl2 (90 ml) at 25°C under room atmosphere: N,N-diisopropylethylamine (10 ml, 56.66 mmol); 1-hydroxybenzotriazole hydrate (1.83 g, 13.51 mmol, 1.0 equivalents); 4-methoxybenzylamine (2.04 g, 14.86 mmol, 1.1 equivalents); and N-(3-dimethylaminopropyl)-N′-ethyl-carbodiimide hydrochloride (5.18 g, 27.02 mmol, 2.0 equivalents). After 48 h, macroporous triethylammonium methylpolystyrene carbonate (4.4 g, 13.51 mmol, 3.077 mmol/g, 1.0 equivalents) was added to the solution, which was then stirred for 3 h at 25°C under room atmosphere. The solution was vacuum-filtered next, and the filtrate was separated with citric acid (1.0 M in water) and CH2Cl2. The organics were dried over MgSO4 and concentrated in vacuo to produce a dark yellow solid. The solid was purified by column chromatography (silica gel, fixed 1:2 EtOAc/hexanes) to afford 2.5 g (7.33 mmol, 54%) of the title compound as a bright yellow solid. Analytical LC/MS (J-Sphere80-S4, 3.0 × 50 mm, 4.0-min gradient, 5%[CH3CN]: 95%[0.1% trifluoroacetic acid/H2O] to 100%[CH3CN]): 2.773 min, >99% (214 nm and ELSD), M + 1 peak m/e342.12; 1H NMR (400 MHz, DMSO-d6) δ 7.27 (d, J = 8.8 Hz, 2H), 6.89 (s, 1H), 6.86 (d, J = 8.8 Hz, 2H), 6.34 (br s, 2H), 5.80 (s, 1H), 4.53 (d, J = 6.0 Hz, 2H), 3.79 (s, 3H), 2.73 (s, 3H), 2.57 (s, 3H); 13C NMR (100 MHz, DMSO-d6) δ 165.9, 159.3, 159.2, 147.7, 143.9, 130.7, 129.3, 123.7, 122.4, 114.4, 98.5, 55.5, 43.3, 24.5, 20.4; high-resolution mass spectroscopy (Q-ToF): m/z calc for C18H19N3O2S [M + H]: 342.1198; found, 342.1276.
VU0152100
The following components were added to a stirred solution of 3-amino-4,6-dimethylthioenol[2,3-b]-pyridine-2-carboxylic acid (2.50 g, 11.26 mmol; ChemBridge Corporation) in CH2Cl2 (90 ml) at 25°C under room atmosphere: N,N-diisopropylethylamine (10 ml, 56.66 mmol); 1-hydroxybenzotriazole hydrate (1.52 g, 11.26 mmol, 1.0 equivalents); piperonylamine (1.87 g, 12.38 mmol, 1.1 equivalents); and N-(3-dimethylaminopropyl)-N′-ethyl-carbodiimide hydrochloride (4.32 g, 22.52 mmol, 2 equivalents). After 48 h, macroporous triethylammonium methylpolystyrene carbonate (3.66 g, 11.26 mmol, 3.077 mmol/g, 1.0 equivalents) was added to the solution, which was then stirred for 3 h at 25°C under room atmosphere. The solution was vacuum-filtered next, and the filtrate was separated with citric acid (1.0 M in water) and CH2Cl2. The organics were dried over MgSO4 and concentrated in vacuo to produce a dark yellow solid. The solid was purified by column chromatography (silica gel, fixed 1:2 EtOAc/hexanes) to afford 2.0 g (5.63 mmol, 50%) of the title compound as a yellow solid. Analytical LC/MS (J-Sphere80-S4, 3.0 × 50 mm, 4.0 min gradient, 5%[CH3CN]: 95%[0.1% trifluoroacetic acid/H2O] to 100%[CH3CN]): 2.740 min, >99% (214 nm and ELSD), M + 1 peak m/e 356.10; 1H NMR (400 MHz, DMSO-d6) δ 8.38 (s, 1H), 7.18 (s, 1H), 6.88 (s, 1H), 6.84 (d, J = 8.0 Hz, 1H), 6.78 (d, J = 8.0 Hz, 1H), 5.98 (br s, 2H), 5.97 (s, 2H), 4.30 (d, J = 5.2 Hz, 2H), 2.77 (s, 3H), 2.57 (s, 3H); 13CNMR (100 MHz, DMSO-d6) δ 179.9, 164.8, 161.7, 158.0, 153.4, 147.4, 133.8, 122.4, 121.9, 120.5, 108.0, 107.9, 100.8, 92.8, 42.1, 22.4, 20.0; high-resolution mass spectroscopy (Q-ToF): m/z calc for C18H17N3O3S [M + H]: 356.0991; found, 356.1069.
Cell Culture
Chinese hamster ovary (CHO K1) cells stably expressing rat (r)M1 were purchased from the American Type Culture Collection (ATCC, Manassas, VA) and cultured according to their recommendations. CHO cells stably expressing human (h) M2, hM3, and hM5 were generously provided by A. Levey (Emory University, Atlanta, GA); rM4 cDNA provided by T. I. Bonner (National Institutes of Health, Bethesda, MD) was used to stably transfect CHO-K1 cells purchased from the ATCC using Lipofectamine 2000. To make stable hM2 and rM4 cell lines for use in calcium mobilization assays, cell lines were cotransfected with a chimeric G protein (Gqi5) using Lipofectamine 2000. rM2, hM3, and hM5 cells were grown in Ham’s F-12 medium containing 10% heat-inactivated fetal bovine serum, 2 mM GlutaMax I, 20 mM HEPES, and 50 µg/ml G418 sulfate. hM2-Gqi5 cells were grown in the same medium supplemented with 500 µg/ml hygromycin B. Stable rM4 cells were grown in Dulbecco’s modified Eagle’s medium containing 10% heat-inactivated fetal bovine serum, 2 mM GlutaMax I, 1 mM sodium pyruvate, 0.1 mM nonessential amino acids, 20 mM HEPES, and 400 µg/ml G418 sulfate; rM4-Gqi5 cells were grown in the same medium supplemented with 500 µg/ml hygromycin B.
CHO cells stably expressing rM1, hM3, or hM5 were plated at a seeding density of 50,000 cells/100 µl/well. CHO cells stably coexpressing hM2/Gqi5 and rat M4/Gqi5 were plated at a seeding density of 60,000 cells/100 µl/well. HEK293/G protein-regulated inwardly rectifying K+ (GIRK) cells stably expressing the human M4 receptor were grown as described in Niswender et al. (2008) and plated at 60,000 cells/100 µl/well. For calcium mobilization or GIRK-mediated thallium flux assays, cells were incubated in antibiotic-free medium overnight at 37°C/5% CO2 and assayed the next day.
Calcium Mobilization Assay
Cells were loaded with calcium indicator dye [2 µM Fluo-4 acetoxymethyl ester (50 µl/well) prepared as a stock in DMSO and mixed in a 1:1 ratio with 10% Pluronic acid F-127 in assay buffer (1× Hanks’ balanced salt solution supplemented with 20 mM HEPES and 2.5 mM probenecid, pH 7.4)] for 45 min at 37°C. Dye was removed and replaced with the appropriate volume of assay buffer. All compounds were serially diluted in assay buffer for a final 2× stock in 0.6% DMSO. This stock was then added to the assay plate for a final DMSO concentration of 0.3%. Acetylcholine (EC20 concentration or full dose-response curve) was prepared at a 10× stock solution in assay buffer before addition to assay plates. Calcium mobilization was measured at 25°C using a FLEXstation II (Molecular Devices, Sunnyvale, CA).
Cells were preincubated with test compound (or vehicle) for 1.5 min before the addition of the agonist, acetylcholine. Cells were then stimulated for 50 s with a submaximal concentration (EC20) or a full dose-response curve of acetylcholine. The signal amplitude was first normalized to baseline and then as a percentage of the maximal response to acetylcholine.
GIRK-Mediated Thallium Flux Assay
Cells were incubated with 80 µl/well of 1.7 µM benzothiazole coumarin-acetoxymethyl ester indicator dye [prepared as a stock in DMSO and mixed in a 1:1 ratio with 10% Pluronic acid F-127 in assay buffer (1× Hanks’ balanced salt solution supplemented with 20 mM HEPES)] for 1 h at room temperature in the dark. The dye was then replaced with 40 µl of assay buffer. Test compounds were prepared as described above. Acetylcholine (EC20 concentration or full dose-response curve) was prepared as a 5× stock solution in thallium buffer (pH 7.3, 12 mM thallium sulfate, 1 mM MgSO4, 1.8 mM CaSO4, 5 mM glucose, and 10 mM HEPES) to which 125 mM NaHCO3 was added immediately before use. Thallium flux was measured at 25°C using the FLEXstation II, as described above for calcium mobilization assays. The slope of the fluorescence increase was obtained over a 10-s window beginning at 5 s after agonist/thallium addition. The signal amplitude was first normalized to baseline and then as a percentage of the maximal response to acetylcholine.
Radioligand Binding Studies
All binding reactions were carried out essentially as described previously (Shirey et al., 2008) using 25 µg of membrane protein prepared from rM4 expressing CHO cells and 0.1 nM [3H]NMS (GE Healthcare) in a final volume of 1 ml. Nonspecific binding was determined in the presence of 1 µM atropine.
Ancillary Pharmacology Assays
Before conducting in vivo experiments, VU0152099 and VU0152100 were submitted to MDS Pharma Services (King of Prussia, PA) (www. mdsps.com) and evaluated in the LeadProfiling Screen, a radioligand binding assay panel employing 68 GPCRs, ion channels, transporters, and enzymes, to ensure a clean ancillary pharmacology profile. VU0152099 also was submitted to GPCR Profiler Service (Millipore Corporation, Billerica, MA) where it was evaluated for agonist, antagonist, and allosteric potentiator activity against a panel of 16 GPCRs in a functional screening paradigm.
Jet Milling
Both VU0152099 and VU0152100 were Jet-milled, to afford uniform nanoparticles, before vehicle formulation and in vivo studies employing a model 00 Jet-O-Mizer with a high-yield collection module from Fluid Energy Processing and Equipment Company (Hatfield, PA) (see: www.fluidenergype.com).
In Vivo Pharmacokinetic Profiling and Behavioral Studies
All experiments were conducted in accordance with the National Institutes of Health regulations of animal care covered in Principles of Laboratory Animal Care (National Institutes of Health publication 85-23, revised 1985) and were approved by the Institutional Animal Care and Use Committee.
Amphetamine-Induced Hyperlocomotion
Animals
All behavioral studies were conducted using male Sprague-Dawley rats (Harlan Sprague-Dawley, Inc., Indianapolis, IN) weighing 270 to 300 g. Subjects were housed in pairs in a large colony room under a 12-h light/12-h dark cycle (lights on at 6:00 AM) with food and water provided ad libitum. Test sessions were performed between 6:00 AM and 6:00 PM. Dose groups consisted of 8 to 16 rats per dose group. All doses of VU0152099 and VU0152100 refer to the salt form and were injected in a 1.0 ml/kg volume. Each compound was dissolved in 10% Tween 80 and double deionized water with the pH adjusted to approximately 7.0 using 1 N NaOH.
Apparatus
Amphetamine-induced hyperlocomotor activity studies were conducted using a SmartFrame Open Field System (Kinder Scientific., San Diego, CA) equipped with 32 horizontal (x- and y-axes) infrared photobeams located 1 cm above the floor of the chamber. Changes in ambulation or locomotor activity were measured as the number of total photobeam breaks, expressed in 5-min intervals, and were recorded with a Pentium I computer equipped with the Motor Monitor System software (Kinder Scientific).
Procedure
Rats were placed in the open-field chambers for a 30-min habituation interval (data not shown), followed by a pretreatment with vehicle or a 56.6 mg/kg i.p. dose of either VU0152099 or VU0152100 for an additional 30 min. Next, all rats received an injection of 1 mg/kg s.c. amphetamine, and locomotor activity was measured for an additional 60 min. Data were analyzed by a one-way ANOVA with comparison with the vehicle + amphetamine control group using Dunnett’s test. Calculations were performed using JMP version 5.1.2 (SAS Institute Inc., Cary, NC) statistical software.
Rotorod Test
The effects of VU0152100 on motor performance were evaluated using a rotorod (Columbus Instruments, Columbus, OH). All rats were given an initial training trial of 120 s, followed by two additional training trials of 85 s, approximately 10 min apart, using a rotorod (7.5 cm in diameter) rotating at a constant speed of 20 revolutions/min. After initial training trials, a baseline trial of 85 s was conducted, and any rats that did not reach the 85-s criteria were excluded from the study. Rats were then pretreated for 30 min i.p. with vehicle or dose of VU0152100, specifically 30, 56.6, or 100 mg/kg, and then the time each animal remained on the rotorod was recorded; animals not falling off of the rotorod were given a maximal score of 85 s. Data were analyzed by a one-way ANOVA, with comparison to the vehicle control group using Dunnett’s test. Calculations were performed using JMP version 5.1.2 (SAS Institute Inc.) statistical software.
Brain and Plasma Exposure Pharmacokinetic Profiling
Male Sprague-Dawley rats (Harlan Sprague-Dawley, Inc., Indianapolis, IN) weighing 225 to 250 g were fasted overnight before dosing. Compounds were dissolved at a concentration of 56.6 mg/ml in 10% Tween 80 and double deionized water, with the pH adjusted to approximately 7.0 using 1 N NaOH, and sonicated until a uniform homogenous solution was obtained. The dose was administered intraperitoneally at 56.6 mg/kg per compound. Three animals were used for each time point. The rat blood and brain were collected at 0.5, 1, 2, and 4 h. Animals were euthanized and decapitated, and the brains were removed and frozen on dry ice. Trunk blood was collected in EDTA Vacutainer tubes, and plasma was separated by centrifugation and stored at −80°C until analysis.
For the brain sample preparation, frozen whole-rat brains were weighed (1.5–1.8 g) and placed in 3 ml of ice-cold solution of acetonitrile and methanol (1:1, volume) with a synthetic internal standard (50 ng/ml) and homogenized using a Sonic Dismembrator model 100 (Thermo Fisher Scientific, Waltham, MA) at maximal speed for 2 min. A 1-ml aliquot of each homogenate was placed next into 1.5-ml centrifuge tubes and centrifuged at 16,000 rpm for 5 min. Finally, 100 µl of supernatant was injected into LC-MS-MS.
Plasma samples (100 µl) were combined with 200 µl of ice-cold solution of the internal standard (100 ng/ml) in acetonitrile with 0.1% formic acid. After vortexing for 1 min, the mixture was centrifuged at 16,000 rpm for 5 min in a bench-top Spectrafuge 16M Microcentrifuge (Labnet, Woodbridge, NJ). The supernatant (100 µl) was injected again into LC-MS-MS.
For the LC-MS-MS analysis, the LC separation was carried out on a Luna ODS column (5 µm, 2.1 mm × 5 cm; Phenomenex, Torrance, CA) at a flow rate of 0.3 ml/min. The gradient started with 80% solvent A (0.1% formic acid in water) and 20% solvent B (0.1% formic acid in CH3CN), held for 1 min, increased to 100% B in 4 min, and held for 1 min. Mass spectrometry was carried out using a ThermoFinnigan TSQ Quantum Ultra (Thermo Fisher Scientific, Waltham, MA) mass spectrometer in positive ion mode. The software Xcalibur version 2.0 was used to control the instrument and collect data. The electrospray ionization source was fitted with a stainless steel capillary (100 µm i.d.). Nitrogen was used as both the sheath gas and the auxiliary gas. The ion transfer tube temperature was 300°C. The spray voltage, tube lens voltage, and pressure of sheath gas and auxiliary gas were optimized to achieve maximal response using the test compounds mixing with the mobile phase A (50%) and B (50%) at a flow rat of 0.3 ml/min. Collision-induced dissociation was performed on the test compounds and internal standards under 1.0 mTorr of argon. Selected reaction monitoring was carried out using the transitions from m/z 356 to 205 at 30 eV for VU0152099, m/z 342 to 205 at 27 eV for VU0152100, and m/z 310 to 223 at 25 eV for our internal standard.
The calibration curves were constructed by spiking known amounts of test compounds in blank brain homogenates and plasma. The samples went through the same extraction steps as described above. A linear response was achieved from 50 ng/ml to 100 µg/ml in both matrices. Compound exposure following administration was determined by calculating AUC0-∞ using the trapezoidal method.
Results
Chemical Lead Optimization
For the chemical optimization of VU10010, we undertook a diversity-oriented synthesis approach to explode structure-activity relationships (SAR) with a variety of hypothesis-driven structural changes to the lead compound. The rationale for this approach for the optimization of VU10010 is that SAR for allosteric ligands is often “flat” or “shallow”, with subtle structural modifications leading to a complete loss of activity, and often only one portion of an allosteric ligand is amenable to change. Therefore, a multidimensional diversity-oriented synthesis library approach provides the best opportunity to quickly identify productive SAR as opposed to a lead optimization strategy based on classical, single compound synthesis (Lindsley et al., 2004; Zhao et al., 2007). One explanation for the lack of central activity observed with VU10010 could be the result of the poor physiochemical properties alone or in combination with P-glycoprotein (P-gp) efflux. P-gp is an efflux transporter with broad substrate specificity present on the luminal membrane of epithelial cells comprising the blood-brain barrier, which is known to impair the brain penetrability of a number of drugs. The β-aminoamide motif 3 present in VU10010 represents a potential P-gp liability, which could be removed by cyclization to analogs such as 4 (Fig. 2A). Alternatively, P-gp susceptibility could also be diminished by electronically attenuating the basicity of the amine moieties by the incorporation of distal fluorine atoms. Utilizing solution-phase parallel synthesis (Fig. 2B), we synthesized small 12 to 24-member focused libraries around each of the 10 scaffolds, 4 and 7 through 15 (Fig. 2C), which were then purified by mass-directed preparative HPLC to analytical purity (>98%). This collection of VU10010 analogs incorporated CF3 moieties (scaffolds 9 and 10) to electronically attenuate potential P-gp susceptibility, deletion of the β-amino moiety (scaffold 14), or replacement of the β-amino moiety with an isosteric methyl group (scaffold 13). Other scaffolds explored the deletion of substituents on the pyridine nucleus (scaffold 11), incorporation of an additional nitrogen atom to afford a pyrimidine nucleus (scaffold 12), or removal of the pyridine nitrogen atom in VU10010 (scaffold 15). Finally, scaffold 7 focused on maintaining the core structure of VU10010 but explored alternative amides selected to improve physiochemical properties and lower the log P value.
Fig. 2.

Chemical optimization of VU10010 using a diversity-oriented approach to achieve soluble, centrally penetrant M4 positive allosteric modulators. A, β-aminoamide as a potential P-gp liability in series 3 and cyclization strategy to diminish this liability in series 4. B, solution-phase parallel synthesis of libraries of VU10010 analogs. Commercial heterocyclic carboxylic acids 5 (X, Y = C or N) were coupled to 12 different amines (HNR1R2) to afford focused libraries of VU10010 analogs 7 to 15 in yields ranging from 15 to 99%. C, generic structures of analogs of VU10010 evaluated in the chemical lead optimization program in an effort to develop soluble, brain penetrant M4 positive allosteric modulators.
Screening Paradigm for Analog Libraries
As observed with positive allosteric modulators of class C GPCRs, SAR around VU10010 was relatively flat, possibly due to a shallow binding pocket (Lindsley et al., 2004; Zhao et al., 2007). An EC20 triage screen, employing a functional fluorescence-based Ca2+ assay in CHO K1 cells stably coexpressing the rat M4 mAChR and the chimeric G protein, Gqi5, quickly eliminated all VU10010 analogs with the exception of those in library 7 (built around scaffold 7) (Fig. 3). Within library 7, all aliphatic and nonbenzyl amides were inactive, and only benzyl and heteroaryl methyl congeners of VU10010, compounds 7a through 7p, retained M4 PAM activity (Table 1). Analogs were synthesized as described previously (Shirey et al., 2008). To identify compounds that potentiated agonist activation of M4, we determined the response to an EC20 concentration of ACh in the absence and presence of test compound. The potency of each compound was determined by preincubating cells with vehicle or increasing concentrations of test compound followed by the addition of an EC20 concentration of ACh to yield CRCs. Subtle substitution changes on the arene ring lost activity 5 to 10-fold in terms of M4 EC50 and/or -fold shift of the ACh CRC (Table 1). For instance, compound 7d, in which the 4-Cl moiety of VU100010 is moved to the 3-position, results in a loss in potency of over 9-fold (EC50 = 3.7 µM). Likewise, the unsubstituted phenyl congener 7a retains M4 PAM activity (EC50 = 630 nM), but the -fold shift diminishes to 8.6-fold versus the 47-fold shift observed for VU10010 (Shirey et al., 2008). In general, functionalized benzyl amides, as well as pyridyl methyl congeners (compounds 7f and 7g), were well tolerated, providing selective M4 PAMs with EC50 values ranging from 380 nM to 3.7 µM and with -fold shifts of the ACh dose-response curve from 8.6 to 70-fold.
Fig. 3.

Screening paradigm for analog libraries 2 to 12 allowing for the rapid triage of inactive analogs. A representative library of 61 analogs (scaffold 7) were tested at a single concentration (10 µM) for their ability to potentiate an EC20 concentration of ACh in CHO K1 cells stably coexpressing the rat M4 mAChR and the chimeric G protein, Gqi5. Calcium mobilization was measured using a FLEXstation II, as described under Materials and Methods. Of those tested, 16 compounds (denoted by an asterisk) were selected for further evaluation. The response to an EC20 concentration of ACh alone is shown in the bar on the far left, and this level of activity is indicated by the solid line spanning the panel. Thus, test compounds increasing the % Max ACh response above this level are considered potentiators of the M4 mAChR. VU10010 was included as a positive control. Bars represent the mean ± S.E.M. of three or more determinations, each performed in duplicate.
TABLE 1. Structures, activities, and ACh CRC -fold shifts of M4 PAM analogs 7.
Subtle substitution changes on the arene ring lost 5 to 10-fold in terms of M4 EC50 and/or -fold shift of the ACh CRC. For instance, compound 7d, in which the 4-Cl moiety of VU10010 is moved to the 3-position, results in a loss in potency of over 9-fold (EC50 = 3.7 µM). Likewise, the unsubstituted phenyl congener 7a retains M4 PAM activity (EC50 = 630 nM); however, the -fold shift diminishes to 8.6-fold vs. the 47-fold shift observed for VU10010 (Shirey et al., 2008). Compounds 7o (VU0152099) and 7p (VU0152100) retained M4 PAM activity comparable to VU10010 (EC50 values of 403 ± 117 and 380 ± 93 nM, respectively).
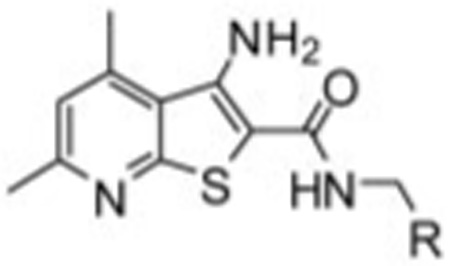 | |||
|---|---|---|---|
| Compound | R | Rat M4 EC50a | Rat M4 Ach -Fold Shift |
| µM | |||
| 7a | 0.63 | 8.6 | |
| 7b | 0.83 | 11.8 | |
| 7c | 1.83 | N.D. | |
| 7d | 3.70 | N.D. | |
| 7e | 2.63 | N.D. | |
| 7f | 2.04 | N.D. | |
| 7g | 2.88 | N.D. | |
| 7h | 1.44 | N.D. | |
| 7i | 1.80 | N.D. | |
| 7j | 2.96 | N.D. | |
| 7k | 3.04 | N.D. | |
| 7l | 0.88 | N.D. | |
| 7m | 1.12 | N.D. | |
| 7n | 0.72 | 13.7 | |
| 7o | 0.40 | 29.7 | |
| 7p | 0.38 | 70.1 | |
N.D., not determined.
EC50 values and -fold shifts are an average of at least three determinations.
VU0152099 and VU0152100 Are Potent Positive Allosteric Modulators of M4 in Two Independent in Vitro Assays
Two compounds were selected for further evaluation of their ability to potentiate the M4-mediated calcium response. VU0152099 (compound 7o) (Fig. 4A) and VU0152100 (compound 7p) (Fig. 4D) retained M4 PAM activity (EC50 values of 403 ± 117 and 380 ± 93 nM, respectively) comparable to VU10010, and in the absence of an ACh EC20, neither VU0152099 nor VU0152100 elicited a response (Fig. 4, B and E). We next determined the effects of maximal concentrations of each compound on the CRC of ACh. Cells were preincubated with a fixed concentration (0.1–30 µM) of test compound and subsequently stimulated with increasing concentrations of ACh. Both VU0152099 and VU0152100 induced a dose-dependent leftward shift of the ACh CRC with maximal shifts of 30-fold observed with 30 µM VU0152099 (Fig. 4C) and 70-fold observed with 10 µM VU0152100 (Fig. 4F).
Fig. 4.

VU0152099 and VU0152100 are potent positive allosteric modulators of rat M4 in a functional calcium mobilization assay. A and D, chemical structure of VU0152099 (7o) (A) and VU0152100 (7p) (D). B and E, potency of VU0152099 (403 ± 117 nM) (B) and VU0152100 (380 ± 93 nM) (E) was evaluated at the rM4 receptor by measuring calcium mobilization in CHO cells stably expressing rM4 and the chimeric G protein Gqi5. A range of concentrations of test compound was added to cells, followed 1.5 min later by addition of an EC20 concentration of ACh (ACh EC20). In the absence of an EC20 concentration of ACh (Vehicle), neither test compound elicited a response. Data were normalized as a percentage of the maximal response to 10 µM ACh and represent the mean ± S.E.M. of three independent experiments. C and F, VU0152099 (C) and VU0152100 (F) potentiate the response of rM4 to ACh, as manifest by a dose-dependent leftward shift in the ACh CRC. At the highest concentrations tested, VU0152099 (30 µM) induced a 30-fold shift, and VU0152100 (10 µM) induced a 70-fold shift in the ACh CRC. Data were normalized as a percentage of the maximal response to 10 µM ACh and represent the mean ± S.E.M. of three to five independent experiments.
Using calcium mobilization to assess the functional activity of VU0152099 and VU0152100 at the M4 receptor requires coexpression of the chimeric G protein, Gqi5, in order to link the Gi/o-coupled M4 receptor to the phospholipase Cβ/Ca2+pathway. As an alternative approach to measure M4 PAM activity, we chose to take advantage of a novel functional assay recently developed in our laboratory that takes advantage of the ability of endogenous Gβγ subunits of Gi/o-coupled GPCRs to alter the kinetics of GIRK channels to conduct the ion thallium (Niswender et al., 2008). For these studies, HEK293 cells stably coexpressing heteromeric GIRK1/2 channels and the human M4 muscarinic receptor were preincubated with test compound and then stimulated with agonist (ACh) in the presence of thallium ion. Both VU0152099 and VU0152100 dose-dependently potentiated the response to an EC20 concentration of ACh with EC50 values of 1.2 ± 0.3 and 1.9 ± 0.2 µM, respectively, and increased the maximal response to ACh to approximately 130% (Fig. 5A). As observed in the calcium mobilization assays described above, both VU0152099 and VU0152100 (10 µM) also enhanced the potency of ACh to induce GIRK-mediated thallium flux, as manifest by a robust (≈30-fold) leftward shift in the ACh CRC from 77 ± 1.2 nM (veh) to 2.09 ± 0.3 nM (VU0152099) and 2.35 ± 0.5 nM (VU0152100) (Fig. 5B). Taken together, these in vitro studies suggest that VU0152099 and VU0152100 are potent positive allosteric modulators that enhance the response of the M4 receptor to the endogenous agonist ACh.
Fig. 5.

VU0152099 and VU0152100 potentiate GIRK-mediated thallium flux in response to ACh in HEK293 cells expressing human M4. A, both VU0152099 (■) and VU0152100 (▲) potentiate hM4-induced GIRK-mediated thallium flux in response to an EC20 concentration of ACh with potencies of 1.2 ± 0.3 and 1.9 ± 0.2 µM, respectively. B, in the presence of 10 µM VU0152099 (▲) and VU0152100 (▼), the ACh CRC for induction of GIRK-mediated thallium flux was leftward shifted (≈30-fold) from 77 ± 1.2 (■, Veh) to 2.09 ± 0.3 nM (▲, VU0152099) and 2.35 ± 0.5 nM (▲, VU0152100). Data were normalized as a percentage of the maximal response to 10 µM ACh and represent the mean ± S.E.M. of three to four independent experiments performed in quadruplicate.
VU0152099 and VU0152100 Bind to an Allosteric Site on the M4 Receptor and Increase ACh Affinity
To further confirm an allosteric mechanism of action by the novel M4 PAMs, we evaluated the effect of VU0152099 and VU0152100 on equilibrium radioligand binding using membranes prepared from cells expressing the rM4 receptor. We first assessed the ability of increasing concentrations of the two M4 PAMs to displace the orthosteric radioligand, [3H]NMS (0.1 nM). Unlike the orthosteric antagonist, atropine, which potently inhibited [3H]NMS binding (Ki = 0.54 ± 0.1 nM), neither M4 PAM displaced [3H]NMS at concentrations up to 30 µM (Fig. 6A), strongly suggesting that VU0152099 and VU0152100 act at a site on the M4 receptor that is distinct from the orthosteric binding site.
Fig. 6.

VU0152099 and VU0152100 bind allosterically and increase ACh affinity at rM4. A, in competition binding studies, neither VU0152099 (■) nor VU0152100 (▲) displaced the orthosteric radioligand, [3H]NMS (0.1 nM), at concentrations up to 30 µM. However, the orthosteric antagonist, atropine (▲), potently inhibited [3H]NMS binding with a Ki of 0.54 ± 0.1 nM. B, in the presence of vehicle alone, an increasing concentration of ACh displaces [3H]NMS (0.1 nM) binding with a Ki of 252 ± 17.9 nM (■). In the presence of a fixed concentration (10 µM) of VU0152099 or VU0152100, the potency of ACh to displace [3H]NMS binding is shifted leftward, yielding Ki values of 10.4 ± 0.91 (▼, VU0152099) and 12.2 ± 0.49 nM (▲,VU0152100), which represent a 25- and 21-fold shift in ACh potency, respectively. Data represent the mean ± S.E.M. of three independent experiments performed in duplicate.
In addition, we evaluated the effect of VU0152099 and VU0152100 on the affinity of ACh for the M4 receptor by assessing the ability of an increasing concentration of ACh to displace [3H]NMS (0.1 nM) binding in the absence or presence of the M4 potentiators. VU0152099 and VU0152100 were found to induce a 20 to 25-fold leftward shift in the potency of ACh to displace [3H]NMS binding to M4, as manifest by a reduction in the ACh Ki from 252 ± 17.9 nM (veh) to 10.4 ± 0.91 (VU0152099) and 12.2 ± 0.49 nM (VU0152100) (Fig. 6B). These data present a possible mechanism whereby these compounds could enhance receptor activation by increasing the affinity of M4 for acetylcholine and are in agreement with data previously determined for VU10010, where a 14-fold decrease in the ACh Ki was reported (Shirey et al., 2008).
VU0152099 and VU0152100 Are Selective for the M4 mAChR Subtype
We next evaluated VU0152099 and VU0152100 in calcium mobilization assays for effects at all mAChR subtypes to determine whether these compounds are selective for M4. Both VU0152099 and VU0152100 were selective for M4 relative to M1, M2, M3, and M5. Thus, neither VU0152099 (Fig. 7A) nor VU152100 (Fig. 7B) had any effect on the ACh dose-response curves at these other mAChR subtypes at concentrations up to 30 µM. To further assess selectivity of these compounds for M4 relative to other potential targets, the activities of VU0152099 and VU0152100 also were evaluated in radioligand binding assays against a large panel of 68 discrete GPCRs, ion channels, transporters, and enzymes (see Supplemental Table 1). These compounds were largely inactive at each of the targets in this panel screen. At concentrations of 10 µM, both compounds were completely inactive at most targets and induced less than 50% displacement of binding for all targets tested, with the single exception of GABAA receptors (assessed by flunitrazepam binding), where VU0152099 displayed 51% displacement. This suggests that VU0152099 may interact with the flunitrazepam site with an IC50 value of approximately 10 µM, which still provides high selectivity for M4 relative to GABAA receptors.
Fig. 7.

VU0152099 (A) and VU0152100 (B) are functionally selective for the M4 mAChR subtype. No shift in the ACh CRC was observed in the presence of 30 µM test compound at CHO K1 cells stably expressing M1, M2-Gqi5, M3, or M5 mAChRs. Calcium mobilization was measured in response to increasing concentrations of ACh following preincubation with either vehicle or test compound (30 µM), as described under Materials and Methods. Assay of M1, M3, and M5 mAChRs took advantage of endogenous coupling to Gq proteins, whereas assay of M2 activity required use of cells coexpressing the chimeric G protein, Gqi5, to allow coupling of this receptor to calcium mobilization. Points represent the mean ± S.E.M. of three independent experiments.
Given that VU0152099 and VU0152100 are allosteric modulators of M4, it is possible that they have activity at similar allosteric sites on other GPCRs. If so, this would not be apparent in the radioligand binding assays discussed above. The finding that these compounds are completely inactive at other mAChR subtypes makes this less likely given that M4 is more closely related to the other mAChR subtypes than to other GPCRs. However, to further evaluate the selectivity of VU0152099 for M4 relative to other family A GPCRs, we contracted with Millipore to determine the effects of this compound on the functional response of a panel of 16 GPCRs (including human M4) to activation by their respective agonists. For these studies, we chose family A GPCR subtypes that are among the closest relatives of mAChRs. We first determined the effects of VU0152099 alone on each receptor and found that these compounds had no agonist activity at any receptor studied (data not shown). We then determined the effects of VU0152099 on full concentration-response curves of agonists of each of these receptors. This allows unambiguous evaluation of whether the compounds possess antagonist activity (either allosteric or orthosteric) or allosteric potentiator activity at these other GPCRs. Consistent with our internal studies, VU0152099 induced a robust potentiation of ACh-induced activation of human M4 but had no potentiator activity at M1 (Supplemental Fig. 1). In addition, VU0152099 had no allosteric potentiator activity at any of the other GPCR subtypes tested (Supplemental Fig. 1). The only significant activity detected for VU0152099 in this functional panel screen was weak antagonist activity at serotonin 2B receptor (Supplemental Fig. 1). Together, these data suggest that VU0152099 and VU0152100 possess clean ancillary pharmacology profiles, which would allow us to pursue the behavioral effects of selective M4 activation in vivo.
VU0152099 and VU0152100 Exhibit Improved Physiochemical and Pharmacokinetic Properties
Before conducting in vivo studies with VU0152099 and VU0152100, pharmacokinetic studies were undertaken to assess brain/plasma ratios following systemic dosing of these compounds. In contrast, to the high log P of VU10010 (4.5), both VU0152099 and VU0152100 possessed log P values of 3.65 and 3.6, respectively, a full order of magnitude less lipophilic than VU10010 (Shirey et al., 2008). As a consequence, both VU0152099 and VU0152100 displayed improved physiochemical properties and afforded homogeneous dosing solutions in multiple vehicles acceptable for in vivo studies. Furthermore, we conducted in vivo exposure (brain and plasma) studies in rats at the dose of 56.6 mg/kg i.p. Both compounds exhibited substantial systemic absorption and brain penetration (Fig. 8). After 56.6 mg/kg i.p. administration, peak brain concentrations for both of the compounds were in the range of 3 to 5 µg/ml. VU0152100 (Fig. 8B) was far superior to VU0152099 (Fig. 8A) in terms of brain penetration, as evident from AUC0-∞ values (Table 2). AUC brain/AUC plasma ratio for VU0152099 was calculated to be 0.39 ± 0.01, whereas the ratio for VU0152100 was determined to be 0.86 ± 0.08 (Table 2). The half-life of the compounds in brain was 1.25 ± 0.02 (VU0152099) and 1.12 ± 0.01 h (VU0152100) (Table 2). Therefore, our earlier concern of P-gp susceptibility within this series was likely unwarranted, and the lack of central activity for VU10010 was most probably due solely to physiochemical properties.
Fig. 8.

Pharmacokinetic profiling of VU0152099 and VU0152100 in rats. Concentration-time profile of VU0152099 (A) and VU0152100 (B) in brain and plasma of male Sprague-Dawley rats following a 56.6 mg/kg i.p. administration of each compound. Blood and brain tissue were collected at 0.5, 1, 2, and 4 h after injection. Samples were extracted as described under Materials and Methods and analyzed by LC-MS-MS. Each time point represents the mean determination ± S.E.M. of three rats.
TABLE 2. Pharmacokinetic analysis of VU0152099 and VU0152100.
AUC(0-∞) and t1/2 values of VU0152099 and VU0152100 in exposure studies in male rats after 56.6 mg/kg i.p. administration. Values represent mean ± S.E.M. (n = 3 rats).
| PK Parameter | VU0152099 | VU0152100 |
|---|---|---|
| Mean AUC (0-∞) brain (ng · h/g) | 4751.80 ± 666.17 | 5726.35 ± 694.68 |
| Mean AUC(0-∞) plasma (ng · h/ml) | 11928.00 ± 1472.36 | 6570.35 ± 235.87 |
| AUC brain / AUC plasma | 0.39 ± 0.01 | 0.86 ± 0.08 |
| t1/2 plasma (h) | 1.66 ± 0.39 | 1.62 ± 0.69 |
| t1/2 brain (h) | 1.25 ± 0.023 | 1.12 ± 0.01 |
VU0152099 and VU0152100 Exhibit in Vivo Activity in Rat
Previous studies using the M1/M4-preferring muscarinic agonist xanomeline produced robust effects in several preclinical models predictive of antipsychotic-like activity, including reversal of amphetamine-induced hyperlocomotion in rats (Stanhope et al., 2001). Based on our initial pharmacokinetic studies suggesting that systemic administration of VU0152099 and VU0152100 provides robust brain levels of these compounds, the effects of VU0152099 and VU0152100 were evaluated in reversing amphetamine-induced hyperlocomotion in rats using a dose of 56.6 mg/kg i.p. for each compound with a 30-min pretreatment interval. As shown in Fig. 9A, both VU0152099 and VU0152100 produced robust decreases in amphetamine-induced hyperlocomotion over the time course tested. In addition, to provide further confirmation that the M4 PAMs have no effect on baseline levels of motor performance, which could complicate the interpretation of the amphetamine-induced hyperlocomotion data, we evaluated the effects of one of the M4 PAMs, specifically VU0152100, after administration alone on performance in the rotorod test (Fig. 9B). As shown, VU0152100 had no effect on performance in the rotorod test, even when tested at a dose of 100 mg/kg, which was higher than that required to observe reversal of amphetamine-induced hyperlocomotion.
Fig. 9.

VU0152099 and VU0152100 inhibit amphetamine-induced hyperlocomotor activity in rats without causing sedation. A, rats were pretreated for 30 min with vehicle or a 56.6 mg/kg dose of either VU0152099 or VU0152100 i.p. (data not shown). All rats received an injection of 1 mg/kg s.c. amphetamine, and locomotor activity was measured for an additional 60 min. Each point represents the mean of 8 to 16 rats. The error bars represent ± S.E.M. and are absent when less than the size of the point. Abscissa, dose of drug in milligrams per kilogram; ordinate, ambulations or total beam breaks per 5-min intervals; *, P < 0.05 versus veh + amphetamine control group, Dunnett’s test. B, lack of effect of VU0152100 on motor performance on the rotorod. After initial training trials, rats were pretreated for 30 min i.p. with vehicle or a dose of VU0152100, specifically 30, 56.6, or 100 mg/kg, and then the time each animal remained on the rotorod was recorded; animals not falling off the rotorod were given a maximal score of 85 s. Abscissa, dose of VU0152100 in milligrams per kilogram; ordinate, time spent on the rotorod in seconds. Each bar graph represents the mean of 8 to 10 rats. The error bars represent ± S.E.M.
Discussion
For decades, the prevailing theory behind the etiology of schizophrenia has been that excessive dopaminergic neurotransmission in the central nervous system is the major contributing factor underlying this severe psychiatric illness. This so-called dopamine hyperfunction hypothesis is based primarily on the observation that stimulation of the endogenous dopamine system (e.g., with amphetamine or cocaine) often leads to transient psychotic symptoms in healthy individuals (Bymaster et al., 2002; Raedler et al., 2007). Furthermore, all clinically relevant antipsychotic drugs, both typical and atypical, possess significant antagonist activity at D2 dopamine receptors (D2Rs) (Carlsson, 1988; Bymaster et al., 2002; Raedler et al., 2007). Thus, the majority of efforts to discover novel therapeutic agents for the treatment of schizophrenia have been aimed at developing therapies that result in some level of D2R blockade or a combination of blockade of D2Rs and other monoamine receptors. Nevertheless, D2R antagonists are only partially effective in treating schizophrenia, in that they only improve the positive symptoms associated with the disease, despite the fact that the negative and cognitive symptoms also markedly affect the quality of life for schizophrenic patients. In addition, these therapies also are often poorly tolerated because of numerous side effects, including sedation, weight gain, sexual dysfunction, diabetes, and Parkinson’s disease-like symptoms. Furthermore, greater than 25% of schizophrenia patients do not respond to these dopamine-based therapies (Hirsch and Barnes, 1995). Thus, although it is evident that dopamine does play a prominent role in the pathogenesis and treatment of schizophrenia, the dopamine hyperfunction hypothesis of schizophrenia fails to account for all aspects of this disorder, and it is increasingly evident that other neurotransmitter systems are probably involved. Based on this, it is unlikely that exclusive focus on discovery and development of antagonists of D2Rs and other monoamine receptors will provide fundamental breakthroughs in the standard of treatment of schizophrenia patients relative to current therapies.
In recent years, the mAChRs have emerged as potential novel targets for the treatment of schizophrenia. This is based on clinical studies demonstrating efficacy of mAChR agonists in treatment of positive symptoms in schizophrenia patients, as well as multiple animal studies suggesting that mAChR agonists could be useful in treatment of cognitive dysfunction in schizophrenia patients (Bymaster et al., 2002). Furthermore, a growing body of evidence from clinical and animal studies involving pharmacological manipulations, post mortem tissue analysis, and brain imaging is consistent with this hypothesis (Raedler et al., 2007). Whereas recent advances suggesting potential utility of mAChR activators in treatment of schizophrenia have been exciting, there have been few selective pharmacological tools available to fully explore this emerging muscarinic hypothesis of schizophrenia. Unfortunately, previous attempts to develop traditional orthosteric agonists that are highly selective for individual mAChR subtypes have been unsuccessful.
The current discovery and optimization of VU0152099 and VU0152100 as highly selective positive allosteric modulators of M4 provides a major advance in establishing a new approach for developing highly selective activators of these receptors. The data presented provide further support for the ability to achieve high subtype selectivity by targeting allosteric sites and provide exciting new data demonstrating that highly selective M4 PAMs have robust activity in at least one animal model that is similar to effects described previously for the nonselective orthosteric mAChR agonist, xanomeline (Stanhope et al., 2001). The finding that VU0152099 and VU0152100 mimic effects of xanomeline in an animal model that has been used to predict antipsychotic activity of new compounds is especially exciting in light of clinical studies demonstrating the clinical efficacy of xanomeline in schizophrenia patients. This raises the exciting possibility that selective activation of M4 may provide a novel approach for the treatment of some symptoms associated with schizophrenia. Furthermore, the discovery of systemically active M4 PAMs suggests that this will be a viable approach for developing selective activators of M1 and other mAChR subtypes.
Whereas the in vitro data for VU0152099 and VU0152100 indicate high pharmacologic selectivity for M4 relative to any other mAChR subtypes or closely related GPCRs, the possibility exists that the observed behavioral effects may be due to an off-target activity not yet identified. In future studies, it will be critical to further validate that the effects of VU0152099 and VU0152100 observed in vivo are mediated by activation of M4 using other tools, including structurally distinct M4 PAMs or selective M4 antagonists, as they become available, and/or M4 knockout mice. Unfortunately, studies in M4 knockout mice will be complex given that these mice display fundamentally different responses to psychomotor stimulants, and compounds will need to be optimized for appropriate pharmacokinetic properties in mice. Furthermore, several behavioral parameters are substantially altered in the M4 knockout mice, including increases in baseline locomotor activity, altered responses to amphetamine, and altered dopamine release in the mesolimbic dopamine circuitry (Gomeza et al., 1999, 2001; Tzavara et al., 2004). These factors will complicate interpretation of studies with M4 knockout mice. However, although it is not possible to definitively establish an exclusive role for M4 in mediating the behavioral effects observed here, previous studies have established similar effects of structurally distinct mAChR agonists (Stanhope et al., 2001). This, coupled with the high selectivities of VU0152099 and VU0152100 for M4 relative to any other mAChR subtype, suggests that M4 is a likely candidate for mediating these effects. In addition, extensive profiling of VU0152099 and VU0152100 showed no functional effects on responses to other closely related family A GPCRs and no significant off-target activity at any of the 68 other GPCRs, ion channels, or enzymes. Thus, although it is impossible to entirely rule out unknown off-target activity, these compounds appear to be much more highly selective for the targeted receptor than is typical for most orthosteric GPCR ligands. As new tools become available for targeting M4 and other mAChR subtypes, they will provide the means to develop a more complete understanding of the roles of each of the individual mAChR subtypes in regulating CNS function.
In addition to their potential relevance for schizophrenia, mAChRs also are thought to regulate motor function by exerting effects on dopaminergic transmission in the basal ganglia (Pisani et al., 2007; Raedler et al., 2007). For instance, multiple electrophysiology studies with M4 knockout mice have led to the suggestion that activation of the M4 mAChR may oppose some actions of dopaminergic neurons on striatal motor function (Calabresi et al., 1998; Sánchez-Lemus and Arias-Montaño, 2006). Again, the lack of highly selective activators of M4 has made it impossible to test this hypothesis directly. Furthermore, previous studies with traditional M4 agonists bypass the action of endogenous ACh in the basal ganglia and do not provide information on the effects of endogenous acetylcholine on motor activity. The finding that highly selective M4 potentiators reverse amphetamine-induced hyperlocomotor activity in rats provides exciting new evidence in support of the hypothesis that endogenous ACh plays an important role in regulating dopaminergic control of motor function. Because these compounds do not activate M4 directly but selectively increase responses of M4 to endogenous ACh, this provides direct evidence that this response can be modulated by endogenous ACh acting on M4 receptors. In addition, this raises the possibility that selective M4 modulators could provide a novel approach to treatment of other disorders involving altered dopaminergic function in the basal ganglia, including Parkinson’s disease and dystonia.
In addition to suggesting the potential roles of M4 in vivo, these data have important implications related to the molecular pharmacology of allosteric modulators of GPCRs. One of the most promising properties of allosteric modulators of GPCRs has been that it is often possible to achieve high selectivity for a targeted GPCR subtype relative to closely related family members (Marino et al., 2003; O’Brien et al., 2003; Kinney et al., 2005; Galici et al., 2006). However, the discovery of these compounds also raises the question of whether allosteric modulators may have broad activity across other GPCRs by interacting at potentially promiscuous allosteric sites. The present finding that VU0152099 and VU0152100 had no major activities across multiple targets in a large panel radioligand binding screen was encouraging in suggesting the high selectivity of these compounds but does not address this critical question. However, the finding that these compounds had no allosteric modulator activity across a panel of 15 other family A GPCR subtypes is exciting and suggests that they are not likely to have activity at a site that is shared across multiple GPCRs. Although it is impossible to rule out activity at other unidentified targets that were not tested, these data suggest that it may be possible to achieve higher subtype selectivity across a range of receptors than has been possible with many orthosteric ligands.
Supplementary Material
Supplemental Material can be found at: http://jpet.aspetjournals.org/cgi/content/full/jpet.108.140350/DC1
Acknowledgment
We thank Drs. A. Levey (Emory University, Atlanta, GA) for stable mAChR cell lines (hM2, hM3, and hM5), T. I. Bonner (NIMH, Bethesda, MD) for the rM4 cDNA construct, and B. Conklin [Gladstone Institute, University of California San Francisco (UCSF), San Francisco, CA] for the chimeric Gqi5 construct. HEK293 cells stably expressing GIRK1, GIRK2, and the human M4 receptor were generously provided by Drs. Huai Hu Chang and Lily Jan (UCSF, San Francisco, CA).
This work was supported by grants from the National Institute of Mental Health (NIMH) and the NINDS, National Institutes of Health. A.E.B. is supported by NIMH Grant 1F32 MH079678-01. C.K.J. was supported by NIMH Grants 5 F32 MH076371-01 and 02. T.M.B. is supported by the Integrative Training in Therapeutic Discovery (ITTD) Grant T90-DA22873 from the Vanderbilt Institute of Chemical Biology, and J.K.S. is supported by NIMH Grant 1 F31 MH80559-01. Vanderbilt is a site in the National Institutes of Health-supported Molecular Libraries Screening Center Network.
ABBREVIATIONS
- mAChR
muscarinic acetylcholine receptor
- VU0152099
3-amino-N-(benzo[d][1,3]dioxol-5-ylmethyl)-4,6-dimethylthieno[2,3-b]pyridine carboxamide
- VU0152100
3-amino-N-(4-methoxybenzyl)-4,6-dimethylthieno[2,3-b]pyridine carboxamide
- VU10010
3-amino-N-(4-chlorobenzyl)-4,6-dimethylthieno[2,3-b]pyridine-2-carboxamide
- ACh
acetylcholine
- HPLC
high-performance liquid chromatography
- CHO
Chinese hamster ovary
- ELSD
evaporative light scattering detector
- LC
liquid chromatography
- MS
mass spectrometry
- CNS
central nervous system
- CRC
concentration-response curve
- GPCRs
G protein-coupled receptors
- M4
muscarinic acetylcholine receptor subtype 4
- [3H]NMS
N-methylscopolamine
- PAM
positive allosteric modulator
- DMSO
dimethyl sulfoxide
- P-gp
P-glycoprotein
- SAR
structure-activity relationship
- AM-807/25050004
3-amino-4,6-dimethylthioenol[2,3-b]-pyridine-2-carboxylic acid
- D2R
D2 dopamine receptors
- r
rat
- h
human
- ANOVA
analysis of variance
- HEK293
human embryonic kidney 293
- GIRK
G protein-regulated inwardly rectifying K+
- Q-ToF
quadrupole time of flight
Footnotes
The online version of this article (available at http://jpet.aspetjournals.org) contains supplemental material.
References
- Bodick NC, Offen WW, Levey AI, Cutler NR, Gauthier SG, Satlin A, Shannon HE, Tollefson GD, Rasmussen K, Bymaster FP, et al. Effects of xanomeline, a selective muscarinic receptor agonist, on cognitive function and behavioral symptoms in Alzheimer disease. Arch Neurol. 1997;54:465–473. doi: 10.1001/archneur.1997.00550160091022. [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Carter PA, Yamada M, Gomeza J, Wess J, Hamilton SE, Nathanson NM, McKinzie DL, Felder CC. Role of specific muscarinic receptor subtypes in cholinergic parasympathomimetic responses, in vivo phosphoinositide hydrolysis, and pilocarpine-induced seizure activity. Eur J Neurosci. 2003a;17:1403–1410. doi: 10.1046/j.1460-9568.2003.02588.x. [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Felder C, Ahmed S, McKinzie D. Muscarinic receptors as a target for drugs treating schizophrenia. Curr Drug Targets. 2002;1:163–181. doi: 10.2174/1568007024606249. [DOI] [PubMed] [Google Scholar]
- Bymaster FP, McKinzie DL, Felder CC, Wess J. Use of M1-M5 muscarinic receptor knockout mice as novel tools to delineate the physiological roles of the muscarinic cholinergic system. Neurochem Res. 2003b;28:437–442. doi: 10.1023/a:1022844517200. [DOI] [PubMed] [Google Scholar]
- Calabresi P, Centonze D, Pisani A, Sancesario G, North RA, Bernardi G. Muscarinic IPSPs in rat striatal cholinergic interneurones. J Physiol. 1998;510:421–427. doi: 10.1111/j.1469-7793.1998.421bk.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlsson A. The current status of the dopamine hypothesis of schizophrenia. Neuropsychopharmacology. 1988;1:179–186. doi: 10.1016/0893-133x(88)90012-7. [DOI] [PubMed] [Google Scholar]
- Felder CC, Porter AC, Skillman TL, Zhang L, Bymaster FP, Nathanson NM, Hamilton SE, Gomeza J, Wess J, McKinzie DL. Elucidating the role of muscarinic receptors in psychosis. Life Sci. 2001;68:2605–2613. doi: 10.1016/s0024-3205(01)01059-1. [DOI] [PubMed] [Google Scholar]
- Galici R, Jones CK, Hemstapat K, Nong Y, Echemendia NG, Williams LC, de Paulis T, Conn PJ. Biphenyl-indanone A, a positive allosteric modulator of the metabotropic glutamate receptor subtype 2, has antipsychotic- and anxiolytic-like effects in mice. J Pharmacol Exp Ther. 2006;318:173–185. doi: 10.1124/jpet.106.102046. [DOI] [PubMed] [Google Scholar]
- Gomeza J, Zhang L, Kostenis E, Felder C, Bymaster F, Brodkin J, Shannon H, Xia B, Deng C, Wess J. Enhancement of D1 dopamine receptor-mediated locomotor stimulation in M(4) muscarinic acetylcholine receptor knockout mice.[see comment] Proc Natl Acad Sci U S A. 1999;96:10483–10488. doi: 10.1073/pnas.96.18.10483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomeza J, Zhang L, Kostenis E, Felder CC, Bymaster FP, Brodkin J, Shannon H, Xia B, Duttaroy A, Deng CX, et al. Generation and pharmacological analysis of M2 and M4 muscarinic receptor knockout mice. Life Sci. 2001;68:2457–2466. doi: 10.1016/s0024-3205(01)01039-6. [DOI] [PubMed] [Google Scholar]
- Hemstapat K, de Paulis T, Chen Y, Brady AE, Grover VK, Alagille D, Tamagnan GD, Conn PJ. A novel class of positive allosteric modulators of metabotropic glutamate receptor subtype 1 interact with a site distinct from that of negative allosteric modulators. Mol Pharmacol. 2006;70:616–626. doi: 10.1124/mol.105.021857. [DOI] [PubMed] [Google Scholar]
- Hirsch S, Barnes TRE. The clinical treatment of schizophrenia with antipsychotic medication. In: Hirsch SR, Weinberger DR, editors. Schizophrenia. Oxford, UK: Blackwell Science; 1995. pp. 443–468. [Google Scholar]
- Kinney GG, O’Brien JA, Lemaire W, Burno M, Bickel DJ, Clements MK, Chen TB, Wisnoski DD, Lindsley CW, Tiller PR, et al. A novel selective positive allosteric modulator of metabotropic glutamate receptor subtype 5 has in vivo activity and antipsychotic-like effects in rat behavioral models. J Pharmacol Exp Ther. 2005;313:199–206. doi: 10.1124/jpet.104.079244. [DOI] [PubMed] [Google Scholar]
- Lindsley CW, Wisnoski DD, Leister WH, O’brien JA, Lemaire W, Williams DL, Jr, Burno M, Sur C, Kinney GG, Pettibone DJ, et al. J Med Chem. Vol. 47. 2004. Discovery of positive allosteric modulators for the metabotropic glutamate receptor subtype 5 from a series of N-(1,3-diphenyl-1H-pyrazol-5-yl)benzamides that potentiate receptor function in vivo; pp. 5825–5828. [DOI] [PubMed] [Google Scholar]
- Marino MJ, Conn PJ. Glutamate-based therapeutic approaches: allosteric modulators of metabotropic glutamate receptors. Curr Opin Pharmacol. 2006;6:98–102. doi: 10.1016/j.coph.2005.09.006. [DOI] [PubMed] [Google Scholar]
- Marino MJ, Williams DL, Jr, O’Brien JA, Valenti O, McDonald TP, Clements MK, Wang R, DiLella AG, Hess JF, Kinney GG, et al. Allosteric modulation of group III metabotropic glutamate receptor 4: a potential approach to Parkinson’s disease treatment. Proc Natl Acad Sci U S A. 2003;100:13668–13673. doi: 10.1073/pnas.1835724100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messer WS., Jr Cholinergic agonists and the treatment of Alzheimer’s disease. Curr Top Med Chem. 2002;2:353–358. doi: 10.2174/1568026024607553. [DOI] [PubMed] [Google Scholar]
- Niswender CM, Johnson KA, Luo Q, Ayala JE, Kim C, Conn PJ, Weaver CD. A novel assay of Gi/o-linked G protein-coupled receptor coupling to potassium channels provides new insights into the pharmacology of the group III metabotropic glutamate receptors. Mol Pharmacol. 2008;73:1213–1224. doi: 10.1124/mol.107.041053. [DOI] [PubMed] [Google Scholar]
- O’Brien JA, Lemaire W, Chen TB, Chang RS, Jacobson MA, Ha SN, Lindsley CW, Schaffhauser HJ, Sur C, Pettibone DJ, et al. A family of highly selective allosteric modulators of the metabotropic glutamate receptor subtype 5. Mol Pharmacol. 2003;64:731–740. doi: 10.1124/mol.64.3.731. [DOI] [PubMed] [Google Scholar]
- O’Brien JA, Lemaire W, Wittmann M, Jacobson MA, Ha SN, Wisnoski DD, Lindsley CW, Schaffhauser HJ, Rowe B, Sur C, et al. A novel selective allosteric modulator potentiates the activity of native metabotropic glutamate receptor subtype 5 in rat forebrain. J Pharmacol Exp Ther. 2004;309:568–577. doi: 10.1124/jpet.103.061747. [DOI] [PubMed] [Google Scholar]
- Pisani A, Bernardi G, Ding J, Surmeier DJ. Re-emergence of striatal cholinergic interneurons in movement disorders. Trends Neurosci. 2007;30:545–553. doi: 10.1016/j.tins.2007.07.008. [DOI] [PubMed] [Google Scholar]
- Raedler TJ, Bymaster FP, Tandon R, Copolov D, Dean B. Towards a muscarinic hypothesis of schizophrenia. Mol Psychiatry. 2007;12:232–246. doi: 10.1038/sj.mp.4001924. [DOI] [PubMed] [Google Scholar]
- Sánchez-Lemus E, Arias-Montaño JA. M1 muscarinic receptors contribute to, whereas M4 receptors inhibit, dopamine D1 receptor-induced [3H]-cyclic AMP accumulation in rat striatal slices. Neurochem Res. 2006;31:555–561. doi: 10.1007/s11064-006-9052-8. [DOI] [PubMed] [Google Scholar]
- Scarr E, Sundram S, Keriakous D, Dean B. Altered hippocampal muscarinic M4, but not M1, receptor expression from subjects with schizophrenia. Biol Psychiatry. 2007;61:1161–1170. doi: 10.1016/j.biopsych.2006.08.050. [DOI] [PubMed] [Google Scholar]
- Shekhar A, Potter WZ, Lienemann J, Sunblad K, Lightfoot J, Herrera J, Unverzagt F, Bymaster FP, Felder C. Efficacy of xanomeline, a selective muscarinic agonist, in treating schizophrenia: a double-blind, placebo controlled study. 40th Annual Meeting of American College of Neuropsychopharmacology; 2005 Dec 11–15; Waikoloa, HI. Nashville, TN: American College of Neuropsychopharmacology; 2001. [Google Scholar]
- Shekhar A, Potter WZ, Lightfoot J, Lienemann J, Dubé S, Mallinckrodt C, Bymaster FP, McKinzie DL, Felder CC. Selective muscarinic receptor agonist xanomeline as a novel treatment approach for schizophrenia. Am J Psychiatry. 2008;165:1033–1039. doi: 10.1176/appi.ajp.2008.06091591. [DOI] [PubMed] [Google Scholar]
- Shirey JK, Xiang Z, Orton D, Brady AE, Johnson KA, Williams R, Ayala JE, Rodriguez AL, Wess J, Weaver D, et al. An allosteric potentiator of M4 mAChR modulates hippocampal synaptic transmission. Nat Chem Biol. 2008;4:42–50. doi: 10.1038/nchembio.2007.55. [DOI] [PubMed] [Google Scholar]
- Stanhope KJ, Mirza NR, Bickerdike MJ, Bright JL, Harrington NR, Hesselink MB, Kennett GA, Lightowler S, Sheardown MJ, Syed R, et al. The muscarinic receptor agonist xanomeline has an antipsychotic-like profile in the rat. J Pharmacol Exp Ther. 2001;299:782–792. [PubMed] [Google Scholar]
- Tzavara ET, Bymaster FP, Davis RJ, Wade MR, Perry KW, Wess J, McKinzie DL, Felder C, Nomikos GG. M4 muscarinic receptors regulate the dynamics of cholinergic and dopaminergic neurotransmission: relevance to the pathophysiology and treatment of related CNS pathologies. FASEB J. 2004;18:1410–1412. doi: 10.1096/fj.04-1575fje. [DOI] [PubMed] [Google Scholar]
- Wess J. Molecular biology of muscarinic acetylcholine receptors. Crit Rev Neurobiol. 1996;10:69–99. doi: 10.1615/critrevneurobiol.v10.i1.40. [DOI] [PubMed] [Google Scholar]
- Zhao Z, Wisnoski DD, O’Brien JA, Lemaire W, Williams DL, Jr, Jacobson MA, Wittman M, Ha SN, Schaffhauser H, Sur C, et al. Challenges in the development of mGluR5 positive allosteric modulators: the discovery of CPPHA. Bioorg Med Chem Lett. 2007;17:1386–1391. doi: 10.1016/j.bmcl.2006.11.081. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Material can be found at: http://jpet.aspetjournals.org/cgi/content/full/jpet.108.140350/DC1