

Abstract
Neuropsychiatric disease in systemic lupus erythematosus (NPSLE)3 is a poorly understood, but potentially fatal, disease manifestation. A pathogenetic role for autoantibodies is suspected, but the mechanism is unclear. Since immune complexes in SLE can stimulate IFN-α and there is strong evidence in humans and in mice that IFN-α can cause neuropsychiatric manifestations, we asked whether NPSLE patient serum and/or cerebrospinal fluid (CSF) contain abnormally high IFN-α-inducing activity. In a bioassay containing plasmacytoid dendritic cells and a source of Ag, NPSLE CSF induced significantly higher IFN-α compared with CSF from patients with multiple sclerosis or other autoimmune disease controls. When normalized for IgG concentration, NPSLE CSF was 800-fold more potent at inducing IFN-α compared with paired serum due to inhibitors present in serum. Analysis of Ig-deficient patient serum, depletion of IgG from normal serum, as well as addition of purified IgG to NPSLE CSF and serum in the bioassays revealed that one inhibitor was contained within the IgG fraction itself. In addition to IFN-α, immune complexes formed by CSF autoantibodies produced significantly increased levels of IFN-γ-inducible protein 10 (IP-10/CXCL), IL-8, and MCP-1, all of which have been reported to be elevated in CSF from NPSLE patients. Taken together, these findings are consistent with a two-step model of NPSLE whereby CSF autoantibodies bind to Ags released by neurocytotoxic Abs or other brain cell injury, and the resulting immune complexes stimulate IFN-α and proinflammatory cytokines and chemokines.
Neuropsychiatric disease in systemic lupus erythematosus (NPSLE)3 remains a common, yet poorly understood manifestation of SLE. Neuropsychiatric manifestations vary from mild cognitive defects to life-threatening impairment of brain function. The striking deficit of CNS pathology, specifically the lack of vasculitis or massive cellular infiltrate in patients dying of CNS lupus (1, 2), suggests that the pathogenesis differs from immune complex deposition that is characteristic of lupus glomerulonephritis and vasculitis at other sites. Despite the absence of immune complex vasculitis, there is a large body of inferential data to support a possible pathogenetic role for autoantibodies in NPSLE. These data include reported associations between NPSLE and specific autoantibodies (lymphocytotoxic, anti-neuronal, anti-ribosomal P (anti-P) and anti-N-methyl-d -aspartate receptor (NMDAR) (reviewed in Ref. 3), evidence for intrathecal IgG synthesis (4), and low complement in the cerebrospinal fluid (CSF) of patients with NPSLE (5). Despite reports of associations between specific autoantibodies and CNS manifestations, serum levels of these autoantibodies have not consistently shown a high degree of sensitivity and specificity for NPSLE.
In addition to autoantibodies, several proinflammatory cytokines and chemokines have been detected in the CSF of patients with NPSLE, although the mechanism responsible for stimulation of these inflammatory mediators has not been identified (6–11). One cytokine of considerable interest in SLE is IFN-α. Despite the difficulty in detection of IFN-α in SLE serum, mRNA expression studies in circulating SLE PBMC revealed evidence for exposure to type 1 IFN in ∼50% of patients (reviewed in Ref. 12). In some studies, SLE patients with renal or CNS disease had the highest IFN signature (12). IFN-α generation in SLE is caused, at least in part, by autoantibodies that bind to nucleoprotein particles released from dead and dying cells. These immune complexes are endocytosed by FcγR on plasmacytoid dendritic cells (pDCs) and induce IFN production by nucleic acid activation of TLRs (reviewed in Refs. 13, 14).
The observation that some patients receiving IFN-α as therapy for either cancer or hepatitis C virus infection develop SLE and other autoimmune disorders (15, 16) indicates that IFN-α may promote SLE symptomatology. Approximately a third of patients receiving IFN-α therapy develop CNS manifestations. Depression is most common, but psychotic features, manic/hypomanic episodes, confusion, focal neurological deficits, and seizures are also observed (reviewed in Ref. 17). Further emphasizing a possible link between intrathecal IFN-α and neuropsychiatric disease, patients with Aicardi-Goutières syndrome develop mental retardation, cerebral atrophy, calcification of the basal ganglia associated with CSF lymphocyte pleocytosis, and increased levels of CSF IFN-α (18). Recent studies reveal that most cases of this syndrome are caused by mutations in DNA/RNA editing or repair enzymes, presumably leading to excess DNA or DNA/RNA hybrids stimulating the production of IFN-α (19, 20). Finally, selective overexpression of IFN-α in the CNS in a transgenic mouse model convincingly demonstrated that local IFN-α production induces many NPSLE-like features (21). Mice that expressed extremely high levels of IFN-α in the CNS developed seizures and severe behavioral disturbances with high mortality, whereas mice with lower IFN-α expression exhibited more subtle learning disabilities. These findings suggest that different clinical manifestations may be observed depending on the concentration of intrathecal IFN-α.
To explore whether serum or CSF from patients with NPSLE contained unusually high interferogenic (IFG) activity (the ability of serum or CSF to induce IFN-α in the presence of an IFN-producing cell), we compared IFG activity in patients with NPSLE and controls. We found significantly higher IFG activity in the CSF, but not the serum, of NPSLE patients. The average IFG activity in NPSLE patient CSF was 800-fold higher than paired serum, which was, in part, explained by an inhibitory effect of serum factors such as IgG. Furthermore, CSF autoantibodies in the presence of cellular Ags and the appropriate responder cells could induce most of the cytokines (IFN-γ-inducible protein 10 (IP-10), IL-8, IL-6, and MCP-1) previously detected in NPSLE patients' CSF.
Materials and Methods
Reagents
Affinity-purified ribosomal P protein was obtained from Arotec Diagnostics. Poly(I:C) was purchased from InvivoGen. CpG 2216 was synthesized by Invitrogen (5′-ggGGGACGATCGTCgggggG-3′; g indicates phosphorothioate linkage). Human purified IgG was obtained from Jackson ImmunoResearch Laboratories and had <0.06 EU/ml by Limulus amebocyte lysate clot assay (Associates of Cape Cod) after Triton X-100 treatment. mAb to IFN-α was from PBL Biomedical Laboratories, and control mouse IgG1 was from eBioscience. Human IFN-β was obtained from the National Institute of Allergy and Infectious Diseases Reference Reagent Repository (operated by KamTek).
Patients
All SLE patients fulfilled the American College of Rheumatology 1982 revised criteria for the classification of SLE (22), and the diagnosis of NPSLE was based on the case definition studies of the 19 NPSLE syndromes proposed by the American College of Rheumatology that also include exclusion criteria (Ref. 23 and the appendix contained therein). The clinical and serological features of the NPSLE patients are described in Table I. NPSLE and other autoimmune disease controls (OAID) were patients hospitalized in Jichi Medical University Hospital: 22 patients with NPSLE (21 women, 1 man; mean age ± SD, 32.9 ± 13.7 years), 12 patients with SLE and no CNS manifestations (6 women, 6 men; mean age ± SD, 39.5 ± 15.1 years), and 17 OAID (13 women, 4 men; mean age ± SD, 49.8 ± 18.1 years) with CNS symptoms. OAID CSF samples (numbers in parentheses) were from patients with dermatomyositis (1), adult-onset Still's disease (1), rheumatoid arthritis (2), periarteritis nodosa (1), vasculitis (2), Sjögren's syndrome (4), sarcoidosis (1), Behçet's syndrome (2), ulcerative colitis (1), antiphospholipid syndrome (1), or polymyalgia rheumatica (1). Serum and CSF were obtained at presentation and were stored at −70°C. Multiple sclerosis (MS) patient CSF (11 women, 13 men; mean age ± SD, 41.5 ± 9.8 years) was obtained from the Human Brain and Spinal Fluid Resource Center, Veterans Affairs West Los Angeles Healthcare Center, and from Richard Nash, Fred Hutchison Cancer Research Center (Seattle, WA). Serum from untreated patients with common variable immune deficiency (CVID, n = 3) and X-linked agammaglobulinemia (XLA, n = 1) were kindly provided by Charlotte Cunningham-Rundles, Mount Sinai School of Medicine (New York, NY), and Troy Torgerson, Seattle Children's Hospital (Seattle, WA). The concentrations of IgG in these sera ranged from <100 μg to 2.5 mg/ml. Normal CSF was purchased from Arotec Diagnostics. All samples were collected with the review board approval of the respective institutions. IgG was depleted from normal sera by incubation with protein A-Sepharose CL-4B (GE Healthcare Bio-Sciences) or immobilized protein G plus (Pierce Biotechnology) for 1 h at 4°C. Following depletion, residual IgG concentrations were 0.6–1 mg/ml.
Table I. Clinical and serological features of NPSLE+ patients.
| Patient No. |
Age/ Gender |
SLEDAI Score |
Drug Treatmenta (per Day) |
Neuropsychiatric Syndrome |
IFN-α Inducedb |
Anti-RNPc | Anti-Smc | Anti-Roc | Anti-Lac | Anti-dsDNAc | Anti-Ribo Pc |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | 38/F | 22 | None | Psychosis | + | − | − | + | + | + | + |
| 2 | 47/F | 26 | PSL 40 mg | Grand mal seizures | − | − | − | − | − | + | − |
| 3 | 31/F | 24 | BM 1.5 mg | Grand mal seizures, acute confusional state | + | − | + | − | − | + | + |
| 4 | 49/F | 11 | BM 1.5 mg | Mood disorder | + | − | − | + | + | + | − |
| 5 | 16/F | 11 | PSL 20 mg | Aseptic meningitis | ++ | − | + | + | + | − | − |
| 6 | 18/F | 18 | PSL 10 mg | Grand mal seizures, acute confusional state | − | + | + | + | − | + | + |
| 7 | 26/F | 7 | PSL 10 mg | Aseptic meningitis | + | + | + | + | + | + | + |
| 8 | 33/F | 12 | None | Mood disorder | ++ | + | + | + | + | − | − |
| 9 | 39/F | 16 | None | Anxiety disorder | ++ | − | − | + | − | + | + |
| 10 | 43/F | 12 | PSL 10 mg | Anxiety disorder | ++ | − | − | + | + | + | − |
| 11 | 16/F | 16 | BM 1.5 mg, Cs 75 mg | Acute confusional state | − | + | − | − | − | + | + |
| 12 | 27/F | 12 | PSL 45 mg | Acute confusional state | ++ | + | + | + | − | − | + |
| 13 | 26/F | 13 | None | Psychosis | + | − | + | + | + | + | − |
| 14 | 26/F | 17 | PSL 15 mg | Aseptic meningitis | + | + | + | − | − | + | + |
| 15 | 31/F | 24 | PSL 9 mg | Aseptic meningitis | + | + | − | − | − | + | − |
| 16 | 65/F | 21 | DM 2 mg | Acute confusional state | ++ | − | − | + | − | + | + |
| 17 | 36/M | 22 | None | Acute confusional state | − | − | − | + | − | + | + |
| 18 | 62/F | 25 | Spt, PSL 50 mg after therapy | Acute confusional state | ++ | bl | + | + | − | + | + |
| 19 | 23/F | 33 | None | Seizure disorders, acute confusional state | ++ | + | + | + | − | + | − |
| 20 | 23/F | 16 | Spt, PSL 100 mg | Seizure disorders, acute confusional state, cerebrovascular disease | ++ | bl | + | + | − | − | − |
| 21 | 29/F | 26 | Spt, PSL 40 mg after therapy | Psychosis | − | bl | bl | + | − | + | + |
| 22 | 18/F | 26 | Spt, PSL 5 mg, MZR 100 mg after therapy | Seizure disorders, acute confusional state | + | + | + | + | − | + | + |
PSL, prednisolone; BM, betamethasone; Cs, cyclosporine; DM, dexamethasone; Spt, steroid pulse therapy (methylprednisolone 0.5 g (patient 22) or 1 g (patients 18, 20, and 21)/day); MZR, mizoribine.
IFN-α induced after stimulation of IFN-primed PBMCs with 20% NPSLE+ CSF and nuclear extract (80 μg/ml): −, <100 pg/ml; +, 100−500 pg/ml; ++, >500 pg/ml.
Serum autoantibodies in patient samples: −, negative for autoantibody specificity; +, positive for autoantibody specificity; bl, baseline positive for autoantibody specificity.
Autoantibody and IgG ELISAs
CSF and serum anti-P levels were measured by ELISA using recombinant ribosomal P0 protein and purified ribosomal P protein (Arotec Diagnostics), respectively. Patient serum and CSF IgG were quantified using a sandwich ELISA using Abs and standards from Jackson ImmunoResearch Laboratories. Serum anti-Sm, anti-ribonucleoprotein (RNP), anti-SS-A, and anti-SS-B Ab levels were quantified using the MESACUP-2 test (Medical & Biological Laboratories) according to the manufacturer's instructions. Sera were considered positive for index levels of equal and more than 30 (anti-Sm), 22 (anti-RNP), 30 (anti-Ro/SS-A), 25 (anti-La/SS-B). Sm, RNP, Ro, and La levels in five patient sera were quantified using the QUANTA Plex ENA profile 5 kit (INOVA Diagnostics) according to the manufacturer's instructions. Measurements were comparable to results with MESACUP-2 when sera levels were quantified using both tests. dsDNA serum levels were determined by Recombigen ELISA anti-dsDNA kit (Mitsubishi Kagaku Iatron).
Preparation of responder cells and cell extracts
PBMC were prepared from healthy human donors using Ficoll-Paque density gradient centrifugation. pDCs were purified from PBMC by negative selection using a pDC isolation kit according to the manufacturer's instructions (Miltenyi Biotec), with purities >95% in each experiment. HeLa cell nuclear and ribosome extracts were made as described previously (24, 25). In certain experiments, freeze-thawed U937 cells (26) (1% (v/v)) were used as Ag, and similar levels of IFN-α were induced compared with nuclear extract (NE). HeLa and U937 cells were determined to be free of mycoplasma contamination using e-Myco Mycoplasma PCR detection kit (iNtRON Biotechnology) and extracts had <0.06 EU/ml endotoxin by Limulus amebocyte lysate clot assay (Associates of Cape Cod).
Preparation of primary human astrocytes and microglia
Cell cultures were prepared from brains of legally aborted human fetuses (12- to 15-wk gestation using the protocol of Satoh and Kim (27)). In brief, brain tissue freed from blood vessels and meninges was trypsinized, triturated with a fire-polished pipette, and washed in Hanks' buffer. The resulting cell suspension was cultured in DMEM supplemented with 5% horse serum, 100 U/ml penicillin, and 100 μg/ml streptomycin at 37°C in a 5% CO2/95% air incubator. For microglial cells, the mixed cultures were supplemented with 10 ng/ml GM-CSF (PeproTech). After 9–21 days, microglial cells were separated from the underlying astrocytic monolayer by gentle agitation using their differential adhesive properties. Microglia cultures routinely consist of >95% microglial cells as determined by Iba1 staining. The astrocytes were plated into poly-l -lysine-coated culture flasks at 6 × 106 cells/flask in DMEM supplemented as above with G5 supplement (Invitrogen, 1/100). Astrocyte purity assessed by glial fibrillary acidic protein staining was >90%. Freeze-thawed material was made by four cycles of freezing astrocytes at −70°C and thawing at 37°C and is referred to as a necrotic extract (26).
PBMC and microglia stimulation
Cells were plated in 96-well plates at 2.5 × 104 microglia/well or 5 × 105 PBMC/well in 125 μl with (primed) or without (unprimed) 500 U/ml universal type I IFN (IFN-α A/D; PBL Biomedical Laboratories) and 2 ng/ml GM-CSF in cell culture medium as described (28). Test serum or CSF samples were added at various dilutions with or without a source of autoantigen to cultured cells and supernatants were collected after 22–24 h. In some experiments, anti-CD32 (Serotec), blood DC Ag-2 (BDCA-2; Miltenyi Biotec), mouse IgG1 isotype control (R&D Systems), or bafilomycin A1 (Sigma-Aldrich) were added to PBMC 1 h before the addition of test samples and Ag. Cell extracts were treated with 8 μg/ml DNase-free RNase (Roche) or 500 U/ml RNase-free DNase (Sigma-Aldrich) for 3 h at 37°C or 15 min at room temperature, respectively, before incubation with PBMC and test samples. Potential toxicity of added components was monitored using the WST-1 assay (Calbiochem).
Cytokine detection
IFN-α levels in supernatants were quantified by an in-house ELISA using commercially available Abs and human IFN-α standard (PBL Biomedical Laboratories). Universal type I IFN used for priming PBMC/pDCs did not react with the Abs used for ELISA. The detection limit of the ELISA was 9.8 pg/ml. Other inflammatory mediators (IL-6, IL-8, IP-10, MCP-1, MIP-1α, TNF-α, IL-12p40, and IL-10) were measured in supernatants using a Milliplex human cytokine immunoassay kit (Millipore). In some assays, IL-8 and IP-10 were measured by ELISA kits from BioLegend and R&D Systems, respectively. Background readings given by NE alone were subtracted from test samples. IFN-α and IP-10 concentrations in patient CSF were quantified by a SearchLight custom multiplex protein array (Pierce Biotechnology, Inc.) with a sensitivity of 0.8 and 2.8 pg/ml, respectively.
Data presentation and analysis
Results below the level of detection were given the value of 1 rather than 0. Statistical significance between groups was determined by a Mann-Whitney U test, an unpaired t test, or a Wilcoxon signed-rank test. Correlations between variables were evaluated by Spearman's rank correlation test. A p value of <0.05 was considered significant. Graphs and statistical analyses were performed using Prism software (version 4, Graphpad Software) or SigmaStat (version 3.11, Systat Software).
Results
CSF from neuropsychiatric lupus (NPSLE+) patients induces high concentrations of IFN-α
Because most SLE sera contain multiple autoantibodies, and because apoptotic/necrotic cells would be expected to release nuclear as well as cytoplasmic Ags, we asked whether serum from NPSLE+ patients contained autoantibodies against nuclear Ags that would generate higher IFN-α compared with control NPSLE− sera. Since little or no IFN-α was induced in the absence of added Ag, all bioassays contained a source of Ag. As shown in Fig. 1A, when a NE was used as a source of Ags, most NPSLE+ serum generated high IFN-α, but levels were similar to NPSLE− serum (p > 0.05). Since anti-ribosomal P protein Abs (anti-P) have been associated with NPSLE (29), we quantified IFN-α following serum incubation with intact ribosomes. The higher frequency of anti-P in these patients' sera is explained by increased frequency of anti-P in Asian populations (30) and possible referral bias. In the presence of ribosomal Ag, most anti-P-containing sera (4 of 7 NPSLE−, 9 of 13 NPSLE+) induced IFN-α production >100 pg/ml. There was, however, no significant difference between IFN-α concentrations induced by NPSLE+ vs NPSLE− patients (Fig. 1B, p> 0.05).
Figure 1.
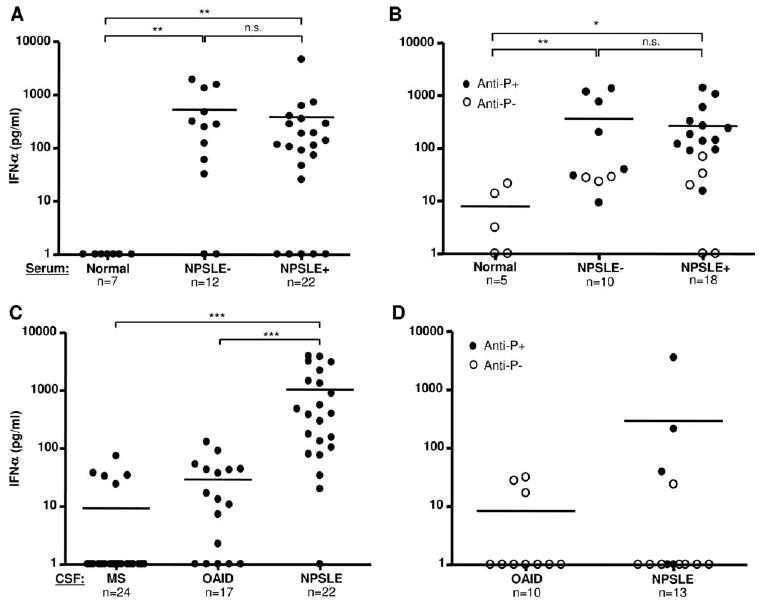
CSF from NPSLE+ patients induces high concentrations of IFN-α. A and B, Serum obtained from normal donors, NPSLE−, or NPSLE+ patients was incubated at a 1/10 dilution with HeLa NE (80 μg/ml) (A) or ribosomes (50 μg/ml) (B) in the presence of type I IFN-primed PBMC. After 24 h, IFN-α concentrations in the supernatants were measured by ELISA. C, CSF (1/5 dilution) from patients with MS, OAID, or NPSLE+ were incubated with NE (80 μg/ml) and added to IFN-primed PBMC as above (standard bioassay). D, CSF from patients with OAID or NPSLE+ was incubated with affinity-purified ribosomal P (10 μg/ml) and IFN-α concentrations were tested as above. IFN-α levels below the level of detection (<10 pg/ml) were given a value of 1. Anti-P+ or anti-P− refers to whether serum contains anti-ribosomal P autoantibodies. Horizontal lines represent mean values. n.s., not significant; *, p < 0.05; **, p < 0.01; ***, p < 0.001; Mann-Whitney U test.
Since serum autoantibodies may not accurately reflect autoantibody concentrations or activities in the CNS, and since IFN-α generation in CSF is much more relevant to the effects on the brain, we performed a similar analysis of IFG activity from the same NPSLE+ patients shown in Fig. 1A for whom paired CSF was available. When NE was used as a source of Ags, most (17 of 22 or 77%) NPSLE+ CSF induced >100 pg/ml IFN-α (Fig. 1C). Similar levels of IFN-α were observed when NPSLE+ CSF was incubated with necrotic extracts from primary human astrocytes (data not shown). Since we could not perform lumbar punctures on SLE patients without CNS symptoms, we selected two other control groups. MS is a disease of the brain and spinal cord characterized by inflammation of the white matter, is associated with the frequent presence of oligoclonal bands in the CSF, and has recently been shown to respond to B cell depletion (31). The second control group comprised patients with OAID with neuropsychiatric complications (see Materials and Methods). As shown in Fig. 1C, none of the CSF from MS patients (n = 24) and only 1 of 17 (5.9%) OAID controls induced >100 pg/ml IFN-α in the bioassay. The concentrations of IFN-α induced by NPSLE+ CSF were significantly greater than either MS or OAID control groups (p < 0.001). Only two NPSLE patients and no OAID patient generated IFN-α >100 pg/ml in CSF with affinity-purified ribosomal P protein (or intact ribosomes, data not shown) as Ags (Fig. 1D).
To determine whether there were clinical or serological correlates with CSF IFG activity in NPSLE+ patients, we evaluated clinical activity (SLE disease activity index (SLEDAI) score), therapy, and serum autoantibody specificities (Table I). There was no correlation between SLEDAI scores and IFG activity in NPSLE CSF (p > 0.05). Similarly, there was no statistically significant difference in IFG activity in patients on low (<20 mg/day) vs high (>20 mg/day) prednisolone or equivalent steroid dose of betamethasone or dexamethasone (p > 0.05). The small volumes of CSF obtained for this study precluded CSF autoantibody analysis. However, when correlations between specific serum autoantibody levels and IFG activity in CSF were examined, a correlation between anti-RNP levels and IFG activity was observed (p < 0.05), whereas correlations with anti-dsDNA, anti-Sm, anti-Ro, anti-La, and anti-ribosomal P levels did not reach statistical significance.
CSF autoantibodies induce IFN-α by pDCs through FcγRII engagement and endosomal trafficking of predominantly ribonucleoprotein Ags
Several studies with SLE serum have shown that immune complexes formed following incubation with cellular Ags are endocytosed by FcγRIIa (CD32a) on pDCs and stimulate IFN-α production most likely by engaging endosomal TLRs (reviewed in Ref. 13). Using both an agonistic mAb to BDCA-2 that results in suppression of IFN-α production specifically by pDCs (32) as well as direct addition of CSF to isolated pDCs, we verified that autoantibodies in the CSF directly stimulated IFN-α production by pDCs (Fig. 2, A and B). Similarly, blocking experiments with an anti-FcγRII Ab, exposure of Ags to nucleases, and exposing cells to bafilomycin A1, an inhibitor of the endosomal proton-translocating ATPase, revealed that the immune complexes formed by autoantibodies in CSF induced IFN-α through FcγRII-mediated endocytosis and activation of endosomal TLR7 by RNA in pDCs (Fig. 2).
Figure 2.
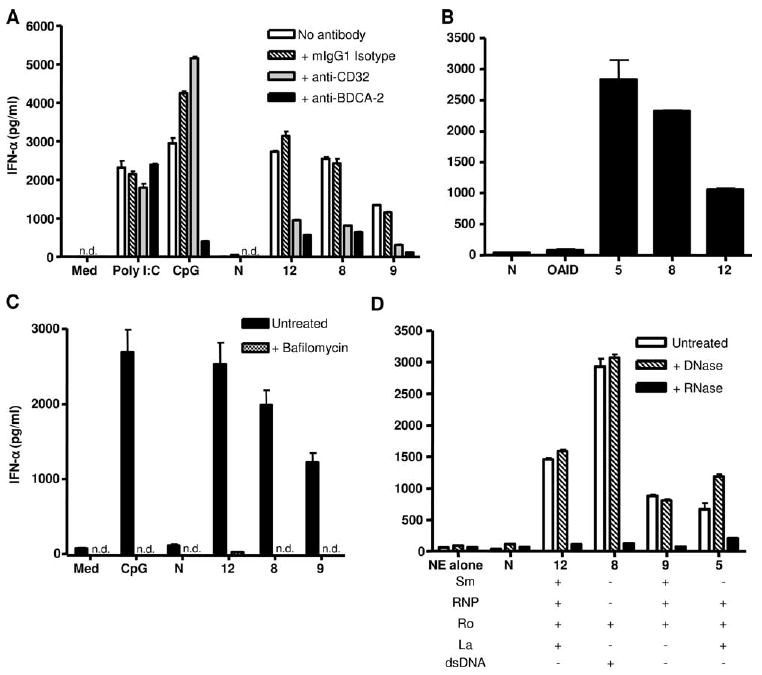
NPSLE+ CSF autoantibodies induce IFN-α by pDCs that is dependent on FcγRII engagement, endosomal transport, and RNA stimulation. A, Type I IFN-primed PBMC were preincubated for 1 h with 0.2 μg/ml anti-CD32, anti-BDCA-2, or mouse IgG1 before the addition of CSF (1/20) and Ag (80 μg/ml NE). Poly(I:C) (50 μg/ml) and CpG 2216 (0.5 μM) were used as controls. B, pDCs were isolated from PBMC by negative selection (>95% pure). Cells (1 × 104) were primed with type I IFN and stimulated with CSF (1/20) and NE (80 μg/ml) as described in A. C, PBMC were preincubated for 1 h with 100 nM bafilomycin A1 before incubation with CSF (1/10) and NE Ag as in A. D, NE was pretreated with RNase (8 μg/ml) or DNase (500 U/ml) before addition with CSF (1/20) to PBMC. Serum anti-Sm, anti-RNP, anti-Ro, and anti-La levels were quantified as described in Materials and Methods. IFN-α was quantified as described in Fig. 1, and the results are shown as the mean ± SD of duplicates. n.d. indicates not detected; N, normal donor; numbers on the x-axis refer to NPSLE patients in Table I. The results shown are representative of two to three independent experiments.
NPSLE+ patient CSF has 800-fold greater IFN-α specific activity compared with serum due to the presence of serum inhibitory factors
Comparison between serum and CSF in Fig. 1, A and C, revealed that, on average, NPSLE+ CSF induced >2-fold more IFN-α compared with serum. This finding is remarkable considering that the average IgG concentration in CSF was ∼600-fold lower than paired serum, and CSF autoantibodies are of lower titer. Anti-P autoantibody levels were >100-fold less in CSF compared with serum (data not shown), but insufficient CSF was available for analysis of all autoantibodies in CSF. To examine this discrepancy more precisely, we calculated the IFN-α “specific activity”, which was derived from the concentration of IFN-α produced (pg/ml) per microgram of serum or CSF IgG from the same patients in the bioassay. As shown in Fig. 3A, the average IFN-α specific activity was 816-fold higher in the CSF (mean, 1501) compared with serum (mean, 1.84) of NPSLE+ patients.
Figure 3.
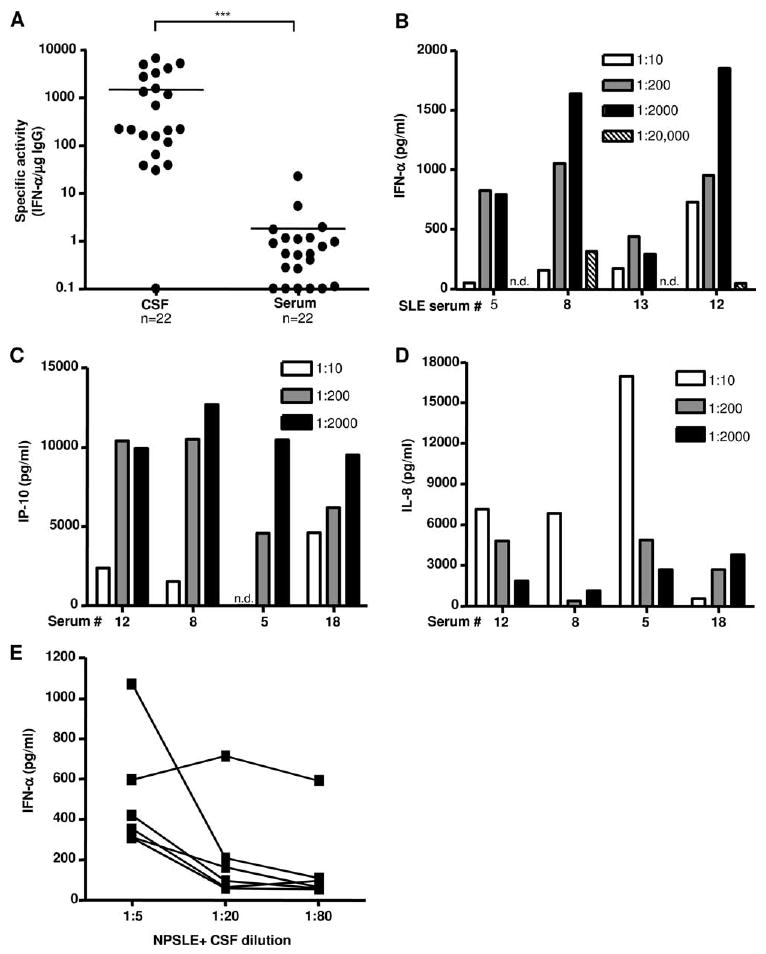
IFN-α induction per μg IgG is >800-fold higher in CSF due to inhibitor(s) of IFN-α production in serum. A, The specific activity (concentration of IFN-α induced per microgram of IgG of test sample) for matched serum and CSF patient samples is shown. Lines represent mean values (***, p < 0.001; Mann-Whitney U test). B, NPSLE+ sera were diluted from 1/10 to 1/20,000 and incubated with NE in the presence of IFN-primed PBMC. IFN-α was quantified after 24 h as in Fig. 1. C and D, NPSLE patient sera were diluted 1/10, 1/200, and 1/2000 and added to unprimed PBMC with NE (80 μg/ml). IP-10 and IL-8 levels were measured in culture supernatants after 24 h by ELISA. Background readings from NE alone control were subtracted from values. E, Six different NPSLE+ patients' CSF was diluted as shown and added to IFN-primed PBMC with NE (80 μg/ml). IFN-α was quantified as in Fig. 1.
To determine whether the difference in IFG activity between CSF and serum was explained by a CSF-enhancing factor or serum-suppressive factor, we performed serial dilutions of serum and showed that in most cases striking increases in IFG activity were observed at 1/200 and 1/2000 dilutions, suggesting that serum contained inhibitory factor(s) (Fig. 3B). Of 21 NPSLE sera tested, 71.4% (15 of 21) induced more IFN-α and 6 induced similar or less IFN-α upon serum dilution. Of six randomly selected normal human sera diluted as in Fig. 3B, none induced IFN-α (>50 pg/ml), indicating that, in the absence of IgG anti-nucleoprotein autoantibodies, normal sera do not stimulate IFN-α production by pDCs. While a similar paradoxical relationship was observed between serum dilution and IP-10 induction, dilution of serum led to reduced IL-8 concentrations in the bioassay, indicating that not all inflammatory mediators are regulated similarly by serum factors (Fig. 3, C and D). Significantly, when CSF from 6 NPSLE patients was diluted 1/5 to 1/80, IFG activity declined in five of six samples (Fig. 3E), indicating that the inhibitory factor(s) were either not present or were present at significantly lower concentrations in most NPSLE CSF.
To confirm that serum contained inhibitor(s) of IFN-α, we next performed mixing experiments. As shown in Fig. 4A, the addition of normal serum to NPSLE+ CSF at equal dilution (1/10) reduced IFN-α induction far in excess of the dilution factor. Similarly, when a low concentration (1/2000 dilution) of a stimulatory NPSLE+ serum was used to induce IFN-α, normal serum added at 1%, 2.5%, or 5% (v/v) inhibited IFN-α production by 20%, 71%, and 94%, respectively, whereas normal CSF added at the same concentrations had a minimal effect (Fig. 4B). Together with the findings in Fig. 3, these observations indicate that serum contains potent inhibitor(s) of IFN-α production that are lacking or decreased in CSF.
Figure 4.
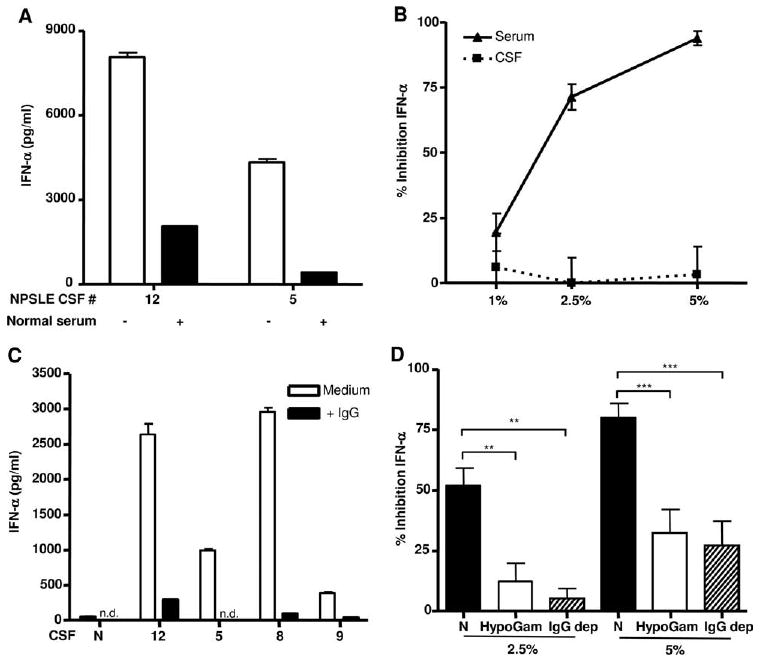
Normal serum and IgG are potent inhibitors of immune complex-stimulated IFN-α production. A, Equal amounts of NPSLE+ CSF and normal serum (both diluted 1/10) were incubated with NE and PBMC and IFN-α quantified as in Fig. 1. B, Normal CSF or serum (1%, 2.5%, or 5% (v/v)) was added to diluted NPSLE serum (1/2000), incubated with NE and PBMCs, and tested for IFN-α production in the bioassay after 24 h. Results are expressed as percentage inhibition of IFN-α production relative to cells that did not receive normal serum or CSF. C, Purified IgG (350 μg/ml) was added to 1/10 diluted normal or four different NPSLE+ patients' CSF, and IFN-α was quantified as above. The results in A and C are representative of two to three independent experiments, and in B, inhibition of IFN-α is expressed as the mean percentage ± SEM of three to six independent experiments. n.d. indicates not detected. D, Serum from normal individuals (N), four patients with hypogammaglobulinemia (HypoGam, associated with either X-linked agammaglobulinemia or common variable immunodeficiency), or three normal sera depleted of IgG (IgG dep) were incubated at 2.5% or 5% (v/v) with stimulatory NPSLE+ serum (1/2000), U937 freeze-thawed Ag (1% (v/v)), and primed PBMC. Results are shown as the mean percentage ± SEM of four independent experiments. **, p < 0.01; ***, p < 0.001; Mann-Whitney U test.
IgG contributes to suppression of IFN-α induced by SLE immune complexes
As the main component of i.v. Ig (IVIG), IgG is known to suppress inflammatory responses in vitro and in vivo (33). We therefore compared the effects of serum and purified IgG on inhibition of IFN-α. As shown in Fig. 4A, serum inhibited IFN-α production stimulated by immune complexes. At equivalent concentrations to that found in serum, purified IgG also inhibited CSF immune complex-induced IFN-α (Fig. 4C). Similar results were obtained with IVIG and with serum immune complex-stimulated IFN-α (data not shown). These observations strongly suggest that at least one inhibitor in serum is IgG. To further test this conclusion, we compared the inhibitory effect of normal serum compared with serum obtained from four patients with humoral immunodeficiency states (CVID or XLA (hypogammaglobulinemia), with IgG reduced by 80% or more) before IVIG therapy. As shown in Fig. 4D, sera with very low IgG levels were significantly less efficient at inhibition of IFN-α stimulated by NPSLE+ serum immune complexes (p < 0.01). Finally, we depleted IgG to <20% of the concentration found in normal sera using protein A or G and also observed a significant reduction in IFN-α inhibition (Fig. 4D, p < 0.01). The fact that inhibition was not completely reversed in these experiments indicates that other serum inhibitory factors are present.
CSF autoantibodies induce other cytokines associated with NPSLE
In addition to IFN-α, many other inflammatory mediators (IM) have been detected in the CSF of patients with NPSLE. These include IP-10 (CXCL10), IL-6, IL-8, MIP-1α, and MCP-1 (7-11). To determine whether in addition to IFN-α, CSF autoantibodies could induce IM associated with NPSLE, we quantified, using a multiplex assay, the five IM mentioned above and three IM (INF-α, IL-10, and IL-12p40) not usually implicated in NPSLE. In this experiment, PBMC responder cells were unprimed to avoid induction of IM that may occur by priming alone. The concentrations of cytokines and chemokines in CSF without (baseline) and with the addition of NE in the presence of PBMC as responder cells are shown in Fig. 5, A and B, respectively. IL-10, IL-12p40, and INF-α were low at baseline and were not significantly increased following the addition of Ag (Fig. 5A, p > 0.05). MIP-1α was only increased in 1 of 14 NPSLE CSF samples at baseline but was induced in another 4 upon addition of Ag. The average increase for this chemokine was not statistically significant. In contrast, IP-10, IL-8, and MCP-1 were stimulated significantly above baseline by CSF immune complexes (Fig. 5B, p < 0.01). Since IP-10 is known to be induced by type 1 and type 2 IFNs, and since we observed that the concentration of immune complex-induced IFN-α correlated with that of IP-10 (r = 0.88, p < 0.001), we tested whether a neutralizing mAb against IFN-α affected IP-10 production in the standard bioassay. As shown in Fig. 5C, the neutralizing mAb substantially reduced IP-10 production, indicating that IP-10 in CSF could be a consequence of type 1 IFN stimulation. This possibility was further supported by the observation that incubation of primary human microglia, the resident macrophage type cell in the CNS, with either IFN-α or IFN-β induced IP-10 as well as IL-6 (especially IFN-β), but only low concentrations of MIP-1α (Fig. 5D). By using an ultrasensitive chemiluminescence method, we detected >2 pg/ml IFN-α in 15 of 19 NPSLE+ CSF tested, whereas IFN-α was below the level of detection (0.8 pg/ml) in 3 normal CSF. Of relevance, the concentration of IP-10 was highly significantly correlated with IFN-α (r = 0.916, p < 0.001) in the NPSLE CSF, again supporting the experimental evidence presented that IP-10 in CSF is stimulated by IFN-α.
Figure 5.
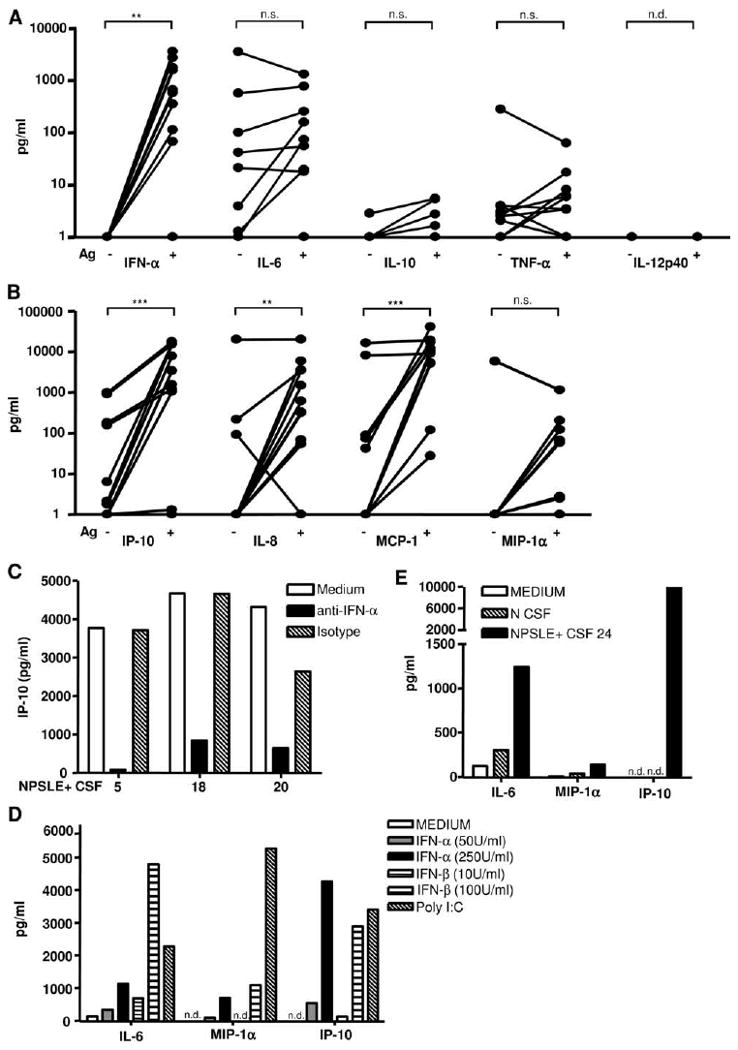
CSF from NPSLE patients induces other inflammatory mediators associated with NPSLE by activation of PBMC or microglia. A and B, NPSLE+ CSF (1/10) was incubated with unprimed PBMCs in the presence or absence of added nuclear Ag. Cytokines (A) and chemokines (B) were quantified after 24 h using a multiplex assay as described in Materials and Methods. IFN-α was measured by ELISA as in Fig. 1. Values below the level of detection were given a value of 1. Results with and without the addition of Ag were compared by the Wilcoxon signed-rank test (n.s., not significant; **, p < 0.01; ***, p < 0.001; n.d., not detected). C, NPSLE+ CSF (1/10) was incubated with NE and added to unprimed PBMC with or without a neutralizing mAb to IFN-α or mouse IgG1 isotype control (3 μg/ml), and IP-10 was measured as described above. D and E, Primary human microglia were plated as described in Materials and Methods and stimulated with IFN-α, IFN-β, orpoly(I:C) (50 μg/ml) (D) or normal (N) or NPSLE+ CSF (1/10) with nuclear Ag (80 μg/ml) (E). IL-6, IP-10, and MIP-1α were quantified as described above after 24 h.
Unlike MIP-1α, IL-6 levels were increased in most of the baseline CSF samples but, on average, the immune complexes did not significantly augment IL-6 concentrations using PBMC as responder cells. To address the possibility that immune complexes could act on cell types resident in the CNS, we used primary human microglia as responders and observed that NPSLE CSF could stimulate IL-6 and IP-10 production by microglia in the presence of Ag (Fig. 5E), although IFN-α induction was not detected (data not shown). Thus, most of the inflammatory mediators implicated in NPSLE could be induced by CSF in the presence of Ag and responder cells. Significantly, cytokines not usually implicated in NPSLE (IL-12p40, IL-10, and INF-α) were not induced by the CSF immune complexes.
Discussion
The main findings in the present study are that immune complexes formed by CSF autoantibodies were potent inducers of IFN-α as well as IP-10 (CXCL10), IL-8, and MCP-1, all of which have been reported to be elevated in CSF from NPSLE patients (6–11). Furthermore, the strong correlation between CSF IFN-α and IP-10 induction, as well as the ability of a neutralizing anti-IFN-α to inhibit IP-10 production, suggests that the presence of IP-10 in CSF could be explained by stimulation by IFN-α. A second important observation was that normal serum, but not normal CSF, could attenuate IFN-α stimulation by immune complexes, which was, in part, explained by the inhibitory effect of normal IgG (see below).
Markers of neuronal and astrocyte damage are markedly elevated in the CSF of NPSLE patients (34). Therefore, patients with NPSLE have both Ags and autoantibodies to form immune complexes to stimulate IM in the CNS. There are many potential sources of apoptotic or necrotic cells in NPSLE, such as cell damage induced by neurocytotoxic autoantibodies that now includes anti-ribosome P (35) and anti-NMDAR Abs (36) discussed above. Additionally, injury to endothelial cells by as yet poorly defined mechanisms or ischemic thrombosis associated with anticardiolipin Abs may release cellular Ags. Regardless of the source of Ag, the presence of CSF autoantibodies that bind to cellular Ags serves as a potentially powerful amplifier of inflammation in the brain. While low CSF IgG concentrations and limited volumes of CSF precluded identification of specific autoantibodies that stimulate IFN-α in CSF, we observed that the IFG activity in CSF correlated with serum anti-RNP, but not with other known autoantibody specificities. Additionally, four sera that tested negative for anti-dsDNA had among the highest IFG activity in CSF (Table I), and IFN-α induction was RNase sensitive (Fig. 2D). These observations suggest that autoantibodies targeted to RNA-containing Ags, rather than DNA-containing Ags, may be especially relevant to the IFG activity detected in NPSLE CSF, although studies of larger numbers of NPSLE patients and direct analysis of CSF will be needed to validate this observation. This finding is of interest since plasma containing Abs targeting RNA-containing protein Ags have previously been associated with the ability to induce high levels of IFN response genes in WISH cells (37), and targeted deletion of TLR7 that interacts with RNA, but not deletion of TLR9 that interacts with DNA, attenuated disease in a mouse model of SLE (38).
IFN-α was originally detected in the CSF by Winfield et al. (4) and has been directly implicated as a causative factor in NPSLE by Shiozawa et al. (6). In the latter study, IFN-α was detected in the CSF of five of six NPSLE patients and in the microglia and neurons following autopsy analysis of a patient who died from CNS lupus (6). Although a lower frequency of CSF IFN-α detection has been reported in other studies, we used an ultrasensitive chemiluminescence method and detected >2 pg/ml IFN-α in most of the NPSLE+ CSF tested, whereas IFN-α was below the level of detection (0.8 pg/ml) in three normal CSF. The concentrations of IP-10 were highly significantly correlated with IFN-α in the NPSLE CSF, strongly supporting the experimental evidence presented that IP-10 in CSF is stimulated by IFN-α. Although pDCs have not been studied in the brains of NPSLE patients, elevated numbers of pDCs have been detected in the CSF of all neuroinflammatory diseases examined (39). Alternatively, IM could be produced by CNS resident cells, and it is interesting that astrocytes are capable of IFN-α and IP-10 production in Aicardi-Goutières syndrome (40).
The mechanisms responsible for IFN-mediated neuropsychiatric disease following systemic administration of IFN-α therapy are uncertain, but there is considerable evidence that this effect could be direct. Type I IFN receptors are found in the glia as well as neurons (41). It has been suggested that IFNs alter brain function through alterations in serotonin, noadrenalin, the hypothalamic-pituitory-adrenal access or by generating toxic metabolites through the kynurenine pathway (17, 41). It is also possible that the neuronal effects are secondary to release of another factor or factors by nonneuronal cells since most cell types express type 1 receptors and there are several hundred IFN response genes. Finally, since only about a third of patients develop CNS manifestations on pharmacologic IFN therapy, additional host factors must determine susceptibility to the CNS effects.
The IFG activity of CSF obtained from NPSLE+ patients was much higher than that observed in other disease controls, suggesting that induction of IFN-α as well as other cytokines contribute to the pathogenesis of disease. In fact, the results reported herein show that almost all cytokines previously implicated in NPSLE could be induced by CSF autoantibodies in the presence of cell debris and appropriate responding cells. Some (IP-10 and IL-6), but not all, of these inflammatory mediators could be directly induced by type I IFNs depending on the responder cell type. This study therefore provides a strong mechanistic link between the subset of CSF autoantibodies directed against nucleoproteins and the inflammatory mediators described previously in NPSLE. Rather than excluding a role for direct Ab-mediated cytotoxic effects, these results suggest a two-step model of NPSLE where certain cytotoxic autoantibodies release nucleoproteins and other autoantibodies bind to form immune complexes, which then become powerful amplifiers of inflammation in the brain.
The NPSLE patients included in this study were heterogeneous with regard to their clinical manifestations (Table I), so it is, at present, unclear whether CSF IFG activity is more commonly associated with one or more subtypes. Since there are 19 different specific NPSLE subtypes (23), large multicenter studies comprising several hundred NPSLE patients will be needed to address this important question in the future. Similarly, knowledge regarding association with disease subtypes is also important since our studies have implications for therapy of NPSLE, including targeting of IFN-α and consideration of IVIG administration. IVIG has successfully been used to treat anecdotal cases of NPSLE (42) as well as other neuroinflammatory and neurodegenerative disorders (43, 44).
The striking difference in the IFG activity in CSF compared with serum normalized for IgG concentrations was shown to be due to inhibitor(s) present in normal serum. Inhibitor(s) were absent or present in low concentrations in NPSLE CSF since dilution of CSF generally led to reduction in IFG activity, and addition of normal CSF to bioassays failed to attenuate IFG responses. In view of the 600-fold lower IgG concentration in CSF and the known inhibitory effect of IgG in IVIG in attenuating inflammation (33), we examined the role of IgG as a possible inhibitory factor. Serum with spontaneous deficiencies of Ig (patients with CVID or XLA) or normal serum depleted of IgG had a significantly lower inhibitory activity for IFN-α compared with normal serum. Furthermore, addition of IgG to PBMC cultures also attenuated IFN-α stimulation by either NPSLE+ CSF or serum. Taken together, these findings indicate that IgG is one functional inhibitor of IFG activity in serum, consistent with the results of Bave et al. (45). Interestingly, the lack of IgG in serum does not fully abrogate inhibition, suggesting that other inhibitors are present. Note that IgG is unlikely to inhibit IFN-α production simply by competition for FcγR since the only FcγR detected on pDCs is FcγRII (45), a low-affinity receptor that should not bind monomeric IgG in preference to immune complexes. Furthermore, cell depletion experiments have revealed that the inhibitory effect is mediated, in part, by cells other than pDCs (D. M. Santer and K. B. Elkon, unpublished observations). Finally, since high concentrations of IgG are necessary for inhibition of IFN-α, we propose that a biochemically distinct subfraction of IgG, possibly arising through differential glycosylation (46), may be responsible for the inhibitory effect observed in these studies.
Acknowledgments
We are grateful to the Birth Defects Research Laboratory, University of Washington, the Human Brain and Spinal Fluid Resource Center (sponsored by National Institute of Neurological Disorders and Stroke/National Institute of Mental Health, National Multiple Sclerosis Society, and the Department of Veterans Affairs), Richard Nash, Troy Torgerson, and Charlotte Cunningham-Rundles for supplying materials and samples for these studies. We thank Daniel Kim for expert technical assistance, and Grant Hughes, Edward Clark, David Martin, and Dan Stetson for helpful discussions.
Footnotes
This work was supported by grants from the National Institutes of Health (National Institute of Arthritis and Musculoskeletal and Skin Diseases; AR48796 and AR054980), the Lupus Research Institute, and the Dana Foundation. D.M.S. is supported by a Natural Sciences and Engineering Research Council of Canada (NSERC) postgraduate scholarship.
Abbreviations used in this paper: NPSLE, neuropsychiatric systemic lupus erythematosus; NMDAR, N-methyl-d-aspartate receptor; anti-P, anti-ribosomal P protein Abs; CSF, cerebrospinal fluid; pDC, plasmacytoid dendritic cell; IFG, interferogenic; IP-10, IFN-γ-inducible protein 10; OAID, other autoimmune disease; MS, multiple sclerosis; CVID, common variable immune deficiency; XLA, X-linked agammaglobulinemia; RNP, ribonucleoprotein; NE, nuclear extract; BDCA-2, blood DC Ag-2; SLEDAI, systemic lupus erythematosus disease activity index; IVIG, intravenous immunoglobulin; IM, inflammatory mediators.
Disclosures: The authors have no financial conflicts of interest.
References
- 1.Johnson RT, Richardson EP. The neurological manifestations of systemic lupus erythematosus. Medicine. 1968;47:337–369. doi: 10.1097/00005792-196807000-00002. [DOI] [PubMed] [Google Scholar]
- 2.Hanly JG. Neuropsychiatric lupus. Rheum Dis Clin North Am. 2005;31:273–298. doi: 10.1016/j.rdc.2005.01.007. [DOI] [PubMed] [Google Scholar]
- 3.Zandman-Goddard G, Chapman J, Shoenfeld Y. Autoantibodies involved in neuropsychiatric SLE and antiphospholipid syndrome. Semin Arthritis Rheum. 2007;36:297–315. doi: 10.1016/j.semarthrit.2006.11.003. [DOI] [PubMed] [Google Scholar]
- 4.Winfield JB, Shaw M, Silverman LM, Eisenberg RA, Wilson HA, 3rd, Koffler D. Intrathecal IgG synthesis and blood-brain barrier impairment in patients with systemic lupus erythematosus and central nervous system dysfunction. Am J Med. 1983;74:837–844. doi: 10.1016/0002-9343(83)91075-6. [DOI] [PubMed] [Google Scholar]
- 5.Small P, Mass MF, Kohler PF, Harbeck RJ. Central nervous system involvement in SLE: diagnostic profile and clinical features. Arthritis Rheum. 1977;20:869–878. doi: 10.1002/art.1780200317. [DOI] [PubMed] [Google Scholar]
- 6.Shiozawa S, Kuroki Y, Kim M, Hirohata S, Ogino T. Interferon-α in lupus psychosis. Arthritis Rheum. 1992;35:417–422. doi: 10.1002/art.1780350410. [DOI] [PubMed] [Google Scholar]
- 7.Jonsen A, Bengtsson AA, Nived O, Ryberg B, Truedsson L, Ronnblom L, Alm GV, Sturfelt G. The heterogeneity of neuropsychiatric systemic lupus erythematosus is reflected in lack of association with cerebrospinal fluid cytokine profiles. Lupus. 2003;12:846–850. doi: 10.1191/0961203303lu472sr. [DOI] [PubMed] [Google Scholar]
- 8.Iikuni N, Okamoto H, Yoshio T, Sato E, Kamitsuji S, Iwamoto T, Momohara S, Taniguchi A, Yamanaka H, Minota S, Kamatani N. Raised monocyte chemotactic protein-1 (MCP-1)/CCL2 in cerebrospinal fluid of patients with neuropsychiatric lupus. Ann Rheum Dis. 2006;65:253–256. doi: 10.1136/ard.2005.041640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Trysberg E, Carlsten H, Tarkowski A. Intrathecal cytokines in systemic lupus erythematosus with central nervous system involvement. Lupus. 2000;9:498–503. doi: 10.1177/096120330000900704. [DOI] [PubMed] [Google Scholar]
- 10.Okamoto H, Katsumata Y, Nishimura K, Kamatani N. Interferon-inducible protein 10/CXCL10 is increased in the cerebrospinal fluid of patients with central nervous system lupus. Arthritis Rheum. 2004;50:3731–3732. doi: 10.1002/art.20598. [DOI] [PubMed] [Google Scholar]
- 11.Fragoso-Loyo H, Richaud-Patin Y, Orozco-Narvaez A, Davila-Maldonado L, Atisha-Fregoso Y, Llorente L, Sanchez-Guerrero J. Interleukin-6 and chemokines in the neuropsychiatric manifestations of systemic lupus erythematosus. Arthritis Rheum. 2007;56:1242–1250. doi: 10.1002/art.22451. [DOI] [PubMed] [Google Scholar]
- 12.Baechler EC, Gregersen PK, Behrens TW. The emerging role of interferon in human systemic lupus erythematosus. Curr Opin Immunol. 2004;16:801–807. doi: 10.1016/j.coi.2004.09.014. [DOI] [PubMed] [Google Scholar]
- 13.Martin DA, Elkon KB. Autoantibodies make a U-turn: the toll hypothesis for autoantibody specificity. J Exp Med. 2005;202:1465–1469. doi: 10.1084/jem.20052228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Marshak-Rothstein A, Rifkin IR. Immunologically active autoantigens: the role of Toll-like receptors in the development of chronic inflammatory disease. Annu Rev Immunol. 2007;25:419–441. doi: 10.1146/annurev.immunol.22.012703.104514. [DOI] [PubMed] [Google Scholar]
- 15.Ronnblom LE, Alm GV, Oberg KE. Autoimmunity after α-interferon therapy for malignant carcinoid tumors. Ann Intern Med. 1991;115:178–183. doi: 10.7326/0003-4819-115-3-178. [DOI] [PubMed] [Google Scholar]
- 16.Ehrenstein MR, McSweeney E, Swane M, Worman CP, Goldstone AH, Isenberg DA. Appearance of anti-DNA antibodies in patients treated with interferon-α. Arthritis Rheum. 1993;36:279–280. doi: 10.1002/art.1780360224. [DOI] [PubMed] [Google Scholar]
- 17.Wichers M, Maes M. The psychoneuroimmuno-pathophysiology of cytokine-induced depression in humans. Int J Neuropsychopharmacol. 2002;5:375–388. doi: 10.1017/S1461145702003103. [DOI] [PubMed] [Google Scholar]
- 18.Lebon P, Meritet JF, Krivine A, Rozenberg F. Interferon and Aicardi-Goutières syndrome. Eur J Paediatr Neurol. 2002;6(Suppl 1):A47–A53. doi: 10.1053/ejpn.2002.0574. discussion A55–58, A77–A86. [DOI] [PubMed] [Google Scholar]
- 19.Crow YJ, Leitch A, Hayward BE, Garner A, Parmar R, Griffith E, Ali M, Semple C, Aicardi J, Babul-Hirji R. Mutations in genes encoding ribonuclease H2 subunits cause Aicardi-Goutières syndrome and mimic congenital viral brain infection. Nat Genet. 2006;38:910–916. doi: 10.1038/ng1842. [DOI] [PubMed] [Google Scholar]
- 20.Crow YJ, Hayward BE, Parmar R, Robins P, Leitch A, Ali M, Black DN, van Bokhoven H, Brunner HG, Hamel BC. Mutations in the gene encoding the 3′-5′ DNA exonuclease TREX1 cause Aicardi-Goutières syndrome at the AGS1 locus. Nat Genet. 2006;38:917–920. doi: 10.1038/ng1845. [DOI] [PubMed] [Google Scholar]
- 21.Campbell IL, Krucker T, Steffensen S, Akwa Y, Powell HC, Lane T, Carr DJ, Gold LH, Henriksen SJ, Siggins GR. Structural and functional neuropathology in transgenic mice with CNS expression of IFN-α. Brain Res. 1999;835:46–61. doi: 10.1016/s0006-8993(99)01328-1. [DOI] [PubMed] [Google Scholar]
- 22.Tan EM, Cohen AS, Fries JP, Masi AT, McShane DJ, Rothfield NF, Schaller JG, Talal N, Winchester RJ. The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum. 1982;25:1271–1277. doi: 10.1002/art.1780251101. [DOI] [PubMed] [Google Scholar]
- 23.American College of Rheumatology. The American College of Rheumatology nomenclature and case definitions for neuropsychiatric lupus syndromes. Arthritis Rheum. 1999;42:599–608. doi: 10.1002/1529-0131(199904)42:4<599::AID-ANR2>3.0.CO;2-F. [DOI] [PubMed] [Google Scholar]
- 24.Elkon KB, Parnassa AP, Foster CL. Lupus autoantibodies target the ribosomal P proteins. J Exp Med. 1985;162:459–471. doi: 10.1084/jem.162.2.459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim D, Peck A, Santer D, Patole P, Schwartz SM, Molitor JA, Arnett FC, Elkon KB. Induction of interferon-α by scleroderma sera containing autoantibodies to topoisomerase: I. Association of higher interferon-alpha activity with lung fibrosis. Arthritis Rheum. 2008;58:2163–2173. doi: 10.1002/art.23486. [DOI] [PubMed] [Google Scholar]
- 26.Lovgren T, Eloranta ML, Bave U, Alm GV, Ronnblom L. Induction of interferon-α production in plasmacytoid dendritic cells by immune complexes containing nucleic acid released by necrotic or late apoptotic cells and lupus IgG. Arthritis Rheum. 2004;50:1861–1872. doi: 10.1002/art.20254. [DOI] [PubMed] [Google Scholar]
- 27.Satoh JI, Kim SU. Differential expression of heat shock protein HSP27 in human neurons and glial cells in culture. J Neurosci Res. 1995;41:805–818. doi: 10.1002/jnr.490410611. [DOI] [PubMed] [Google Scholar]
- 28.Lovgren T, Eloranta ML, Kastner B, Wahren-Herlenius M, Alm GV, Ronnblom L. Induction of interferon-alpha by immune complexes or liposomes containing systemic lupus erythematosus autoantigen- and Sjögren's syndrome autoantigen-associated RNA. Arthritis Rheum. 2006;54:1917–1927. doi: 10.1002/art.21893. [DOI] [PubMed] [Google Scholar]
- 29.Bonfa E, Golombek SJ, Kaufman LD, Skelly S, Weissbach H, Brot N, Elkon KB. Association between lupus psychosis and anti-ribosomal P protein antibodies: measurement of antibody using a synthetic peptide antigen. N Engl J Med. 1987;317:265–271. doi: 10.1056/NEJM198707303170503. [DOI] [PubMed] [Google Scholar]
- 30.Teh LS, Lee MK, Wang F, Manivasagar M, Charles PJ, Nicholson GD, Hay EM, Isenberg DA, Amos N, Williams BD. Antiribosomal P protein antibodies in different populations of patients with systemic lupus erythematosus. Br J Rheumatol. 1993;32:663–665. doi: 10.1093/rheumatology/32.8.663. [DOI] [PubMed] [Google Scholar]
- 31.Hauser SL, Waubant E, Arnold DL, Vollmer T, Antel J, Fox RJ, Bar-Or A, Panzara M, Sarkar N, Agarwal S, Langer-Gould A, Smith CH. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N Engl J Med. 2008;358:676–688. doi: 10.1056/NEJMoa0706383. [DOI] [PubMed] [Google Scholar]
- 32.Dzionek A, Sohma Y, Nagafune J, Cella M, Colonna M, Facchetti F, Gunther G, Johnston I, Lanzavecchia A, Nagasaka T. BDCA-2, a novel plasmacytoid dendritic cell-specific type II C-type lectin, mediates antigen capture and is a potent inhibitor of interferon α/β induction. J Exp Med. 2001;194:1823–1834. doi: 10.1084/jem.194.12.1823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nimmerjahn F, Ravetch JV. Anti-inflammatory actions of intravenous immunoglobulin. Annu Rev Immunol. 2008;26:513–533. doi: 10.1146/annurev.immunol.26.021607.090232. [DOI] [PubMed] [Google Scholar]
- 34.Trysberg E, Nylen K, Rosengren LE, Tarkowski A. Neuronal and astrocytic damage in systemic lupus erythematosus patients with central nervous system involvement. Arthritis Rheum. 2003;48:2881–2887. doi: 10.1002/art.11279. [DOI] [PubMed] [Google Scholar]
- 35.Matus S, Burgos PV, Bravo-Zehnder M, Kraft R, Porras OH, Farias P, Barros LF, Torrealba F, Massardo L, Jacobelli S, Gonzalez A. Antiribosomal-P autoantibodies from psychiatric lupus target a novel neuronal surface protein causing calcium influx and apoptosis. J Exp Med. 2007;204:3221–3234. doi: 10.1084/jem.20071285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.DeGiorgio LA, Konstantinov KN, Lee SC, Hardin JA, Volpe BT, Diamond B. A subset of lupus anti-DNA antibodies cross-reacts with the NR2 glutamate receptor in systemic lupus erythematosus. Nat Med. 2001;7:1189–1193. doi: 10.1038/nm1101-1189. [DOI] [PubMed] [Google Scholar]
- 37.Kirou KA, Lee C, George S, Louca K, Peterson MG, Crow MK. Activation of the interferon-α pathway identifies a subgroup of systemic lupus erythematosus patients with distinct serologic features and active disease. Arthritis Rheum. 2005;52:1491–1503. doi: 10.1002/art.21031. [DOI] [PubMed] [Google Scholar]
- 38.Christensen SR, Shupe J, Nickerson K, Kashgarian M, Flavell RA, Shlomchik MJ. Toll-like receptor 7 and TLR9 dictate autoantibody specificity and have opposing inflammatory and regulatory roles in a murine model of lupus. Immunity. 2006;25:417–428. doi: 10.1016/j.immuni.2006.07.013. [DOI] [PubMed] [Google Scholar]
- 39.Pashenkov M, Huang YM, Kostulas V, Haglund M, Soderstrom M, Link H. Two subsets of dendritic cells are present in human cerebrospinal fluid. Brain. 2001;124:480–492. doi: 10.1093/brain/124.3.480. [DOI] [PubMed] [Google Scholar]
- 40.van Heteren JT, Rozenberg F, Aronica E, Troost D, Lebon P, Kuijpers TW. Astrocytes produce interferon-alpha and CXCL10, but not IL-6 or CXCL8, in Aicardi-Goutières syndrome. Glia. 2008;56:568–578. doi: 10.1002/glia.20639. [DOI] [PubMed] [Google Scholar]
- 41.Dafny N, Yang PB. Interferon and the central nervous system. Eur J Pharmacol. 2005;523:1–15. doi: 10.1016/j.ejphar.2005.08.029. [DOI] [PubMed] [Google Scholar]
- 42.Levy Y, Sherer Y, Ahmed A, Langevitz P, George J, Fabbrizzi F, Terryberry J, Meissner M, Lorber M, Peter JB, Shoenfeld Y. A study of 20 SLE patients with intravenous immunoglobulin: clinical and serologic response. Lupus. 1999;8:705–712. doi: 10.1191/096120399678841007. [DOI] [PubMed] [Google Scholar]
- 43.Dalakas MC. Role of IVIg in autoimmune, neuroinflammatory and neurodegenerative disorders of the central nervous system: present and future prospects. J Neurol. 2006;253(Suppl 5):V25–V32. doi: 10.1007/s00415-006-5004-0. [DOI] [PubMed] [Google Scholar]
- 44.Linker RA, Gold R. Use of intravenous immunoglobulin and plasma exchange in neurological disease. Curr Opin Neurol. 2008;21:358–365. doi: 10.1097/WCO.0b013e3282ff5b8f. [DOI] [PubMed] [Google Scholar]
- 45.Bave U, Magnusson M, Eloranta ML, Perers A, Alm GV, Ronnblom L. FcγRIIa is expressed on natural IFN-α-producing cells (plasmacytoid dendritic cells) and is required for the IFN-α production induced by apoptotic cells combined with lupus IgG. J Immunol. 2003;171:3296–3302. doi: 10.4049/jimmunol.171.6.3296. [DOI] [PubMed] [Google Scholar]
- 46.Nimmerjahn F, Ravetch JV. The antiinflammatory activity of IgG: the intravenous IgG paradox. J Exp Med. 2007;204:11–15. doi: 10.1084/jem.20061788. [DOI] [PMC free article] [PubMed] [Google Scholar]