

Abstract
The discovery that impaired autophagy is linked to a wide variety of prominent diseases including cancer and neurodegeneration has lead to an explosion of research in this area. Methodologies that allow investigators to observe and quantify the autophagic process will clearly advance our knowledge of how this process contributes to the pathophysiology of many clinical disorders. The recent identification of essential autophagy genes in higher eukaryotes has made it possible to analyze autophagy in mammalian cells that express autophagy proteins tagged with fluorescent markers. This chapter describes such methods using primary cultured neurons that undergo up-regulation of autophagy when trophic factors are removed from their medium. The prolonged up-regulated autophagy, in turn, contributes to the death of these neurons, thus providing a model to examine the relationship between enhanced autophagy and cell death.
Neurons are isolated from the cerebellum of postnatal day 7 rat pups and cultured in the presence of trophic factors and depolarizing concentrations of potassium. Once established, the neurons are transfected with an adeno-viral vector expressing MAP1-LC3 with red fluorescent protein (RFP). MAP1-LC3 is the mammalian homologue of the yeast autophagosomal marker Atg8 and when tagged to GFP or RFP, it is the most widely used marker for autophagosomes. Once expression is stable, autophagy is induced by removing trophic factors. At various time points after inducing autophagy, the neurons are stained with LysoSensor Green (a pH-dependent lysosome marker) and Hoechst (a DNA marker) and subjected to live-cell imaging. In some cases, time-lapse imaging is used to examine the step-wise process of autophagy in live neurons.
II. Introduction
Autophagy is a regulated, catabolic pathway for the turnover of long-lived proteins, macromolecular aggregates, and damaged organelles by lysosomal degradation (Levine and Klionsky, 2004). It also plays a role in clearing the cell of invading bacteria and viruses (Levine and Deretic, 2007; Lee and Iwasaki, 2008). In mammalian cells, the lysosomal pathway of intracellular degradation is divided into three distinct pathways: macroautophagy, microautophagy, and chaperone-mediated autophagy (Cuervo, 2004). Macroautophagy (Mizushima, 2007; Xie and Klionsky, 2007) begins with the formation of a unique double-membrane vesicle (autophagosome) that engulfs cytoplasmic constituents such as proteins, lipids, and damaged organelles, including mitochondria. The outer membrane of the autophagosome then docks and fuses with the lysosome to deliver the sequestered cargo. The inner membrane of the fused vesicle (autolysosome), along with the interior contents of the autophagosome are degraded by lysosomal hydrolases, a process that generates nucleotides, amino acids, and free fatty acids that are recycled to provide raw materials and energy to the cell. Microautophagy (Klionsky et al., 2007) circumvents the autophagosome sequestration step and begins with the direct uptake of cytosolic components by invaginations and pinching off of the lysosomal membrane. As in macroautophagy, the internalized cytosolic components are digested by lysosomal enzymes and the macromolecules are released when the vacuolar membrane disintegrates. In chaperone-mediated autophagy (Majeski and Dice, 2004; Massey et al., 2006), specific chaperone proteins bind to targeted proteins containing a KFERQ sequence and direct these proteins to the surface of the lysosome. These proteins bind to LAMP-2A on lysosomal membranes and are then transported across the membrane with the assistance of chaperone proteins where they are degraded by the lysosomal proteases.
In eukaryotic cells, macroautophagy (hereon referred to as autophagy) occurs constitutively at low levels in all cells to perform housekeeping functions such as degradation of proteins and destruction of dysfunctional organelles. Dramatic up-regulation of autophagy occurs in the presence of external stressors such as starvation, hormonal imbalance, oxidation, extreme temperature, and infection; as well as, internal needs such as cellular remodeling and removal of protein aggregates (Levine and Kroemer, 2008). Mediators of phosphoinositide-3 (PI3) kinase (Type III) signaling pathways and trimeric G proteins play major roles in regulating the formation of autophagosomes (Backer, 2008). Conversely, the Type I PI3K/Akt signaling pathway inhibits autophagy in response to insulin and other growth factor signals (Meijer and Codogno, 2004). Target of rapamycin (TOR) kinase, a highly conserved environmental sensor protein downstream of PI3K, is a predominant inhibitor of autophagy (Meijer and Codogno, 2004). The eukaryotic initiation factor 2 kinase, Gcn2, and its downstream target, Gcn4, a transcriptional transactivator of autophagy genes, induces autophagy under conditions of cellular starvation (Natarajan et al., 2001; Tallóczy et al., 2002). In the nervous system, autophagy is unresponsive to starvation, but is induced by removal of trophic factors and the presence of aggregate-prone cytosolic proteins, damaged organelles, or prions, factors that contribute to neurodegenerative disease (Levine and Yuan, 2005). Although autophagy can be induced as a protective mechanism for neurons to clear unwanted proteins and damaged organelles, depending on the extent and duration of the insult, it can also lead to autophagic cell death. The regulatory events that switch autophagy from a protective event to a cell death mechanism are poorly defined, but important to understand if autophagic mechanisms are to be manipulated for therapeutic gain.
Recent studies in our laboratory have shown that trophic factor withdrawal (TFW) leads to the death of both granule neurons and Purkinje neurons in postnatal cerebellar cultures. Granule neurons (95-97% of total cells) die by a classic intrinsic apoptotic pathway, whereas, Purkinje neurons (3-5% of total cells) die by an autophagic cell death pathway (Florez-McClure et al., 2004). Thus, under the same culture conditions and the same neurotoxic stimulus, the mechanisms mediating cell death in the two types of neurons are very distinct. Why Purkinje neurons choose an autophagic mechanism to die under these experimental conditions is not clear, although this type of cell demise mimics the death of Purkinje neurons in the Lurcher mouse carrying a point mutation in the delta2 glutamate receptor (Zuo et al., 1997). The Lurcher mouse demonstrates extensive Purkinje neuron degeneration followed by secondary loss of granule neurons in the cerebellum. Lurcher Purkinje neurons demonstrate ultrastructural features of autophagy at a time when they begin to degenerate and more recent biochemical studies have linked autophagy as the cause of death (Selimi et al., 2003; Yue et al., 2002). In this report, we describe our model system for studying autophagy in cultured Purkinje neurons and the methods that we have developed for observing and quantifying autophagy via live-cell fluorescence imaging.
III. Cultured cerebellar Purkinje neurons as a model to study neuronal autophagy
III a. Primary culture
Primary rat cerebellar neurons are isolated from 7-day old Sprague Dawley rat pups as described previously (D’Mello et al., 1993). The day before plating, coverslips in a 24-well plate are treated with poly-D-lysine (40 μg/ml; Sigma-Aldrich, St. Louis, MO, catalog number, P0899) and placed in a 37°C (5% CO2) incubator for 1 hr. Poly-D-lysine is then aspirated and coverslips are left to dry overnight in the tissue culture hood. The following day, laminin (1 μg/mL; BD Biosciences Discovery Labware, Bedford, MA, catalog number 354232) is added to the coverslips for at least 1 hr at room temperature. Prior to plating cells, the laminin is aspirated and coverslips are rinsed twice with plating media. Neurons are plated at a density of 2.0×106 cells/ml in basal modified Eagle’s medium (BME) containing 10% fetal bovine serum, 25 mM KCl, 2 mM L-glutamine, and penicillin (100 U/ml)-streptomycin (100 μg/ml; Invitrogen, Gaithersburg, MD, catalog number 15140-122). The cultures are grown in a 5% CO2 incubator at 37°C.
Cytosine arabinoside (10 μM, Sigma-Aldrich, St. Louis, MO, catalog number C6645) is added to the culture medium 24 hr after plating to limit the growth of non-neuronal cells.
After 5 days in culture, neurons are infected with adeno-viral vector expressing MAP1-LC3 labeled with red fluorescent protein (RFP-LC3, a kind gift from Aviva Tolkovsky, Cambridge University) at a multiplicity of infection of 100 for 24 hr.
Autophagic cell death is induced by removing the plating medium and replacing it with TFW medium (serum-free BME containing 5 mM KCl).
III b. Immunostaining
Purkinje neurons in cerebellar cultures are identified by immunostaining with a polyclonal antibody to calbindin D-28k (Chemicon, Temecula, CA, catalog number AB1778). For immunocytochemical staining experiments, we use the following protocol:
Neuronal cultures are plated on poly-D-lysine/laminin-coated glass cover slips at 2.0×106 cells/ml
Following incubation in control medium or TFW media for 24-48 hr, the neurons are fixed with 4% paraformaldehyde in phosphate-buffered saline (PBS) for 30 min at room temperature.
The neurons are then permeabilized and blocked with PBS, pH 7.4 containing 0.2% Triton X-100 and 5% BSA for 1 hr at room temperature.
Cells are incubated with primary antibody (Calbindin D-28K, 1:250; Chemicon, Temecula, CA, catalog number, AB1778) overnight at 4°C diluted in PBS containing 0.1% Triton X-100 and 2% BSA.
Primary antibodies are then removed and the cells are washed at least six times with PBS at room temperature for at least 1 hr.
The neurons are then incubated with the appropriate Cy3-conjugated secondary antibody (diluted 1:500) and DAPI (1 μg/ml) for 1 hr at room temperature.
The cells are washed six more times with PBS at room temperature, and coverslips are adhered to glass slides with mounting medium (0.1% p-phenylenediamine in 75% glycerol in PBS). Slides are sealed with clear nail polish and stored at -20°C prior to imaging.
III c. Fluorescence imaging and cell counts of cultured Purkinje neurons
Imaging is performed using a Zeiss (Thornwood, NY) Axioplan 2 microscope equipped with a Cooke Sensicam deep-cooled CCD camera. Images are captured on the CY3 and DAPI channels and analyzed with the Slidebook (4.2) software program (Intelligent Imaging Innovations Inc., Denver, CO). The number of calbindin-positive cells are counted in 152 fields using a 63X oil objective that are randomly selected by following a fixed grid pattern over the coverslip. The total area counted per coverslip is 14.6mm2 or ∼13% of the coverslip. Cells counted as Purkinje neurons have large rounded cell bodies, elaborate processes and larger, oval nuclei (in comparison to smaller, rounder nuclei of granule neurons). At least three coverslips are counted per experimental condition. The numbers are averaged and expressed as a percentage of the average number counted in the appropriate controls. This is repeated for at least three independent experiments. Results shown in Figure 1 demonstrate that TFW induces the death of Purkinje neurons by a mechanism distinct from granule neurons that does not involve nuclear condensation, but is associated with extensive cytoplasmic vacuolation. The cytoplasmic vacuoles are identified as autophagosomes and autolysosomes by monodansylcadaverine staining and electron transmission microscopy (Florez-McClure et al., 2004) (see also the chapter by Vázquez and Colombo in this volume).
Figure 1. Trophic factor withdrawal induces a non-apoptotic death of Purkinje neurons.
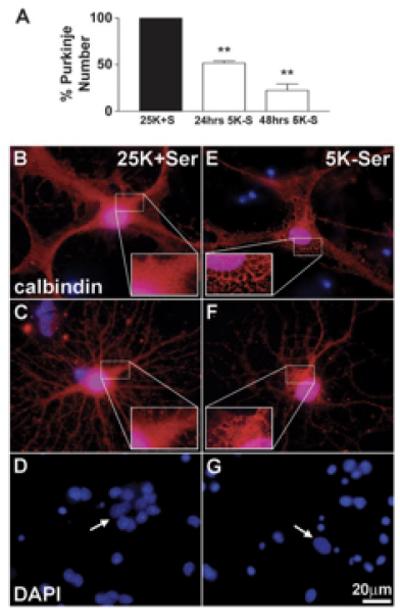
(A) Calbindin-positive Purkinje cells plotted as a percent of control. Following 24 hr or 48 hr of TFW, 51.7± 2.2% and 22.3 ± 7.0% of Purkinje neurons survive, respectively, as compared to controls (** indicates significant difference from 25K+S control at p<0.01). (B-G) Cells fixed and stained with polyclonal antibodies against Calbindin D-28K (in red) and the nuclear dye, DAPI (in blue). The remaining Purkinje neurons showed markedly different morphology than control cells, characterized by extensive cytoplasmic vacuolation (compare control cells B,C to trophic factor-deprived cells E,F). In contrast to granule neurons, which demonstrated substantial nuclear condensation and fragmentation characteristic of apoptosis (compare D to G), Purkinje neurons showed no obvious signs of nuclear condensation or fragmentation (compare nuclei indicated by arrows in D and G). This figure was previously published in Florez-McClure et al., (2004), and is reprinted with permission from The Society for Neuroscience, J. Neuroscience.
IV. Characterization of autophagic vacuole size and number in Purkinje neurons
To visualize vacuole size and number, we use the following protocol:
Cerebellar neurons are cultured and infected with RFP-LC3 as described above.
Twenty-four hours later, autophagic cell death is induced by removing the plating medium and replacing it with either control medium or TFW medium for various time points (0-24 hr).
After the appropriate treatment times, cells are stained at 37°C with Hoechst (20 ng/ml) for 15 min to visualize cellular nuclei, and LysoSensor Green (2 μM) for 5 min to visualize lysosomes.
The cells are then washed three times for 5 min with 37°C phenol red-free control medium (Dulbecco’s Modified Eagle Medium (D-MEM) #31053 containing 10% fetal bovine serum, 25mM KCL, 2 mM L-glutamine, and penicillin (100 U/ml)-streptomycin (100 μg/ml; Invitrogen, Gaithersburg, MD) or TFW medium (serum free D-MEM containing 5 mM KCL) to remove nonspecifically bound dyes.
The individually timed coverslips to be imaged are attached to a 35mm dish using Vaseline petroleum jelly and submerged in 3 mL of the appropriate phenol red-free medium.
The dish is then fitted into a pre-warmed 37°C heated stage, which maintains the temperature at 37°C. Imaging is performed as described above. Images of Lysosensor Green, RFP-LC3, and Hoechst fluorescence are captured on the FITC, Cy3 and DAPI channels, respectively, using the 63X water immersion objective. The length of exposure on the DAPI channel is calibrated by the Slidebook program (Slidebook 4.2, Intelligent Imaging Innovations Inc., Denver, CO). Collected images are used to measure the diameters of all the visible RFP-positive and LysoSensor Green-positive vacuoles in at least seven Purkinje neurons per condition using the Slidebook ruler tool as described in further detail in Figure 2.
Figure 2. Schematic for measuring the size of autophagy-positive vacuoles.

Autophagosome and lysosome vacuole size are quantified by measuring the diameters of all visible RFP-LC3- and LysoSensor Green-positive vacuoles. At least 7 images are collected via live cell microscopy per treatment. Using the Slidebook magnification tool, images are increased in size so that the diameter (μm) of each vacuole can be easily measured using the ruler tool. The diameters of all visible vacuoles in 7-12 Purkinje neurons per treatment are measured, which reflects diameter measurements of ∼200 vacuoles per treatment. The total number of vacuoles are then counted and categorized as small (<0.75), medium (0.75-1.5) large (1.5-2.25) or extra large (>2.25) based on their diameter, and the size distribution is graphed as percent of total vacuoles within the indicated size ranges. Images shown represent a 6 hr treatment of cerebellar cultures in TFW medium. Scale bar represents, 5 μm.
Data collected from vacuole size measurements are used to create an autophagic vacuole size distribution profile (n=2 for at least three independent experiments). The autophagosome size distribution profile of Purkinje neurons exposed to 0, 6 and 24 hr of TFW is shown in Figure 3A. In healthy control Purkinje neurons, ∼90% of RFP-LC3-positive vacuoles are smaller than 0.75 μm in diameter. Following 6 hr of TFW, there is a significant shift in the size distribution resulting in a decrease of smaller vacuoles (<0.75 μm) and increase of medium size autophagosomes (0.75 - 1.5 μm) (Figure 3 p**<0.01 and p###<0.001 compared to their respective controls). This shift in autophagosome size during TFW was also observed in lysosomes as determined with lysosomal diameter measurements (Figure 3B). The increase in the size of autophagic and lysosomal vacuoles present in dying Purkinje neurons is negatively correlated with survival (Florez-McClure et al., 2004). Furthermore, addition of 3-methyladenine at the time of TFW blocks the change in vacuolization size and decreases cell death (Florez-McClure et al., 2004). The addition of rapamycin, the specific inhibitor for mTOR, induces the formation of large autophagosomes (data not shown).
Figure 3. Accumulation of Autophagic Vacuoles in Purkinje Neurons.

Purkinje neurons were maintained in control medium or TFW medium for 24 hr in the absence and presence of bafilomycin A1. Autophagosome and lysosome vacuole size were quantified by measuring the diameters of all RFP-LC3-positive vacuoles (A) and LysoSensor Green-positive vacuoles (B) in 7-12 Purkinje neurons per treatment. The size distribution was graphed as percent of total vacuoles within the indicated size ranges. For bafilomycin A1-treated conditions, the total number of vacuoles per Purkinje neuron was also determined. **p<0.01 and ***p<0.001 compared to <0.75 μm control, ##p<0.01 and ###p<0.001 compared to 0.75-1.5 μm control. one-way ANOVA, Tukey’s post hoc test.
Measuring the vesicle size profile is a good indicator of autophagy induction. Activation of autophagy by TFW results in the formation of vesicles greater than 0.75 μm with significant increases in vesicles between .75-1.5 μm. It is important to keep in mind that these measurements are not limited to autophagosomes, as RFP-LC3-positive vacuoles may also reflect autolysosomes that have not yet degraded LC3. However, this is not an accurate representation of the total autophagosome population as these vesicles are continuously being degraded. Total autophagosome numbers can be obtained in the presence of lysosomal protease inhibitors or drugs that block autophagosome degradation such as, bafilomycin A1, a vacuolar ATPase inhibitor that disrupts lysosomal acidification (Fass et al., 2006). Comparing the vesicle size distribution in the absence and presence of bafilomycin A1 reveals a further increase in larger vesicles greater than 1.5 μm, which under normal autophagy conditions are rapidly degraded (Figure 3A; see 6 hr TFW +Baf A1). The inclusion of bafilomycin A1 is therefore necessary when quantifying the total number of autophagic vacuoles formed per treatment condition and when making any inferences on autophagic flux.
Changes in autophagic flux between two treatments can also be determined by measuring the vesicle size distribution profile in the absence and presence of bafilomycin A1. For example, if the total numbers of autophagic vacuoles between two treatment conditions (in the presence of bafilomycin A1) are similar, but (in the absence of bafilomycin A1) the large vesicles (>1.5 μm) in one condition are significantly reduced, this could indicate an increase in autophagic turnover as the larger autolysosomes are being more rapidly degraded. However, if the total number of vesicles between each treatment condition is significantly different in the presence of bafilomycin A1, this would indicate a difference in the formation rate of autophagic vacuoles between the two treatments.
V. Using co-localization of fluorescent tags to measure autophagosome-lysosome fusion
A distal step in the autophagic pathway is the fusion of the autophagosome with the lysosome and the degradation of both the bulk cytoplasm within the autophagosome and the autophagosome itself. In the described Purkinje neuron autophagy model, fusion can be visualized via live cell imaging as the co-localization of RFP-LC3 with LysoSensor Green.
Cerebellar cultures are infected with RFP-LC3 (multiplicity of infection 100, 24 h) and subjected to a trophic factor withdrawal time course (0-24 h).
Cells are stained with Hoechst and LysoSensor Green and imaged via live cell imaging as described above.
Quantitation of fluorescence signals from Purkinje neurons is assessed using Slidebook (4.2).
The degree of co-localization between RFP-LC3-positive vacuoles and LysoSensor Green-positive vacuoles is quantified using the Pearson’s correlation analysis tool in Slidebook 4.2, which is described in Figure 4 A-D. Pearson’s Correlation calculates the correlation between the intensity distributions of Cy3 (RFP-LC3) and FITC (LysoSensor Green) and expresses the correlation as a r-value between the range of −1 and +1, indicating no co-localization to perfect co-localization, respectively (Manders et al., 1983). The correlation of signal intensities between two channels (Cy3 and FITC) is calculated using the equation below where Ri is the intensity in channel 1 for pixel I, Gi is the intensity in channel 2 for the same pixel and Rav and Gav are the average or mean intensity values over all pixels (Slidebook 4.2).
As shown in Figure 4E, trophic factor withdrawal results in a time-dependent increase in autophagosome-to-lysosome fusion, indicated by the increase in the Pearson’s coefficient (r) from 0.15 to 0.75 over a 24 hr period.
Figure 4. (A-D) Schematic for measuring autophagosome-to-lysosome fusion.

At least 7-12 live cell images are captured per treatment condition (n=2 for at least three independent experiments). Fusion is measured as the degree of co-localization between the two fluorescent flours, Cy3 (RFP-LC3) and FITC (LysoSensor Green), which represents the fused vesicles or autolysosome. For each captured Purkinje neuron, a mask is created in Slidebook by outlining the perimeter of the neuron as shown in D. The Pearson’s correlation coefficient (r) is then determined between the FITC and Cy3 channels by choosing cross channel statistics from the Statistics option under the Mask menu. The degree of co-localized vesicles can be visualized in image D as yellow punctate staining. Images shown represent a 24 hr treatment of cerebellar cultures in TFW medium. Scale bar represents, 5 μm. (E) Purkinje neuron autophagy fusion rate. RFP-LC3-infected cerebellar cultures were subjected to a time course of TFW (0, 6, 10, 16, 24 hr). Coverslips were stained with Hoechst to visualize cellular nuclei and LysoSensor Green to visualize lysosomes, and images were captured via live cell imaging. Co-localization between RFP-LC3-positive vacuoles and Lysosensor Green-positive vacuoles was quantified using Pearson’s correlation coefficient analysis in Slidebook 4.2. The degree of RFP-LC3 and LysoSensor Green co-localization, expressed in Pearson’s correlation coefficient (r) increased with time and at 24 hr was 0.70, indicative of increased fusion. This time-dependent increase in fusion rate correlates with an increased accumulation of large autophagic vesicles at 24 hr TFW (data not shown).
Concluding remarks
The methodologies described herein are directed towards live-cell imaging of autophagy in primary cultured neurons utilizing RFP-tagged LC3 and LysoSensor Green. Examining autophagic vacuole size distribution profiles in combination with autophagosome-lysosome fusion measurements provides useful information regarding autophagic flux. More importantly, these analyses allow for the investigation of the effects of neuroprotective agents on the autophagic pathway and whether such compounds can alter the formation and/or fusion of autophagic vesicles. However, as with any method, there are caveats to consider when selecting methods to measure autophagy. The advantages and disadvantages of using RFP-LC3 and LysoSensor Green to measure autophagy via live-cell imaging are highlighted below.
Immunofluoresence microscopy is imperative when using primary neurons as a cellular model to study autophagy. Primary neuronal cultures derived from different areas of the brain most often comprise a heterogeneous population of cells including both neurons and non-neuronal cell types (glia). Although glia can be easily removed with the addition of drugs that inhibit cellular proliferation (cytosine arabinoside or 5′-DFUR), cultures that are rich in one neuronal cell type are difficult to produce. Therefore, measuring autophagy induction via LC3 immunoblotting in primary cultures raises the concern of neuronal specificity and caution should be used in data interpretation. Live cell imaging of autophagy induction using a fluorescent-tagged LC3 as described in this chapter allows one to selectively measure neurons of interest in a mixed population of cells. The difficulty of studying primary neurons, however, is the lack of efficient methods to introduce exogenous genes into neurons. Primary neurons, which are highly sensitive to culture conditions, are in general extremely difficult and labor-intensive to transfect. Adenovirus (Adv)-mediated gene delivery is a commonly used approach that we have found to be effective in cerebellar neurons. Interestingly, the adenoviral RFP-LC3 used in our studies selectively infects Purkinje neurons over granule neurons providing a unique model system to easily identify and examine Purkinje neuron autophagy.
As emphasized by Bampton et al. (2005), fluorescent LC3 is a more optimal marker of autophagosomes in comparison to monodansylcadaverine, which was shown to label only late acidic autophagosomes or autolysosomes after fusion with the lysosome. More recently, RFP-LC3 has been described as being more stable in comparison to GFP-LC3, which is sensitive to the acidity of lysosomes resulting in loss of the GFP signal (Kimura et al., 2007; Shaner et al., 2005) during autophagosome-to-lysosome fusion. Therefore, RFP-LC3 represents a marker of early to late autophagosomes including autolysosomes, which may not be labeled consistently using GFP-LC3. Combined with a lysosomal marker such as LysoSensor Green, RFP-LC3 allows for the complete visualization of autophagosome maturation into autolysosomes via time-lapse imaging and/or time course analyses.
To date, fusion measurements have been quantified by counting the number of autophagosomes (labeled by GFP-LC3 or mCherry-LC3) that colocalize with lysosomal markers, which is then expressed as a percentage of the total number of autophagosomes. We describe a more rapid method of fusion quantification over an autophagy induction time course using Pearson’s Correlation in Slidebook (4.2) software. Creating masks in Slidebook as described here is a convenient method to define a region of interest for further statistical analysis. Rather than counting the total number of colocalized RFP-LC3/LysoSensor Green vesicles within each neuron, a mask can be created around the area of interest, and the correlation coefficients between the red and green flours are calculated. As a cautionary note, this method is reliable when the captured images are of high quality (without saturation) and have been properly prepared via deconvolution to reduce or eliminate background.
When combined together, autophagic vacuole distribution profiles and autophagosome-lysosome fusion measurements provide detailed information on autophagy induction and flux. Moreover, usage of RFP-LC3 and LysoSensor Green allows for the morphological observation and quantification of autophagosome maturation and fusion with the lysosome. These methods can be used to characterize the process of autophagy in specific neuronal subtypes as well as to characterize drugs that regulate autophagy, which may prove useful in certain neurodegenerative conditions.
Acknowledgements
The authors would like to thank Aviva Tolkovsky for the kind gift of adeno-RFP-LC3. This research was supported by a Merit Award from the Veteran Affairs.
References
- Backer JM. The regulation and function of Class III PI3Ks: novel roles for Vps34. Biochem. J. 2008;410:1–17. doi: 10.1042/BJ20071427. [DOI] [PubMed] [Google Scholar]
- Bampton ET, Goemans CG, Niranjan D, Mizushima N, Tolkovsky AM. The dynamics of autophagy visualized in live cells: from autophagosome formation to fusion with endo/lysosomes. Autophagy. 2004;1:23–36. doi: 10.4161/auto.1.1.1495. [DOI] [PubMed] [Google Scholar]
- Cuervo AM. Autophagy: in sickness and in health. Trends in Cell Biology. 2004;14:70–77. doi: 10.1016/j.tcb.2003.12.002. [DOI] [PubMed] [Google Scholar]
- D’Mello SR, Galli C, Ciotti T, Calissano P. Induction of apoptosis in cerebellar granule neurons by low potassium: inhibition of death by insulin-like growth factor I and cAMP. Proc Natl Acad Sci USA. 1993;90:10989–10993. doi: 10.1073/pnas.90.23.10989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fass E, Shvets E, Degani I, Hirschberg K, Elazar Z. Microtubules support production of starvation-induced autophagosomes but not their targeting and fusion with lysosomes. J Biol Chem. 2006;281:36303–36316. doi: 10.1074/jbc.M607031200. [DOI] [PubMed] [Google Scholar]
- S.S., Laessig TA, Heidenreich KA. The p75 neurotrophin receptor can induce autophagy and death of cerebellar Purkinje neurons. J. Neurosci. 2004;24:4498–4509. doi: 10.1523/JNEUROSCI.5744-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimura A, Noda T, Yoshimori T. Dissection of the autophagosome maturation process by a novel reporter protein, tandem fluorescent-tagged LC3. Autophagy. 2007;3:452–460. doi: 10.4161/auto.4451. [DOI] [PubMed] [Google Scholar]
- Klionsky DJ, Cuervo AM, Seglen PO. Methods for monitoring autophagy from yeast to human. Autophagy. 2007;3:181–206. doi: 10.4161/auto.3678. [DOI] [PubMed] [Google Scholar]
- Lee HK, Iwasaki A. Autophagy and antiviral immunity. Curr. Opin. Immunol. 2008;20:23–29. doi: 10.1016/j.coi.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Deretic V. Unveiling the roles of autophagy in innate and adaptive immunity. Nat. Rev. Immunol. 2007;7:767–777. doi: 10.1038/nri2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Klionsky DJ. Development by self-digestion: molecular mechanisms and biological functions of autophagy. Dev. Cell. 2004;6:463–477. doi: 10.1016/s1534-5807(04)00099-1. [DOI] [PubMed] [Google Scholar]
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132:27–42. doi: 10.1016/j.cell.2007.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine B, Yuan J. Autophagy in cell death: an innocent convict? J. Clin. Invest. 2005;115:2679–2688. doi: 10.1172/JCI26390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Majeski AE, Dice JF. Mechanisms of chaperone-mediated autophagy. Int. J. Biochem. Cell Biol. 2004;36:2435–2444. doi: 10.1016/j.biocel.2004.02.013. [DOI] [PubMed] [Google Scholar]
- Manders EMM, Verbeek FJ, Aten JA. Measurement of co-localisation of objects in dual colour confocal images. J.Microscopy. 1993;169:375–382. doi: 10.1111/j.1365-2818.1993.tb03313.x. [DOI] [PubMed] [Google Scholar]
- Massey AC, Zhang C, Cuervo AM. Chaperone-mediated autophagy in aging and disease. Current Topics in Developmental Biology. 2006;73:205–235. doi: 10.1016/S0070-2153(05)73007-6. [DOI] [PubMed] [Google Scholar]
- Meijer AJ, Codogno P. Regulation and role of autophagy in mammalian cells. Int. J. Biochem. Cell Biol. 2004;36:2445–2462. doi: 10.1016/j.biocel.2004.02.002. [DOI] [PubMed] [Google Scholar]
- Mizushima N. Autophagy: process and function. Genes Dev. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- Natarajan K, Meyer MR, Jackson BM, Slade D, Roberts C, Hinnebusch AG, Marton MJ. Transcriptional profiling shows that Gcn4p is a master regulator of gene expression during amino acid starvation in yeast. Mol. Cell. Biol. 2001;21:4347–4368. doi: 10.1128/MCB.21.13.4347-4368.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selimi F, Lohof AM, Heitz S, Lalouette A, Jarvis CI, Bailly Y, Mariani J. Lurcher GRID2-induced death and depolarization can be dissociated in cerebellar Purkinje cells. Neuron. 2003;37:813–819. doi: 10.1016/s0896-6273(03)00093-x. [DOI] [PubMed] [Google Scholar]
- Shaner NC, Steinbach PA, Tsien RY. A guide to choosing fluorescent proteins. Nat Methods. 2005;2:905–909. doi: 10.1038/nmeth819. Review. [DOI] [PubMed] [Google Scholar]
- Tallóczy Z, Jiang W, Virgin HW, IV, Leib DA, Scheuner D, Kaufman RJ, Eskelinen E-L, Levine B. Regulation of starvation- and virus-induced autophagy by the eIF2α kinase signaling pathway. Proc. Natl. Acad. Sci. U. S. A. 2002;99:190–195. doi: 10.1073/pnas.012485299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie Z, Klionsky DJ. Autophagosome formation: core machinery and adaptations. Nat. Cell Biol. 2007;9:1102–1109. doi: 10.1038/ncb1007-1102. [DOI] [PubMed] [Google Scholar]
- Yue Z, Horton A, Bravin M, DeJager PL, Selimi F, Heintz N. A novel protein complex linking the delta 2 glutamate receptor and autophagy: implications for neurodegeneration in lurcher mice. Neuron. 2002;35:921–933. doi: 10.1016/s0896-6273(02)00861-9. [DOI] [PubMed] [Google Scholar]
- Zuo J, De Jager PL, Takahashi KA, Jiang W, Linden DJ, Heintz N. Neurodegeneration in Lurcher mice caused by mutation in delta2 glutamate receptor gene. Nature. 1997;388:769–773. doi: 10.1038/42009. [DOI] [PubMed] [Google Scholar]