

Abstract

Molecular docking is the most practical approach to leverage protein structure for ligand discovery, but the technique retains important liabilities that make it challenging to deploy on a large scale. We have therefore created an expert system, DOCK Blaster, to investigate the feasibility of full automation. The method requires a PDB code, sometimes with a ligand structure, and from that alone can launch a full screen of large libraries. A critical feature is self-assessment, which estimates the anticipated reliability of the automated screening results using pose fidelity and enrichment. Against common benchmarks, DOCK Blaster recapitulates the crystal ligand pose within 2 Å rmsd 50−60% of the time; inferior to an expert, but respectrable. Half the time the ligand also ranked among the top 5% of 100 physically matched decoys chosen on the fly. Further tests were undertaken culminating in a study of 7755 eligible PDB structures. In 1398 cases, the redocked ligand ranked in the top 5% of 100 property-matched decoys while also posing within 2 Å rmsd, suggesting that unsupervised prospective docking is viable. DOCK Blaster is available at http://blaster.docking.org.
Introduction
Molecular docking has had important recent successes1−12 and is now widely used in industry and academia. But whereas other techniques in computational biology such as homology modeling(13) and sequence database searching(14) have been successfully deployed on a proteomic scale, docking has remained manually intensive. Docking programs are challenging to use, with many parameters to be chosen, file formats to be manipulated, and decisions to be made at both the preparation and analysis stages. Even in expert hands, there are targets for which docking simply fails to recapitulate experimentally known binding information. These barriers to entry have diminished the impact of the technique by making it less accessible to biologically oriented nonexperts and challenging even for specialists to deploy on a large scale.
One approach to make docking accessible to more investigators, and to make it more systematic even for experts, is to automate it. We have therefore investigated an expert system, DOCK Blaster, which aims to emulate experts at all stages of the docking process. Ideally, DOCK Blaster could start from as little as a PDBa code and from that launch a full screen of a large compound library to find novel ligands. To do so it must overcome substantial challenges in preparing a target site for docking, it must explore variation in the sampling and often scoring, and it must conduct control calculations to judge the quality of the screen. For instance, the automated procedure must recognize common cofactors, metals, post-translational modifications, and solutes to identify the ligand and separate it from the receptor. Second, “hot spots” for docking must be identified for a wide range of binding site sizes and shapes. Third, parameters must be assigned to receptor atoms in a robust way that can cope not only with cofactors, metals, and post-translational modifications but also with unforeseen moieties for which no dictionary is available. Because an expert would normally experiment with several variations in sampling and scoring, an automated system should do so also, picking the best parameters to use for a full database screen. Finally, the entire docking process should be integrated from end to end so as to recover from simple problems, continue as far as possible, and end gracefully should an unrecoverable error occur.
Here we describe DOCK Blaster, a fully automated docking system including self-assessment. The method is tested for pose-fidelity, the ability to reproduce experimentally observed poses within some tolerance limit, and enrichment, the ability to enrich actives from among a database of decoys, where a decoy is a member of the database that does not bind to the target. We have used three of the most common benchmarking sets: the Astex-85 set,(7) having 85 high-quality crystal structures of therapeutically relevant targets and “drug-like” ligands, the GOLD-114 benchmark,(15) derived from the most widely used docking benchmarks,16,17 and the DUD set,(27) 38 protein targets for which sets of annotated actives and corresponding property-matched decoys are available for each target. Property-matched decoys have similar physical properties but different topologies that one would not expect should be recognized by the protein, a key requirement for a high score. Whereas we find that the automated method is typically outperformed by an expert, its performance is nevertheless respectable.
In principle, such an automated procedure could bring docking to a much larger community and could be used to explore targets on a proteomic scale. For the first goal to provide more good than harm, it is important to provide what may often be naïve users with an automated self-assessment of the docking results. We investigate methods to do so using pose fidelity and enrichment based on a single observed crystallographic ligand. To investigate the plausibility of the second goal, we describe here docking screens on 7755 protein targets where each screen is performed against a feasibility library of 100 molecules.
Methods
DOCK Blaster is composed of an expert system engine and a web-enabled user interface (Figure 1). The docking program used is DOCK 3.5.54,28,29 a version of UCSF DOCK. The DOCK Blaster pipeline is composed of six modules (Figure 2): (a) the parser, which identifies the receptor and ligand from a PDB file, (b) the scrutinizer, which attempts to correct for problems, such as incomplete or disordered residues on the receptor, (c) the preparer, which protonates the receptor, calculates “hot spots” and scoring grids, assigns atomic parameters, including these for cofactors, post-translational modifications and metals, and prepares the ligand, decoys, and any actives and inactives for docking, (d) the calibrator, which uses supplied data to assess docking performance and suggests optimal docking parameters, (e) the docker, which manages a full database screen on the computer cluster, and (f) the assessor, which prepares reports to interpret database screening results. We take up each module in turn.
Figure 1.

The DOCK Blaster web interface, available starting from http://blaster.docking.org/.
Figure 2.
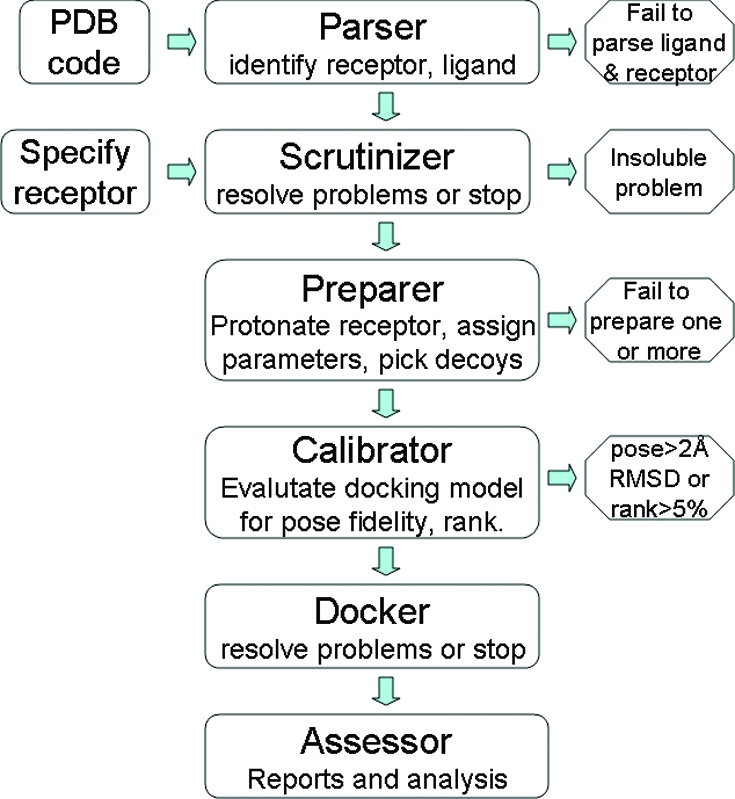
DOCK Blaster pipeline schematic. Left: two starting points for DOCK Blaster. Center: six main modules of the automatic docking pipeline. Right: four places in which automatic docking can fail.
a. The Parser
This expert system starts with a PDB code. The file is retrieved directly from the PDB Web site with no preprocessing. The Parser uses dictionaries of common cofactors, ions, post-translational modifications, and solutes to identify and separate the ligand from the receptor. If the ligand itself is a common solute or cofactor, a three letter code of the ligand must be specified. If more than one ligand is available, the Parser asks the user to pick one. In fully automated screens, such ambiguity stops the procedure. If no ligand can be identified, the Parser also halts the calculation with an error message. A future version may use automatic binding site identification software30−32 to identify the binding site in this case. The Parser produces files that are ready to be used by the scrutinizer in the next step and may be accessed online at http://blaster.docking.org/parser.shtml.
b. The Scrutinizer
The Scrutinizer takes as input a target structure and a specification of the binding site, which may be either a docked ligand in mol2 format or atoms in or near the binding site in PDB format. The Scrutinizer checks that the receptor and ligand are properly formatted, and that at least one atom of the ligand is within a binding site on the protein. This step also attempts to flesh out incomplete residues and pick the first of any disorder models present. The Scrutinizer can take input directly from the parser (module a, above) or from the job preparation page, http://blaster.docking.org/start.shtml.
c. The Preparer
The Preparer is an expert system that performs actions necessary before docking can begin. This includes maturing the receptor model by removal of ordered water molecules, protonation of receptor atoms to a united atom model, and assignment of AMBER atom types(33) including to metals, cofactors, and post-translational modifications that are effectively part of the receptor. Subsequently docking “hot spots” are calculated by sphgen,(34) while van der Waals and electrostatics scoring grids are calculated by chemgrid(35) and Delphi,(36) respectively. To correct for ligand desolvation, solvmap(37) is used to calculate a solvent occlusion grid. To prepare a dockable database of the ligand and any actives and inactive controls, the PDB format ligand is converted to SMILES using OpenEye’s OEChem(38) to eliminate bias. The molecule is then processed using the standard ZINC protocol,(39) which aims to enumerate all physiologically relevant protonated and tautomeric forms of the molecule. In parallel, property-matched decoys for the ligand are found from ZINC using the DUD protocols27,40 for enrichment and ranking.
Four parameter sets are evaluated by performing sampling and scoring each in two different ways. DOCK Blaster uses two different sampling schemes called “coarser” and “finer” to sample fewer or more ligand orientations by adjusting the bins used to generate initial orientations. The “coarser” scheme uses 45 “hot spots” and wider bins and generally runs somewhat faster, whereas “finer” uses 55 “hot spots”, narrower bins, and is often slower. Two scoring schemes called “polarized” and “normal” are also used. The “normal” scheme uses standard AMBER 94 partial atomic charges on the protein, while “polarized” increases the dipoles on selected polar atoms in residues within 3.5 Å of the crystallographic ligand without changing the net charge. In prospective studies involving experimental testing of novel inhibitors, we have found such limited polarization useful.41−44 Each ligand configuration that passes a rapid steric fit filter is scored for electrostatic and van der Waals complementarity and adjusted for partial ligand desolvation due to solvent occlusion.(37) The high-scoring ligand conformation is minimized with 100 steps of simplex rigid-body minimization. More details of these schemes and other technical details may be found on the DOCK Blaster documentation Web site, http://disi.docking.org/DOCK_Blaster:Technical_Details.
d. The Calibrator
This subsystem evaluates how well docking works as judged by pose fidelity to the crystal structure and by enrichment versus decoys. In doing so, this tool selects the best docking parameters for a full database screen. It first redocks the ligand using all four docking parameter sets. It evaluates pose fidelity using the rmsd of all non-H atoms to the crystallographic pose and calculates the rank of the redocked ligand among the 100 property-matched decoys, which are also docked using all four docking parameter sets. If actives and inactives were supplied, as is the case for the DUD benchmark, these are also docked and ROC plots are calculated.
A concise calibration docking report allows users to judge which parametrization, if any, is best for prospective docking (Figure 3). Outcomes are color coded to indicate successful (in green), unsuccessful (in red), and borderline (in yellow) results. Particularly for borderline cases, the user may wish to inspect the scores and poses of each ligand and decoy before selecting the best parametrization for prospective docking.
Figure 3.

Calibration docking report, containing pose fidelity (Å, rmsd) and enrichment (% rank) of the redocked crystallographic ligand compared to 100 property matched decoys using four parametrizations, separated by a forward slash (/) in each cell. Successful runs are in green, unsuccessful ones in red, and marginal ones in yellow.
e. The Docker
This module screens (potentially very large) ZINC subsets on a computer cluster. It is normally only invoked after the calibrator has established that docking is viable. The most successful set of docking parameters from the calibration phase is selected and used. The Docker manages the screen across multiple CPUs, combining the results when the entire library has been searched.
f. The Assessor
When database docking is complete, the Assessor prepares reports including full purchasing information, annotation of physical properties, annotation of known activities derived from public data sources, and similarity to annotated compounds, other top hits, and other purchasable compounds. Docked structures may be viewed in the context of the binding site using PyMol,(45) Chimera(46) or JMol.(47) These reports may be browsed online or downloaded in tab-delimited format for processing by analysis software such as Excel.
DOCK Blaster is accessible via a web-based interface at http://blaster.docking.org (Figure 1). It is hosted on a cluster of 700 CPU cores managed using the Sun Grid Engine with access to 30 terabytes of RAID-6 storage. DOCK Blaster is integrated with ZINC,(39) a public access database of commercially available compounds for library screening, and the DUD Decoy Maker,27,40 which are also accessible separately. We do preserve the results of some screens, which we may use for methods development and analyses.
DOCK Blaster was tested using ligand-bound crystal structures from the PDB,(48) including some of the most widely used benchmarking standards: Astex-85,(7) GOLD-114(15), and DUD-38(27) (release 2, Oct 2006). We have used only the 38 structures in DUD for which a ligand-bound crystal structure is available. We have used only postremediation(49) PDB structures for which a single small (<500 Da) organic ligand is bound, having 2.5 Å resolution or better, and no polynucleotides.
Results
DOCK Blaster is a new tool for automatic docking and is available for free anonymous public access at http://blaster.docking.org. DOCK Blaster can start from the structure of a target and a specification of the binding site or simply from a PDB code in many cases. To assess the performance of our automatic high throughput docking procedure retrospectively, we turned to common benchmarks in the field: Astex-85,(7) GOLD-114,(15) and DUD-38.(27) We began by retrieving the files directly from the remediated(49) PDB and used the parser to automatically separate the ligand from the receptor. In all but a few cases, the parser succeeded well enough in this task to proceed to docking (). Occasionally the ligand could not be identified automatically. One reason was that the ligand was a common solute such as citrate. These could be rescued by simply specifying the three letter code of the ligand to the parser. The average total CPU time per target, from preparation to docking the ligand and its 100 decoys to final analysis of the results, all automated, was 92 min.
Astex-85 Benchmarking
We began by using DOCK Blaster to automatically dock to targets in the Astex-85 set.(7) In all but one case, the ligand was automatically separated from the protein starting from the PDB code alone (). For 1LF7, we were required to specify the ligand (citrate, CIT) because it is a common solute. Of the 84 jobs started automatically, 83 finished normally and produced a docked pose and score for the ligand and its decoys (). One ligand failed during conversion to SMILES (1U4D).
We asked how well automatic docking could recapitulate the crystal structure using each of four docking parameter sets. Of the 83 protein−ligand pairs that yielded a docked ligand score, 51 were within 2 Å rmsd of the crystal structure ( , and http://data.docking.org/2009/AstexTables.doc). Some parametrization schemes were more successful than others. The best was “finer/polarized” with 49 successes, followed by “finer/normal” with 46, “coarser/polarized” with 37, and the least successful being “coarser/normal” with 34 successes. The “finer” schemes having more spheres generally outperformed the “coarser” schemes, although this was not always true. The polarized dipoles in the electrostatics of “polarized” were slightly more successful than the “normal” scheme having standard AMBER94 charges on the protein.
For the 51 protein−ligand complexes docked within 2 Å rmsd of the crystal pose, we then asked how well the redocked crystallographic ligand ranked compared to around 100 property-matched decoys (Tables 1 and 2 and http://data.docking.org/2009/AstexTables.doc). The well-posed ligand also ranked in the top 5% of the property matched decoys in 29 cases, suggesting that these are suitable for automatic prospective docking. Just beyond the 2 Å cutoff, there were 11 cases in which the ligand posed close, within 3 Å, and a further 20 cases where the ligand never got even as close as 3 Å rmsd to the crystallographic ligand.
Table 1. Docking Successes with DOCK Blastera.
| Outcome | Astex-85 | Gold-114 | DUD-38 | PDB-9050 |
|---|---|---|---|---|
| success | 29 | 27 | 15 | 1398 |
| ligand never ranks well | 44 | 67 | 21 | 6357 |
| ligand fails to dock automatically | 2 | 20 | 2 | 1295 |
Number of targets where fully automatic docking with DOCK blaster is successful, as judged by both good pose fidelity (< 2.0 Å RMSD) and rank (top 5%) versus about 100 property-matched decoys.
Table 2. Docking Success Depends on Docking Parametersa.
| parameter choice | DOCK score | ScreenScore | FlexX | PLP | PMF |
|---|---|---|---|---|---|
| all | 18 | 18 | 16 | 18 | 10 |
| any | 29 | 36 | 33 | 38 | 22 |
Success is defined as ligand within 2 Å RMSD of the crystal pose and rank in the top 5% of about 100 property-matched decoys. Astex-85 benchmark. For instance, whereas 29 targets succeeded for pose fidelity and rank using any of four parameter sets and the standard DOCK scoring function, only 18 of these succeeded with all parameter sets.
For 18 targets, the results were excellent regardless of which docking parameter set was chosen; we could find no common theme among the targets to which to attribute this good fortune (Figure 4). There were three nuclear receptors (1M2Z, 1N46, 1SQN), six kinases (1KE5, 1OPK, 1YWR, 1V4S, 1PMN, 1T46), and seven other enzymes (1HWI, 1OF6, 1LPZ, 1Q4G, 1JLA, 1R9O, 1VCJ). There was no obvious pattern in the physical or chemical properties of the ligands to explain why these targets worked so reliably. We did notice some patterns among the less successful docking calculations, however.
Figure 4.

Ligands from the Astex-85 benchmarks that redock with good pose fidelity and good rank (top 5% of property matched decoys) with all parameter sets used. (A) Nuclear hormone receptors (B) kinases (C) other enzymes.
Five targets achieved good poses with all parametrizations, yet the ligand never ranked well (Figure 5 and http://data.docking.org/2009/AstexTables.doc, class 4): 1IA1, 1J3J, 1TZ8, 1XM6, and 1XOQ. These cases are interesting because pose fidelity is often used as a metric for docking success. Poor ranks despite good poses can arise when either the ligand scores unexpectedly poorly or the decoys score unexpectedly well. For instance, many decoys for 1IA1, dihydrofolate reductase (DHFR), contained guanidinium and other groups that might well bind to DHFR and in turn received excellent scores. Because the ligands looked competitive to us, we attribute the failure to achieve a good rank as being due to highly competitive decoys rather than a failure of the ligand itself, as it also received a good score. Future work will investigate whether the decoy selection procedure might be improved to remove viable-looking ligands that are not currently eliminated by our fingerprint-based dissimilarity metric.
Figure 5.

Ligands from the Astex-85 benchmarks that redock with good pose fidelity but poor rank compared to property-matched decoys.
Failure to achieve even a good pose against perhaps 20−30% of targets is common in docking studies, so we were not much surprised to note 31 targets where the ligand did not dock even close (within 3 Å rmsd) to a satisfactory pose (Figure 6 and http://data.docking.org/2009/AstexTables.doc, class 6). Many of these pose fidelity failures could be attributed to one of four common causes: (a) insufficient conformational sampling of the ligand, particularly of aliphatic ring puckering, (b) symmetric or pseudosymmetric molecule, (c) critical missing water molecules, and (d) ligand pose dominated by electronic (orbital) effects. These issues are common to all docking methods and protocols and are therefore expected for this study. In the scripted, automatic context of DOCK Blaster, some of these problems could be addressed, for instance by using a rescoring method to allow for relaxation of ring puckering or an approach that samples explicit water molecule positions.(50) Other failures, such as the plausible reversed docking pose of a pseudosymmetrical molecule in 1YV3, could be better addressed using a density-based pose-assessment method.(51) Some failures are probably simply beyond the capabilities of an orbital-naïve method like DOCK, such as 1P2Y, a P450, where the ligand pose appears to be under electronic rather than steric control. Whereas some of these failures may be addressed by improvements in the protocol, we worry about a tendency to build in too much “expertise” that leads to over fitting. Some of these problem targets will likely remain beyond the capability of full automation, at least in the near term.
Figure 6.

Ligands from the Astex-85 benchmark that do not achieve good pose fidelity under any circumstances.
With four parameter sets to choose from, we wondered whether one of them was more successful at achieving good pose fidelity than the others. To answer this, we plotted the frequency and cumulative frequency of pose fidelity for the best parameter set for each case (Figure 7) and also for each parameter set separately (). We found that all parameter sets achieved a pose within 2 Å rmsd at least 50% of the time. The “finer” schemes performed slightly better at 60%. These graphs confirm that all parameter sets are roughly comparable at achieving good ligand poses, that each is best some of the time, and none of the parametrizations is either much better or much worse than average.
Figure 7.

Histogram and cumulative frequency of pose fidelity (Å, rmsd) using the best parameter set. Astex-85 benchmark.
We wondered whether better pose fidelity could be used to predict rank. If it were, good pose fidelity as measured by rmsd could be used to select the best docking parameters. We plotted pose fidelity (Å, rmsd) versus rank using the parameters that led to the best pose fidelity in each case (Figure 8) and also for each parameter set separately (). Poor pose fidelity was generally associated with poorly ranked compounds, although there was one notable exception, beta II tryptase (2BM2), where the ligand docked backward but still managed to get a good score compared to its decoys. However, good pose fidelity was entirely uncorrelated with rank, regardless of which parameter set was used. It would thus appear that pose fidelity cannot be used to predict rank and thus whether the model is suitable for prospective docking.
Figure 8.

Plot of pose fidelity (Å, rmsd) versus % rank compared to about 100 property matched decoys (log scale). For Astex-85 benchmark. In each case, the parameter set that gave the best pose fidelity was used.
We were curious about whether ligand rank was sensitive to the scoring function used. To explore this, we rescored the ligand and all its decoys as docked using DOCK with four widely used, different scoring functions (Table 2 and http://data.docking.org/2009/RescoringTables.doc), in each case without any change to the receptor or ligand structure. Three of the scoring functions ranked the ligand substantially higher compared to the decoys, resulting in between 4 and 9 more successes. Only PMF resulted in fewer successes than DOCK.
GOLD-114 Benchmarking
Because we used the Astex-85 set to tune our protocol and parameter choices, we worried about overfitting.(52) To control for protocol bias, we turned to the GOLD-114 benchmark(15) for which we did not allow ourselves to adjust the automatic procedure or its parameters in any way. A total of 75 targets were successfully separated into ligand and receptor automatically from the PDB code alone (), and a further 19 cases (94 total) could be run if the ligand three letter code was also specified. Of the 94 calculations that started automatically, all but two yielded a docked pose and score for the ligand. Of the 20 targets that failed to produce a docked ligand, three failed during ligand identification and separation, five failed to prepare the ligand for docking, and 12 tried but failed to dock and score the ligand.
We asked how well automatic docking could recapitulate the crystal structure when the docking protocol could not be modified. Of the 92 that yielded a docked and scored ligand, 58 posed within 2 Å rmsd of the crystal structure ( and http://data.docking.org/2009/GOLDTables.doc). All parameter sets performed nearly equivalently, and no single one was as successful as combining the best result from all four. When the docked pose was within 2 Å rmsd, 27 of these also managed to rank in the top 5% of about 100 property matched decoys (Table 1 and http://data.docking.org/2009/GOLDTables.doc).
DUD Benchmarking
Worrying about depending on a single positive control as the sole basis for judging docking performance, we turned to a benchmark for which more actives were available. We used DOCK Blaster to dock to targets in the DUD-38 benchmark(27) for which the ligand pose was crystallographically observed. All targets could be handled by the parser and submitted to the DOCK Blaster queue. One ligand failed to be converted to SMILES automatically (1CKP), but the rest of the 37 targets completed normally yielding a redocked and scored ligand.
We began by assessing performance as before using a single crystallographic ligand and its automatically generated decoys, asking how well automatic docking could recapitulate the crystal structure. Of the 37 protein−ligand pairs that yielded a docked ligand score, 23 posed within 2 Å rmsd of the crystallographic ligand ( and http://data.docking.org/2009/DUDTables.doc). When the docked pose was within 2 Å rmsd, 15 of these also ranked the ligand in the top 5% of about 100 property matched decoys (Table 1 and http://data.docking.org/2009/DUDTables.doc).
As part of DUD, these targets benefited from between 20 and 600 additional annotated actives and their corresponding property-matched decoys with which to test DOCK Blaster. We began by checking that the fully automatic docking results using DOCK Blaster were at least comparable to the expert docking reported in the DUD paper (Figure 9 and ). If automatic docking were consistently different from an expert, the conclusions of this study could be undermined. For instance, an expert can experiment with the placement of specific water molecules, modification of receptor parameters, or docking parameters and do so repeatedly until the results converge. Whereas fully automated docking often produced results somewhat inferior to an expert, we thought it was close in all but five cases.
Figure 9.

Receiver operator characteristic (ROC) plots comparing enrichment by DOCK Blaster (magenta) versus an expert (dark blue) against four targets from the DUD benchmark. (A) COX-2 (B) RXR alpha (C) ER-agonist (D) SAHH. Random enrichment shown by a thin gray line.
A critical question was how predictive the single molecule metric used thus far was of the multimolecule DUD metric (Table 3 and http://data.docking.org/2009/). For simplicity we rated each metric as “good” or “bad” to indicate whether the docking results looked compelling enough to use for a discovery project. For the single ligand metric, “good” meant both pose fidelity within 2 Å rmsd and rank strictly in the top 5% of the property matched decoys. For the multiligand (DUD) metric, “good” meant that either the adjusted logAUC(53) was greater than 10, or that EF1, the fraction of ligands found by the first percentile, was greater than 10%, or that EF5 was greater than 20%. Our results show that in 26 of 36 or 72% of cases where automatic docking completed normally, the single ligand metric of docking success predicted the multiligand assessment. This result allows us to deploy the single ligand metric as a reasonable predictor of overall enrichment.
Table 3. Correlation of Docking Success Assessment between the Single Molecule Metric and Multiple Molecule Metric for the DUD-38 Benchmark. a.
| single molecule metric |
||
|---|---|---|
| multiple molecule metric (DUD) |
good | bad |
| good | 12 | 6 |
| bad | 4 | 14 |
Success for the single molecule metric has pose fidelity within 2 Å RMSD and rank in the top 5% of property-matched decoys. Success for the DUD metric is adjusted logAUC > 10 or EF1 > 10% or EF5 > 20%. Diagonal: 26. Off diagonal: 10. Automatic docking failure: 2. Correlation: 72%.
PDB Benchmarking
A common criticism of docking is that it often seems to encounter unforeseen problems with each new target, and correspondingly one never knows in advance whether docking will “work”. Equipped with a fully automated system and single-ligand based assessment tools, we turned to a larger set of targets to investigate the prevalence of situations that are not well handled by our scripts. Of the 9050 eligible PDB targets submitted to the parser, 7755 produced a docked ligand structure that could be scored and ranked (Table 4). Of these, 3056 had the ligand redock within 2 Å rmsd of the crystal structure (http://data.docking.org/2009/PDB3056.xls). About 100 property-matched decoys were generated automatically for each ligand, docked, and used to calculate the rank of the redocked ligand using each of four parameter sets. In 1398 of these cases, the ligand also scored in the top 5%, representing 18% of the 7755 targets that were viable for automatic docking.
Table 4. Attrition of PDB Structures Subjected to Fully Automated Docking Using DOCK Blastera.
| count | as % of previous row |
|
|---|---|---|
| ligand identified, DOCK Blaster job started | 9050 | |
| ligand docked and scored | 7755 | 86 |
| pose good (<2 Å rmsd) | 3056 | 39 |
| may be useful prospectively (<2 Å rmsd; rank top 5%) | 1398 | 46 |
Aug 1, 2008 version, of the PDB having 52000 x-ray crystal structures in total.
Discussion
Recognizing the potential benefit of automated high throughput docking to interpret the growing number of protein structures, we have developed an automatic docking system and assessed its performance in retrospective tests. Five results emerge from this study. First, DOCK Blaster, an automatic docking system, is now available for free public use and can produce useful results starting with as little as a PDB code. Second, useful results as judged by good pose fidelity and rank compared to property matched decoys were achieved in around 30% of cases against common benchmarks. Third, surprisingly, pose fidelity as measured by rmsd is not predictive of rank and is therefore not a useful metric of docking success by itself.(54) Fourth, a single ligand metric using the crystallographic ligand is predictive of the multiligand enrichment over property-matched decoys 72% of the time. Finally, DOCK Blaster has been deployed on a large scale and produced screening results that deserve further consideration for experimental testing in 1398 of 7755 cases drawn from the PDB.
The result that will have perhaps the greatest pragmatic impact is the creation of the DOCK Blaster server itself. Because DOCK Blaster can start from as little as a PDB code, can produce results like an expert in some cases, and can self-evaluate, it is suitable for use by nonexperts and for large-scale experiments. When docking performs poorly or fails completely, this can usually be deduced automatically from failures in pose fidelity and rank-to-decoys statistics. Thus the prospective user of this system can estimate whether the docking results are likely to be useful for discovery, should be ignored completely, or perhaps fall somewhere in between. Of course, any system of this complexity is bound to have flaws, and some projects will simply not work. Still, it is our hope that DOCK Blaster will be useful for nonexperts, lowering barriers to entry to the field.
Pose fidelity using DOCK Blaster in retrospective studies is not quite as good as when performed by an expert. Our black-box system achieves pose fidelity within 2 Å rmsd for about 50−60% of targets, compared to 70−80% for expert-guided docking. Perhaps surprisingly, pose fidelity as measured by rmsd deviation did not correlate at all with rank. For enrichment, again our results are not quite as good as reported in other available studies. This should be unsurprising, however, because experts are at liberty to manually curate sites, ligands, and protocols to maximize performance. Useful results, i.e., good pose fidelity and good enrichment, were obtained in 25−40% of benchmark cases. The success rates for Astex-85 was 29 out of 85, for GOLD-114 it was 27 out of 114, and for DUD-38 15 out of 38 were successful for both pose-fidelity and enrichment. Whereas this might not seem a very high success rate, no study has ever held itself to such a stringent standard before, and this was only possible with an automated program.
As a basis for performance assessment, a single ligand with generated property-matched decoys predicts the multiligand assessment three times out of four. Because only one ligand is needed, this has significant advantages for automation, enabling automatic self-assessment of docking results, which would be impractical if a list of actives had to be assembled to benchmark each target. The correlation was largely insensitive to the precise cutoffs used to characterize docking success or to which scoring function was used. Of course, this result is provisional, as it is based on the DUD-38 benchmark and may be revised as larger benchmarks appear.
DOCK Blaster has been deployed on a near-proteome scale, and produced useful results for 1398 of 7755 targets or 18% of the PDB structures that were amenable to this method. Because docking can recapitulate what is already known about the site in these cases, there is a reasonable hope for prospective docking to suggest ligands that might actually bind. Many of these problems were previously known (conformational sampling, ligand representation, structural waters), but their prevalence is now better quantified.
There is a critical need to find new small molecule reagents for biology. At the same time, a rapidly growing backlog of uninterpreted structures has accumulated in the PDB. Yet in an age of plentiful target structures, freely available small molecule databases, many sophisticated docking programs, and fast computers to run them on, many biologically oriented investigators still find docking for ligand discovery daunting. We have developed DOCK Blaster to help nonspecialists find new reagents for biology without the need for an expert. As for experts, DOCK Blaster does not produce compelling results for all targets. But for the nearly 20% of targets we tried where it does produce results deserving of further investigation, DOCK Blaster offers a way to automatically leverage structure for ligand discovery.
Acknowledgments
This work is supported by NIH GM71896 (to J.J.I. and B.K.S.) and the Buchheit Family Foundation. We thank OpenEye Scientific Software (Santa Fe, NM) for the use of Omega, OEChem, Vida, Ogham, QuacPAC, and other tools. We thank Schrodinger Inc. (New York, NY) for the use of Ligprep and Epik, Dr. Peter Ertl for the Java Molecular Editor, molinspiration.com for the use of mitools, Dr. Martin Stahl for the SCORE program, and Dr. W. D. Ihlenfeldt of Xemistry GmbH (Germany) for Catvs. F.C. was supported by the University of Bologna and a Marco Polo fellowship. We are grateful to Drs. Austin Kirschner, Ruth Brenk, Binqing Wei, and David Lorber for scripts. We thank our early users for helpful feedback and Gabriel Rocklin, Dr. Oliv Eidam and Dr. Peter Kolb for reading the manuscript.
Supporting Information Available
Additional figures and tables documenting the performance of DOCK Blaster. This material is available free of charge via the Internet at http://pubs.acs.org. Additional Supporting Information is available at http://data.docking.org/2009/.
Funding Statement
National Institutes of Health, United States
Footnotes
Abbreviations: rmsd, root mean squared deviation; SAR, structure−activity relationship; PDB, Protein Data Bank; ROC, receiver operator characteristic.
Supplementary Material
References
- Shoichet B. K. Virtual screening of chemical libraries. Nature 2004, 432, 862–865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoichet B. K.; McGovern S. L.; Wei B.; Irwin J. J. Lead discovery using molecular docking. Curr. Opin. Chem. Biol. 2002, 6, 439–446. [DOI] [PubMed] [Google Scholar]
- Bottegoni G.; Kufareva I.; Totrov M.; Abagyan R. Four-dimensional docking: a fast and accurate account of discrete receptor flexibility in ligand docking. J. Med. Chem. 2009, 52, 397–406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis I. W.; Baker D. RosettaLigand docking with full ligand and receptor flexibility. J. Mol. Biol. 2009, 385, 381–392. [DOI] [PubMed] [Google Scholar]
- Jain A. N. Effects of protein conformation in docking: improved pose prediction through protein pocket adaptation. J. Comput.-Aided Mol. Des. 2009, 23, 355–374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorgensen W. L. Efficient drug lead discovery and optimization. Acc. Chem. Res. 2009, 42, 724–733. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartshorn M. J.; Verdonk M. L.; Chessari G.; Brewerton S. C.; Mooij W. T.; Mortenson P. N.; Murray C. W. Diverse, high-quality test set for the validation of protein−ligand docking performance. J. Med. Chem. 2007, 50, 726–741. [DOI] [PubMed] [Google Scholar]
- Zhao Y.; Sanner M. F. FLIPDock: docking flexible ligands into flexible receptors. Proteins 2007, 68, 726–737. [DOI] [PubMed] [Google Scholar]
- Luksch T.; Chan N. S.; Brass S.; Sotriffer C. A.; Klebe G.; Diederich W. E. Computer-aided design and synthesis of nonpeptidic plasmepsin II and IV inhibitors. ChemMedChem 2008, 3, 1323–1336. [DOI] [PubMed] [Google Scholar]
- Cho Y.; Ioerger T. R.; Sacchettini J. C. Discovery of novel nitrobenzothiazole inhibitors for Mycobacterium tuberculosis ATP phosphoribosyl transferase (HisG) through virtual screening. J. Med. Chem. 2008, 51, 5984–5992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiss R.; Kiss B.; Konczol A.; Szalai F.; Jelinek I.; Laszlo V.; Noszal B.; Falus A.; Keseru G. M. Discovery of novel human histamine H4 receptor ligands by large-scale structure-based virtual screening. J. Med. Chem. 2008, 51, 3145–3153. [DOI] [PubMed] [Google Scholar]
- Cavasotto C. N.; Orry A. J.; Murgolo N. J.; Czarniecki M. F.; Kocsi S. A.; Hawes B. E.; O'Neill K. A.; Hine H.; Burton M. S.; Voigt J. H.; Abagyan R. A.; Bayne M. L.; Monsma F. J. Jr. Discovery of novel chemotypes to a G-protein-coupled receptor through ligand-steered homology modeling and structure-based virtual screening. J. Med. Chem. 2008, 51, 581–588. [DOI] [PubMed] [Google Scholar]
- Eswar N.; John B.; Mirkovic N.; Fiser A.; Ilyin V. A.; Pieper U.; Stuart A. C.; Marti-Renom M. A.; Madhusudhan M. S.; Yerkovich B.; Sali A. Tools for comparative protein structure modeling and analysis. Nucleic Acids Res. 2003, 31, 3375–3380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul S. F.; Gish W.; Miller W.; Myers E. W.; Lipman D. J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [DOI] [PubMed] [Google Scholar]
- Moustakas D. T.; Lang P. T.; Pegg S.; Pettersen E.; Kuntz I. D.; Brooijmans N.; Rizzo R. C. Development and validation of a modular, extensible docking program: DOCK 5. J. Comput.-Aided Mol. Des. 2006, 20, 601–619. [DOI] [PubMed] [Google Scholar]
- Nissink J. W.; Murray C.; Hartshorn M.; Verdonk M. L.; Cole J. C.; Taylor R. A new test set for validating predictions of protein−ligand interaction. Proteins 2002, 49, 457–471. [DOI] [PubMed] [Google Scholar]
- Jones G.; Willett P.; Glen R. C.; Leach A. R.; Taylor R. Development and validation of a genetic algorithm for flexible docking. J. Mol. Biol. 1997, 267, 727–748. [DOI] [PubMed] [Google Scholar]
- Verdonk M. L.; Berdini V.; Hartshorn M. J.; Mooij W. T.; Murray C. W.; Taylor R. D.; Watson P. Virtual screening using protein−ligand docking: avoiding artificial enrichment. J. Chem. Inf. Comput. Sci. 2004, 44, 793–806. [DOI] [PubMed] [Google Scholar]
- Kellenberger E.; Rodrigo J.; Muller P.; Rognan D. Comparative evaluation of eight docking tools for docking and virtual screening accuracy. Proteins 2004, 57, 225–242. [DOI] [PubMed] [Google Scholar]
- Perola E.; Walters W. P.; Charifson P. S. A detailed comparison of current docking and scoring methods on systems of pharmaceutical relevance. Proteins 2004, 56, 235–249. [DOI] [PubMed] [Google Scholar]
- Ferrara P.; Gohlke H.; Price D. J.; Klebe G.; Brooks C. L. III. Assessing scoring functions for protein−ligand interactions. J. Med. Chem. 2004, 47, 3032–3047. [DOI] [PubMed] [Google Scholar]
- Kontoyianni M.; McClellan L. M.; Sokol G. S. Evaluation of docking performance: comparative data on docking algorithms. J. Med. Chem. 2004, 47, 558–565. [DOI] [PubMed] [Google Scholar]
- Wang R.; Lu Y.; Fang X.; Wang S. An extensive test of 14 scoring functions using the PDBbind refined set of 800 protein−ligand complexes. J. Chem. Inf. Comput. Sci. 2004, 44, 2114–2125. [DOI] [PubMed] [Google Scholar]
- Xing L.; Hodgkin E.; Liu Q.; Sedlock D. Evaluation and application of multiple scoring functions for a virtual screening experiment. J. Comput.-Aided Mol. Des. 2004, 18, 333–344. [DOI] [PubMed] [Google Scholar]
- Onodera K.; Satou K.; Hirota H. Evaluations of molecular docking programs for virtual screening. J. Chem. Inf. Model. 2007, 47, 1609–1618. [DOI] [PubMed] [Google Scholar]
- Zhou Z.; Felts A. K.; Friesner R. A.; Levy R. M. Comparative performance of several flexible docking programs and scoring functions: enrichment studies for a diverse set of pharmaceutically relevant targets. J. Chem. Inf. Model. 2007, 47, 1599–1608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N.; Shoichet B. K.; Irwin J. J. Benchmarking sets for molecular docking. J. Med. Chem. 2006, 49, 6789–6801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorber D. M.; Shoichet B. K. Flexible ligand docking using conformational ensembles. Protein Sci. 1998, 7, 938–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorber D. M.; Shoichet B. K. Hierarchical docking of databases of multiple ligand conformations. Curr. Top. Med. Chem. 2005, 5, 739–749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tong W.; Wei Y.; Murga L. F.; Ondrechen M. J.; Williams R. J. Partial order optimum likelihood (POOL): maximum likelihood prediction of protein active site residues using 3D Structure and sequence properties. PLoS Comput. Biol. 2009, 5, e1000266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weisel M.; Proschak E.; Kriegl J. M.; Schneider G. Form follows function: shape analysis of protein cavities for receptor-based drug design. Proteomics 2009, 9, 451–459. [DOI] [PubMed] [Google Scholar]
- Laurie A. T.; Jackson R. M. Methods for the prediction of protein−ligand binding sites for structure-based drug design and virtual ligand screening. Curr. Protein Pept. Sci. 2006, 7, 395–406. [DOI] [PubMed] [Google Scholar]
- Pearlman D. A.; Case D. A.; Caldwell J. W.; Ross W. S.; Cheatham T. E. III; Debolt S.; Ferguson D.; Seibel G.; Kollman P. AMBER, a package of computer programs for applying molecular mechanics, normal mode analysis, molecular dynamics and free energy calculations to simulate the structural and energetic properties of molecules . Comput. Phys. Commun. 1995, 91, 1–41. [Google Scholar]
- Kuntz I. D.; Blaney J. M.; Oatley S. J.; Langridge R.; Ferrin T. E. A geometric approach to macromolecule−ligand interactions. J. Mol. Biol. 1982, 161, 269–288. [DOI] [PubMed] [Google Scholar]
- Meng E. C.; Shoichet B. K.; Kuntz I. D. Automated docking with grid-based energy evaluation. J. Comput. Chem. 1992, 13, 505–524. [Google Scholar]
- Nicholls A.; Honig B. A Rapid Finite-Difference Algorithm, Utilizing Successive Over-Relaxation to Solve the Poisson−Boltzmann Equation. J. Comput. Chem. 1991, 12, 435–445. [Google Scholar]
- Shoichet B. K.; Mysinger M. M.. unpublished results,2008.
- OpenEye OEChem 1.6.1; OpenEye Scientific Software: Santa Fe, NM; www.eyesopen.com.
- Irwin J. J.; Shoichet B. K. ZINC—a free database of commercially available compounds for virtual screening. J. Chem. Inf. Model. 2005, 45, 177–182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mysinger M. M.Unpublished automation and refinement of the DUD protocol,2009.
- Powers R. A.; Morandi F.; Shoichet B. K. Structure-based discovery of a novel, noncovalent inhibitor of AmpC beta-lactamase. Structure (Cambridge, MA, U.S.) 2002, 10, 1013–1023. [DOI] [PubMed] [Google Scholar]
- Doman T. N.; McGovern S. L.; Witherbee B. J.; Kasten T. P.; Kurumbail R.; Stallings W. C.; Connolly D. T.; Shoichet B. K. Molecular Docking and High-Throughput Screening for Novel Inhibitors of Protein Tyrosine Phosphatase-1B. J. Med. Chem. 2002, 45, 2213–2221. [DOI] [PubMed] [Google Scholar]
- Babaoglu K.; Simeonov A.; Irwin J. J.; Nelson M. E.; Feng B.; Thomas C. J.; Cancian L.; Costi M. P.; Maltby D. A.; Jadhav A.; Inglese J.; Austin C. P.; Shoichet B. K. A Comprehensive Mechanistic Analysis of Hits from High-Throughput and Docking Screens Against β-Lactamase. J. Med. Chem. 2008, 51, 2502–2511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kolb P.; Rosenbaum D. M.; Irwin J. J.; Fung J. J.; Kobilka B. K.; Shoichet B. K. Structure-based discovery of {beta}2-adrenergic receptor ligands. Proc. Natl. Acad. Sci. U.S.A. 2009, 106, 6843–6848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delano W.The PyMol Molecular Graphics System.
- Pettersen E. F.; Goddard T. D.; Huang C. C.; Couch G. S.; Greenblatt D. M.; Meng E. C.; Ferrin T. E. UCSF Chimera—a visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [DOI] [PubMed] [Google Scholar]
- Gezelter D.Jmol: an open-source Java viewer for chemical structures in 3D. http://www.jmol.org.
- Berman H. M.; Westbrook J.; Feng Z.; Gilliland G.; Bhat T. N.; Weissig H.; Shindyalov I. N.; Bourne P. E. The Protein Data Bank. Nucl. Acid. Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henrick K.; Feng Z.; Bluhm W. F.; Dimitropoulos D.; Doreleijers J. F.; Dutta S.; Flippen-Anderson J. L.; Ionides J.; Kamada C.; Krissinel E.; Lawson C. L.; Markley J. L.; Nakamura H.; Newman R.; Shimizu Y.; Swaminathan J.; Velankar S.; Ory J.; Ulrich E. L.; Vranken W.; Westbrook J.; Yamashita R.; Yang H.; Young J.; Yousufuddin M.; Berman H. M. Remediation of the protein data bank archive. Nucleic Acids Res. 2008, 36, D426–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang N.; Shoichet B. K. Exploiting ordered waters in molecular docking. J. Med. Chem. 2008, 51, 4862–4865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yusuf D.; Davis A. M.; Kleywegt G. J.; Schmitt S. An alternative method for the evaluation of docking performance: RSR vs RMSD. J. Chem. Inf. Model. 2008, 48, 1411–1422. [DOI] [PubMed] [Google Scholar]
- Irwin J. J. Community benchmarks for virtual screening. J. Comput.-Aided Mol. Des. 2008, 22, 193–199. [DOI] [PubMed] [Google Scholar]
- Mysinger M. M.Adjusted LogAUC - the aggregate percentage area between the ROC curve and random curve when plotted on a log scale from 0.1 to 100%.
- Graves A. P.; Brenk R.; Shoichet B. K. Decoys for docking. J. Med. Chem. 2005, 48, 3714–3728. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.