

Abstract
The nonnucleoside reverse transcriptase (RT) inhibitors (NNRTIs) are a therapeutic class of compounds that are routinely used, in combination with other antiretroviral drugs, to treat HIV-1 infection. NNRTIs primarily block HIV-1 replication by preventing RT from completing reverse transcription of the viral single-stranded RNA genome into DNA. However, some NNRTIs, such as efavirenz, have been shown to inhibit the late stages of HIV-1 replication by interfering with HIV-1 Gag-Pol polyprotein processing, while others, such as the pyrimidinediones, have been shown to inhibit both HIV-1 RT-mediated reverse transcription and HIV-1/HIV-2 viral entry. Accordingly, in this review we describe the multiple mechanisms by which NNRTIs inhibit HIV-1 reverse transcription (and in some cases HIV-2 reverse transcription) and other key steps involved in HIV-1/HIV-2 replication.
Keywords: HIV, reverse transcriptase, nonnucleoside, Gag-Pol, viral entry
1. Introduction
Reverse transcription of the HIV single-stranded RNA genome into double-stranded DNA is an essential step in the virus replication life-cycle (Götte et al., 1999). This process is complex and requires the concerted function of two enzyme active sites in HIV reverse transcriptase (RT) (Fig. 1). Reverse transcription is initiated at the 3′-end of cellular lysyl-tRNALys,3, hybridized to the primer binding site (PBS) of the HIV RNA genome, by the RNA-primed RNA-dependent DNA polymerase activity (RDDP) of RT and is elongated until the 5′-end of the HIV-1 RNA is reached (Fig.1, Step 1). The product formed from this reaction is termed minus-strand strong-stop DNA. RT ribonuclease H (RNase H) activity then hydrolyzes the HIV genomic RNA (Fig. 1, Step 2) to allow the nascent DNA to hybridize with the repeat sequence (R) at the 3′end of the HIV genomic RNA (Fig. 1, Step 3). After this strand transfer, the nascent DNA strand is further elongated by RT DNA-primed RDDP activity (Fig. 1, Step 4). RNase H activity is again required to hydrolyze the rest of genomic RNA except for a purine rich sequence, termed the polypurine tract (PPT), which serves as a primer for the initiation of second strand DNA synthesis (Fig. 1, Step 5). RNA-primed DNA-dependent DNA polymerase activity (DDDP) then elongates the PPT primer (Fig. 1, Step 6). Removal of the PPT and tRNA primers by RT RNase H activity (Fig. 1, Step 7) then allows a second strand transfer to take place by interaction of the complementary PBS sequences (Fig. 1, Step 8). HIV-1 RT DNA-primed DDDP activity including strand-displacement activity completes the synthesis of the double stranded proviral DNA precursor. The final product of the complete reaction carries U3-R-U5 long terminal repeats (LTR) at both ends (Fig. 1, Step 9), and serves as a substrate for genomic DNA integration, catalyzed by HIV integrase.
Fig. 1.

Diagram of HIV reverse transcription mediated by the DNA polymerase, RNase H and strand-transfer activities of the viral RT. Each step is described in more detail in the text. RDDP and DDDP refer to the RNA- and DNA-dependent DNA polymerase activities of RT, respectively.
2. RT inhibitors (RTIs)
Due to its essential role in HIV replication, RT is a major target for antiretroviral drug development (Parniak and Sluis-Cremer, 2000). To date, two therapeutic classes of RT inhibitors (RTIs), the nucleoside and nucleotide RTIs (NRTIs) and the nonnucleoside RTIs (NNRTIs), are routinely used in the clinic to treat HIV-1 infection.
NRTIs are analogs of deoxyribonucleosides that lack a 3′-OH group on the deoxyribose sugar/pseudosugar. To exhibit antiviral activity, NRTIs must be metabolically converted by host-cell kinases to their corresponding triphosphate forms (NRTI-TPs). NRTI-TPs inhibit reverse transcription by acting as chain-terminators of DNA synthesis (Goody et al., 1991). Eight NRTIs have been approved for clinical use, namely 2′,3-dideoxyinosine (ddI), (R)-9-(2-phosphonylmethoxypropyl)adenine (TDF), 2′,3′-dideoxycytidine (ddC), (-)-β-2′,3′-dideoxy-3′-thiacytidine (3TC), 5-fluoro-1-[(2R,5S)-2-(hydroxymethyl)-1,3-oxathiolan-5-yl]cytosine (FTC), (1S,4R)-4-[2-amino-6-(cyclopropyl-amino)-9H-purin-9-yl]-2-cyclopentene-1-methanolsuccinate (ABC), 3′-azido-3′-deoxythymidine (AZT), and 2′,3′-deoxy-2′,3′-didehydrothymidine (d4T).
NNRTIs are chemically distinct from nucleosides and, unlike the NRTIs, do not require intracellular metabolism for activity. In general, NNRTIs are a group of small (<600 Da) hydrophobic compounds with diverse structures that specifically inhibit HIV-1 RT, but not HIV-2 RT (De Clercq, 1998). There are however, some exceptions and these are discussed in more detail below. The three NNRTIs approved for clinical use include 11-cyclopropyl-4-methyl-5,11-dihydro-6H-dipyrido[3,2-b:2′,3′-e][1,4]diazepin-6-one (nevirapine), 1-[3-[(1-methylethyl)amino]-2-pyridinyl]-4-[[5-[(methylsulfonyl)amino]-1H-indol-2-yl]carbonyl]-piperazine (delavirdine) and (4S)-6-chloro-4-cyclopropylethynyl-4-trifluoromethyl-1,4-dihydro-benzo[d][1,3]oxazin-2-one (efavirenz) (Fig. 2). Although NNRTIs represent, in terms of chemical structures, a heterogeneous class of inhibitors, they all interact with HIV-1 RT by binding to a single site on the p66 subunit of the HIV-1 RT p66/p51 heterodimer termed the NNRTI binding pocket that is situated approximately 10 Å from the RT DNA polymerase active site and 60 Å from the RT RNase H active site (Fig. 3) (Kohlstaedt et al., 1992).
Fig. 2.

Chemical structures of the NNRTIs described in this review.
Fig. 3.
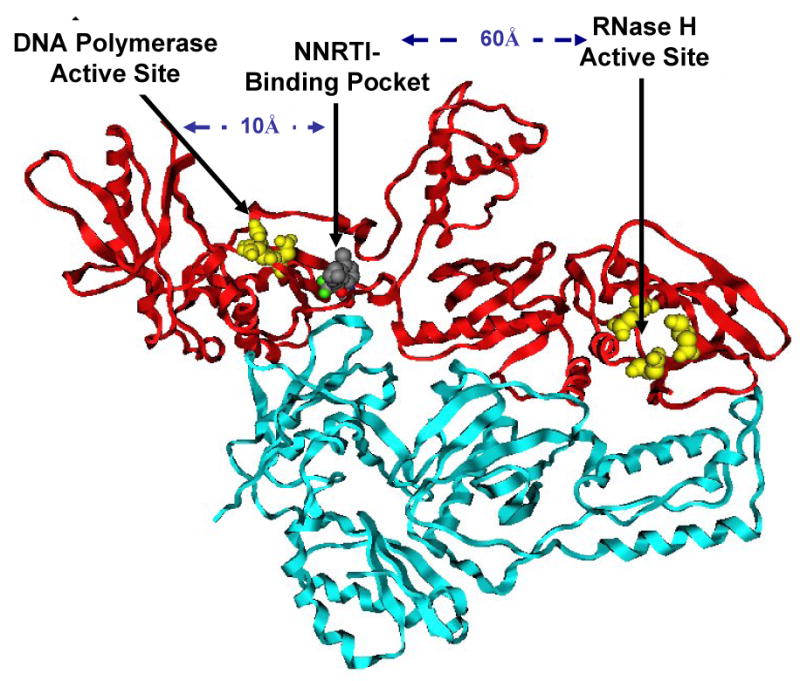
Ribbon representation of the HIV-1 RT in complex with efavirenz (PDB coordinates 1FK9). The p66 and p51 subunits of HIV-1 RT are colored red and blue, respectively. Residues located in the DNA polymerase active site (Asp110, Asp185, Asp186) and the RNase H active site (Glu478, Asp443, Asp498) are indicated with yellow spheres.
3. Inhibition of HIV replication by NNRTIs
NNRTIs act primarily by inhibiting HIV-1 reverse transcription (Fig. 4; Step 3). However, some NNRTIs such as efavirenz and the diarylpyrimidine derivatives dapivirine (TMC 120) and etravirine (TMC 125) (Fig. 2) have been shown to inhibit the late stages of HIV-1 replication by interfering with HIV-1 Gag-Pol polyprotein processing (Fig. 4; Step 7), while others such as 1-(3-cyclopenten-1-yl)methyl-6-(3,5-dimethylbenzoyl)-5-ethyl-2,4,pyrimidinedione (IQP-0410, formerly SJ-3366) have been shown to inhibit both RT-mediated reverse transcription (Fig. 4; Step 3) and viral entry (Fig. 4; Step 2). Accordingly, in this review we will describe the multiple mechanisms by which NNRTIs inhibit HIV-1 reverse transcription and other steps involved in viral replication.
Fig. 4.

The HIV life-cycle. (1) HIV binds to a CD4 receptor and one of two co-receptors (CXCR4 or CCR5) on the surface of the CD4+ T-lymphocyte. (2) The viral envelope then fuses with the host cell membrane. After fusion, the viral capsid is released into the host cell cytoplasm. (3) The HIV RT converts the single-stranded HIV RNA into double-stranded DNA (dsDNA). (4) The proviral dsDNA integrates into the host cell's genome in a process mediated by the viral IN. (5) The replication and transcription machinery of the host cell is involved in provirus replication, and in viral mRNA synthesis. (6) Viral mRNAs are used in the synthesis of the viral proteins Gag, Gag-Pol and Env, as well as accessory proteins such as Nef, Vif, Vpr, and Vpu. (7) Sets of viral protein chains come together with 2 copies of the viral RNA to generate an immature virus particle that pushes out (or buds) from the cell, taking some of the cell membrane with it. (8) The virus then matures, which involves the processing of viral proteins by the HIV PR.
4. Inhibition of HIV-1 reverse transcription by NNRTIs
Steady-state kinetic analyses initially demonstrated that NNRTIs acted as noncompetitive or uncompetitive inhibitors of HIV-1 RT DNA polymerization reactions (De Clercq, 1998). However, the NNRTI kinetic constants for inhibition (IC50 or Ki) appeared to be dependent on the sequence and identity (i.e. DNA/DNA versus RNA/DNA) of the template/primer (T/P) substrate used in the polymerase assay. For example, the IC50 value for a given NNRTI to inhibit RT DNA polymerase activity can vary between 2- and 400-fold depending on the T/P substrate (as well as other conditions) used in the assay (Table 1). In 1999, Quan et al used quantitative PCR to analyze HIV-1 reverse transcription within acutely infected cells treated with nevirapine (Quan et al., 1999). This study showed that minus-strong stop DNA synthesis was only slightly inhibited by nevirapine at concentrations that greatly exceed those necessary for complete suppression of viral replication. In contrast, intermediate-length and full-length reverse transcribed products generated after the first strand transfer decreased gradually as viral DNA strand elongation took place. Based on these observations, the authors suggested that NNRTIs acted in a stochastic manner with longer templates providing more opportunities for the inhibitors to act (Quan et al., 1998; Quan et al., 1999). However, as described below, recent studies have demonstrated that NNRTIs may preferentially target specific steps during reverse transcription. This information is reviewed below.
Table 1.
Inhibitory and binding constants determined for nevirapine, delavirdine and efavirenz using different assay systems.
| Assay | Virus & Cell Type | T/P Substrate | EC50 or IC50 or Kd (nM) | ||
|---|---|---|---|---|---|
| Nevirapine | Delavirdine | Efavirenz | |||
| Antiviral a (EC50) |
LAI P4/R5 | NA | 85.5 ± 27 | 63.3 ± 19 | 1.46 ± 0.29 |
| Antiviral b (EC50) |
NL4-3 HeLa-CD4 | NA | 490 ± 18 | 290 ± 10 | 10 ± 0.5 |
| Antiviral c (EC50) |
HXB2 MT4 | NA | 99 ± 32 | 18 ± 6 | 1.6 ± 0.8 |
| Antiviral c (EC50) |
HXB2 PBMC | NA | ND | ND | 2.1 ± 0.5 |
| Polymerase d (IC50) |
NA | poly(rC)-oligo(dG) | 200 ± 45 | 5.4 ± 0.4 | 7.7 ± 0.9 |
| Polymerase e (IC50) |
NA | poly(rA)-oligo(dT) | 7222 ± 138 | 2180 ± 62 | 94.2 ± 6.3 |
| Polymerase f (IC50) |
NA | Heteropolymeric (RNA/DNA) | 2217 ± 954 | ND | 3.9 ± 0.2 |
| RNase H d (IC50) |
NA | Polymerase-independent | 216 ± 40 | ND | 4.5 ± 0.8 |
| RNase H | NA | Polymerase-dependent | Accelerated h | Accelerated h | Accelerated h |
| Biacore g (Kd) |
NA | NA | 1550 ± 441 | 6.19 ± 0.15 | 0.63 ± 0.34 |
Data derived from Clark et al., 2006;
Data derived from Sato et al., 2006;
Data derived from Hazen et al., 2005;
Data derived from Motakis and Parniak, 2002;
Data derived from Nissley et al., 2007;
Data derived from Hang et al., 2007;
Data derived from Geitmann et al., 2006;
studies by Shaw-Reid et al., 2005, Hang et al., 2007 and Radzio and Sluis-Cremer, 2007 have shown that NNRTIs increase the rate of polymerase dependent RNase H activity. However, quantitative analyses of these findings have not yet been reported.
4.1. Inhibition of the first strand transfer
The first strand transfer reaction is an essential step in reverse transcription (Fig. 1) and requires the co-ordination of both the DNA polymerase and RNase H activities of HIV-1 RT. Although the NNRTI-binding pocket in RT is located ∼ 60Å from the RNase H active site of RT (Fig. 3), several studies have demonstrated that NNRTIs can either partially inhibit or accelerate this activity depending on the mode of RNase H activity (Shaw-Reid et al., 2005; Hang et al., 2007; Radzio and Sluis-Cremer, 2007). For example, Hang et al demonstrated that several structurally diverse NNRTIs all partially inhibited 5′-RNA directed HIV-1 RNase H activity (or polymerase independent RNase H activity) with maximal inhibition of 40-65% (Table 1) (Hang et al., 2007). In contrast, the 3′-DNA directed RNase H activity of RT (or polymerase dependent RNase H activity) is significantly stimulated by NNRTI binding to RT (Shaw-Reid et al., 2005; Hang et al., 2007; Radzio and Sluis-Cremer, 2007). Therefore, strand transfer inhibitory potencies of NNRTIs may be dependent on both DNA polymerase and RNase H inhibition efficiencies. In this regard, Hang et al demonstrated that IC50 values for inhibition of strand transfer for some NNRTIs were significantly lower in strand transfer assays than in polymerase or RNase H assays (Hang et al., 2006). Furthermore, NNRTI resistance mutations affected NNRTI inhibitory potencies in different RT assays in an activity and compound specific manner. For example, the level of efavirenz resistance due to the single mutation Y188L was more than 30-fold higher when determined in strand transfer assays as compared to the polymerase and RNase H assays (Hang et al., 2006).
4.2. Inhibition of plus-strand initiation
Grobler et al demonstrated that NNRTIs potently inhibited plus-strand initiation in vitro under conditions in which little or no inhibition of minus-strand DNA synthesis was observed (Grobler et al., 2007). In addition, they showed that NNRTIs completely abrogated dNTP binding to RT associated with RNA PPT primer/DNA template substrate, but only exerted a modest decrease in the dNTP affinity for RT associated with a DNA PPT primer/DNA template. Götte et al have delineated the temporal events involved in HIV-1 RT initiation of plus-strand DNA synthesis (Fig. 5) (Götte et al., 2001). RT first binds to the RNA PPT primer/DNA template and elongates the primer by 12 nucleotides. The enzyme then goes back and binds the PPT with its 5′-end in the polymerase active site and cleaves the PPT from the nascent DNA. RT then completes elongation of the nascent DNA. As described by Götte et al, and others, RT can bind RNA-DNA hybrid duplexes in different modes depending on the structure of the substrate (Götte et al., 2000; Palaniappan et al., 1998). With substrates that contain a recessed DNA 3′-OH, RT engages the nucleic acid in a polymerase-dependent mode of binding where the 3′-end of the DNA is positioned in the polymerase active site poised to be extended. For RNA-DNA hybrid duplex substrates in which the 3′-end of the DNA is not recessed, RT preferentially engages substrate in a polymerase-independent mode where the 5′-end of the RNA strand, and not the 3′-end, is situated in the polymerase active site. Thus, most recessed RNA oligonucleotides annealed to DNA templates do not act as efficient primers for initiation of polymerization. RNA PPT hybrid duplexes are selectively utilized as primers by RT because binding of the 3′-end of the RNA in the polymerase active site is not as disfavored as with non-RNA PPT primers. In this regard, Grobler et al suggested that NNRTIs affect the enzyme's ability to bind the RNA PPT primer/DNA template in a polymerase dependent mode (Grobler et al., 2007). It should be noted that other studies have also demonstrated that NNRTIs directly affect the RT-T/P equilibrium (Divita et al., 1993; Rittinger et al., 1995).
Fig. 5.

Diagram of the temporal events involved in the initiation of plus-strand DNA synthesis.
5. Kinetic mechanism by which NNRTIs inhibit HIV-1 RT DNA polymerase activity
Most studies describing the mechanism of action of NNRTIs on HIV-1 RT mediated DNA synthesis reactions have been carried out using steady-state kinetics. Because steady-state experiments are unable to resolve kinetic steps which are masked by the rate-limiting step of a reaction (the release of DNA substrate from RT), this approach cannot elucidate the detailed interactions of the drug with RT at the polymerase active site (Kati et al., 1992). In this regard, we, and others, have used the pre-steady-state kinetic approach to provide detailed mechanistic insights into the catalytic events that occur directly at the enzyme's active site in the absence and presence of NNRTIs (Table 2) (Spence et al., 1995; Wang et al., 2004; Xia et al., 2007). These studies show that NNRTI-RT-T/P complexes display a metal-dependent increase in dNTP binding affinity (Kd). For example, the Kd for Mg2+-dTTP, Mn2+-dTTP and Co2+-dTTP are increased 130-, 15.5- and 1.1-fold when nevirapine is complexed to RT (Table 2). In contrast, the NNRTI-RT/T/P complexes exhibit a metal-independent decrease in the maximum rate of dNTP incorporation (kpol). Furthermore, no phosphorothioate elemental effects are evident irrespective of the metal ion used in the assay (Table 2). (Phosphorothioate elemental effects, derived from experiments which compare the rates of incorporation of the natural dNTP substrate versus dNTPαS, are frequently used as a diagnostic for determining whether the chemical step of polymerization reactions is rate-limiting.) These data suggest that the slow rate of dNTP incorporation observed for NNRTI-RT-T/P complexes might not be due to a direct effect of the inhibitor on phosphodiester bond formation, as suggested previously (Spence et al., 1995), but rather an indirect effect through alteration/perturbation of the constellation of amino acids involved in positioning the active site for efficient catalysis (Xia et al., 2007). In this regard, structural studies have demonstrated that inhibitor binding in the NNRTI-binding pocket causes the “primer grip” to be shifted upward by approximately 5 Å in comparison with its position in the RT-T/P binary and RT-T/P-dNTP ternary complexes (Ding et al., 1997). Therefore, the slow rate of dNTP incorporation observed for NNRTI-RT-T/P complexes might be due to changes in the position and conformation of the “primer grip” which significantly slow down the necessary conformational changes that are required to align the substrates and to facilitate phosphodiester bond formation (Xia et al., 2007).
Table 2.
Pre-steady-state kinetic parameters determined for incorporation of dNTP or dNTPαS by RT-T/P and nevirapine-RT-T/P complexes using different metal ion complexes.
| Drug | Metal | dNTP |
Kd (μM) |
kpol (s-1) |
kpol/Kd (s-1 · μM-1) |
Fold change in: | Metal Effect | ||
|---|---|---|---|---|---|---|---|---|---|
| Kd | kpol | kpol/Kd | |||||||
| None a | Mg2+ | dATP | 4 | 33 | 8.25 | - | - | - | - |
| Nevirapine b | Mg2+ | dATP | 0.019 | 0.0087 | 0.46 | 210.5 | 3793 | 17.9 | ND |
| None c | Mg2+ | dTTP | 2.6 | 8.9 | 3.4 | - | - | - | - |
| None c | Mg2+ | Sp-TTPαS | 2.9 | 6.8 | 2.4 | 0.9 | 1.3 | 1.4 | 1.3 |
| Nevirapine c | Mg2+ | dTTP | 0.02 | 0.0060 | 0.30 | 130 | 1483 | 11.3 | - |
| Nevirapine c | Mg2+ | Sp-TTPαS | 0.02 | 0.0057 | 0.29 | 130 | 1561 | 11.7 | 1.1 |
| None c | Mn2+ | dTTP | 3.4 | 5.1 | 1.5 | - | - | - | - |
| None c | Mn2+ | Sp-TTPαS | 4.2 | 3.2 | 0.76 | 0.8 | 1.6 | 2.0 | 1.6 |
| Nevirapine c | Mn2+ | dTTP | 0.22 | 0.0054 | 0.025 | 15.5 | 944 | 60 | - |
| Nevirapine c | Mn2+ | Sp-TTPαS | 0.21 | 0.0028 | 0.013 | 16 | 1821 | 115 | 1.9 |
| None c | Co2+ | dTTP | 21.4 | 2.2 | 0.10 | - | - | - | - |
| None c | Co2+ | Sp-TTPαS | 20.2 | 2.3 | 0.11 | 1.1 | 1.0 | 0.9 | 1.0 |
| Nevirapine c | Co2+ | dTTP | 20.2 | 0.0019 | 9.4×10-5 | 1.1 | 1157 | 1063 | - |
| Nevirapine c | Co2+ | Sp-TTPαS | 22.5 | 0.0013 | 5.8×10-5 | 1.0 | 1692 | 1724 | 1.5 |
Data derived from Kati et al., 1992;
Data derived from Spence et al., 1995;
Data derived from Xia et al., 2007.
6. Inhibition of HIV-2 reverse transcription by NNRTIs
Although HIV-2 RT shows significant amino acid sequence homology to HIV-1 RT, most NNRTIs are completely inactive against HIV-2 (De Clercq, 1998). This lack of activity is primarily due to the residues at codon 181 and 188 (Tyr181 and Tyr188 in HIV-1; Ile181 and Leu188 in HIV-2) which prevent the drugs from binding to HIV-2 RT (Condra et al., 1992). Interestingly, the phenylmethylthiazolylthiourea derivative MSK-076 shows marked activity against both HIV-1 (EC50 = 0.0018 μM) and HIV-2 (EC50 = 0.63 μM) replication (Ren et al., 2000; Auwerx et al., 2004). Steady-state kinetic studies demonstrated that MSK-076 was a non-competitive inhibitor of both HIV-1 and HIV-2 RT (Ren et al., 2000; Auwerx et al., 2004). In cell culture, the drug selected for the A101P and G112E mutations in HIV-2 RT, and for K101E, Y181C and G190R mutations in the HIV-1 RT (Auwerx et al., 2004). Mapping of the resistance mutations to the HIV-1 RT structure ascertained that A101P is located at a position equivalent to the NNRTI-binding site residue Lys101 in HIV-1 RT but that G112E resides close to the DNA polymerase active site (Fig. 6), implying a novel molecular mode of action and mechanism of resistance (Auwerx et al., 2004).
Fig. 6.
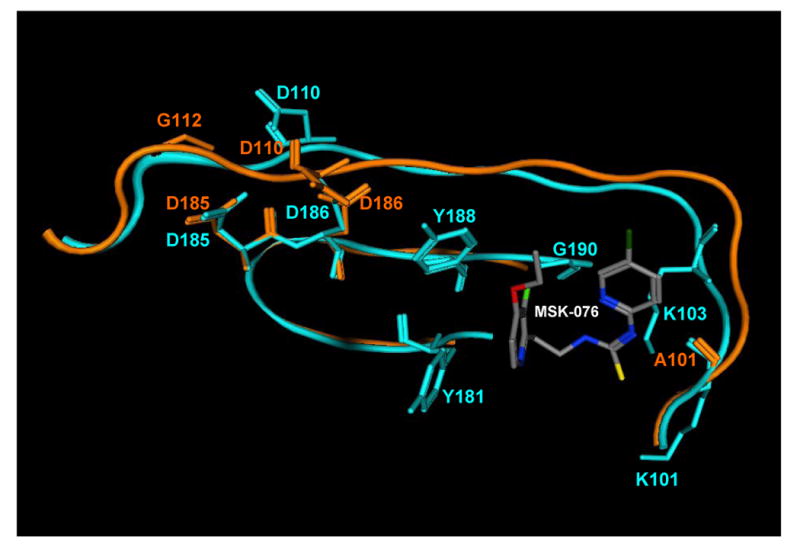
Superimposition of the HIV-1 RT in complex with MSK-076 (blue; PDB co-ordinates 1DTT) with the apo-form of HIV-2 RT (orange; PDB co-ordinates 1MU2), illustrating location of the NNRTI-binding pocket (in HIV-1) and conserved residues in the DNA polymerase active site (Asp110, Asp185, Asp186). Amino acid residues that were mutated in HIV-1 (Lys101, Tyr181 and Gly190) and HIV-2 (Ala101 and Gly112) during selection experiments with MSK-076 are also depicted.
7. Pyrimidinediones inhibit HIV-1 reverse transcription and HIV-1/HIV-2 viral entry
The pyrimidinedione IQP-0410 was found to inhibit both HIV-1 (EC50 ∼ 1 nM) and HIV-2 (EC50 ∼ 150 nM) replication (Buckheit et al., 2001). However, unlike MSK-076, IQP-0410 is a potent inhibitor of HIV-1 RT, but not of HIV-2 RT. Instead, IQP-0410 inhibits viral entry by targeting a conformational epitope that forms following interaction of the viral receptors with CD4 and the chemokine receptors but prior to fusion of the viral and cellular membranes. Structure-activity-relationship evaluations have demonstrated that the molecular features of the pyrimidinediones responsible for the two distinct mechanisms of action are integrated in the structure of the molecule and the substituents required for RT inhibition versus entry inhibition are not distinct but overlapping (Buckheit et al., 2007). In this regard, it is anticipated that IQP-0410 might be expected to provide the same level of protection in patients as the combination of efavirenz and enfuvirtide (a peptide fusion inhibitor), but in one small molecule (Buckheit et al., 2007).
8. NNRTIs with distinct binding sites and mechanisms of action
All of the NNRTIs described above bind in the NNRTI-binding pocket of HIV-1 RT that is situated between the β6-β10-β9 and β12-β13-β14 sheets in the palm subdomain of the p66 subunit of the enzyme. However, there are some NNRTIs that appear to have distinct binding sites and mechanisms of action. These are discussed below.
8.1. [2′,5′-Bis-O-(tert-butyldimethylsilyl)-β-D-ribofuranosyl]-3′spiro-5″-(4″-amino-1″, 2″-oxathiole-2″,2″-dioxide)thymine (TSAO-T) derivatives
TSAO-T (Fig. 2) derivatives represent a unique class of compounds that are potent inhibitors of HIV-1 replication (Camarasa et al., 2004). Although TSAO-T derivatives are highly functionalized nucleosides, they behave as allosteric inhibitors of HIV-1 RT (Balzarini et al., 1992). They were originally thought to target the same non-substrate binding site as all the other NNRTIs. However, TSAO compounds are one of few inhibitors that require amino acids in both the p66 and p51 subunits of RT for optimal interaction with the enzyme (Balzarini et al., 1993). Furthermore, we demonstrated that the N-3-ethyl derivative of TSAO-T destabilizes the inter-subunit interactions of HIV-1 RT (Sluis-Cremer et al., 2000). More recently, we carried out a comprehensive structure-activity-relationship analysis of this class of compounds and identified key functional regions that were responsible for their ability to inhibit RT dimerization (Sluis-Cremer et al., 2006).
8.2. 5-methyl-1-(4-nitro-phenyl)-2-oxo-2,5-dihydro-1H-pyrido[3,2-b]indole-3-carbonitrile (INDOPY)
Steady-state kinetics analyses, binding studies and site-specific footprinting experiments have demonstrated that INDOPY (Fig. 2) binds in the DNA polymerase active site of RT and traps the T/P substrate in the post-translocational state, thereby preventing binding and incorporation of the next complementary nucleotide (Jochmans et al., 2006). As expected, this novel mode of action also translates into a unique resistance profile: INDOPY susceptibility is unaffected by NNRTI or multi-drug NRTI resistance mutations, but exhibits decreased susceptibility to A62V, M184V and Y115F and hypersusceptibility to K65R (Jochmans et al., 2006).
9. NNRTI inhibition of post-integration steps in the HIV-1 life-cycle
The post-integration steps of the HIV life-cycle begin after proviral DNA integration (Fig. 4, Step 4), where singly and multiply spliced mRNAs are transcribed that encode the HIV-1 envelope proteins and regulatory proteins, respectively (Rabson and Graves, 1997), while unspliced mRNAs are translated to form the Pr55gag (Gag) and Pr160gag-pol (Gag-Pol) polyproteins (Fig. 4. Steps 5 and 6) (Swanstrom, 1997). Gag consists of the viral structural proteins matrix (MA), capsid (CA), nucleocapsid (NC), p6 and two spacer peptides termed p1 and p2. Gag-Pol consists of MA, CA and NC in addition to protease (PR), RT and integrase (IN). Following translation, Gag and Gag-Pol are targeted to the host cell plasma membrane, a process that is dependent on the myristoylation of the N-terminus of Gag (Park and Morrow, 1992; Smith et al., 1993). As the newly assembled virions bud from the host cell it is thought that Gag and Gag-Pol polyproteins oligomerize to promote the activation of the viral PR, through the formation of an active homodimer. This results in the sequential cleavage of Gag and Gag-Pol into the mature structural proteins and viral enzymes (Fig. 4, Step 7) (Kaplan et al., 1994; Pettit et al., 1998).
9.1. NNRTIs act as potent inhibitors of HIV-1 RT dimerization
It has been demonstrated that some NNRTIs act as chemical enhancers of HIV-1 RT heterodimerization (Tachedjian et al., 2001; Venezia et al., 2006). To date, efavirenz was found to be the most potent enhancer of RT heterodimerization, whereas nevirapine has a weak effect and delavirdine has no effect at all (Tachedjian et al., 2001). While there doesn't appear to be a correlation between the impact of NNRTI-mediated enhancement of RT heterodimerization and the defects in RT polymerase function (Xia et al., 2007), recent studies have demonstrated effects of some potent NNRTIs, (e.g. efavirenz, dapivirine and etravirine) on the late stages of HIV replication (Tachedjian et al., 2005; Figueiredo et al., 2006).
9.2. Efavirenz accelerates Gag-Pol processing
Gag-Pol expresses a p66 embedded form of the HIV-1 RT. Studies demonstrate that efavirenz is not only a potent enhancer of RT heterodimerization, but it also strongly promotes p66/p66 homodimerization (Tachedjian et al., 2005; Figueiredo et al., 2006). In contrast, nevirapine has no significant effect on p66/p66 homodimerization (Tachedjian et al., 2005; Figueiredo et al., 2006). This observation provided the impetus to evaluate whether efavirenz can mediate an effect on Gag-Pol processing. Using a 90 kDa Pol construct that expresses the last 3 amino acids of NC, the transframe region, PR, RT and the first 43 amino acid of IN in bacteria (Sluis-Cremer et al., 2004), it was shown that efavirenz enhances the processing kinetics of this minimal Pol to the p66 and p51 RT subunits compared to untreated and nevirapine treated bacteria (Fig. 7) (Tachedjian et al., 2005). An explanation for this observation is that efavirenz is promoting the oligomerization of the minimal Pol construct by binding to p66 resulting in activation of the HIV-1 PR. Yeast two-hybrid analyses of Pol polyproteins that includes a knock-out active site mutation in PR and are truncated in the integrase domain, confirmed that efavirenz mediates a concentration dependent increase in Pol/Pol dimerization (Figueiredo et al., 2006).
Fig. 7.
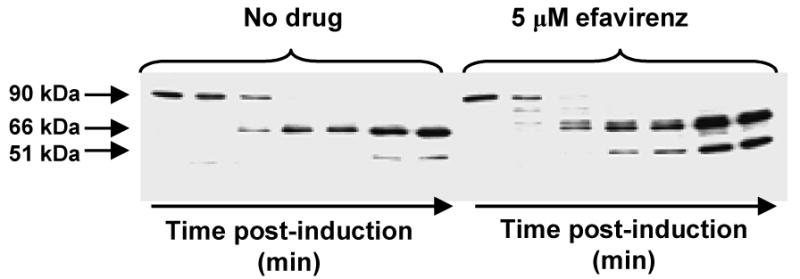
Western blot analysis of sequential processing products from a model Pol construct, pol-pET28a(+), expressed in E. coli strain BL21(DE3)pLysS and grown in the absence or presence of 5 μM efavirenz. Samples were taken at 60, 75, 90, 105, 120, 150 and 180 min post-induction, respectively. Western blots were probed with mAB 5B2 (anti-RT). As is clearly evident, more p66/p51 RT is generated in the efavirenz reaction than in the absence of drugs.
9.3. Potent NNRTIs inhibit the late stages of HIV-1 replication
A recent study evaluated the impact of potent NNRTIs in HIV-1 transfected 293T and HeLa cells (Figueiredo et al., 2006). Treatment of these cells with efavirenz, dapivirine and etravirine, but not nevirapine and delavirdine, resulted in a dramatic increase in the processing of intracellular Gag and Gag-Pol polyproteins (Figueiredo et al., 2006). This enhancement of polyprotein processing was associated with a decrease in viral particle production. Enhanced Gag and Gag-Pol processing was even more dramatic when cells were transfected with a myristoylation-defective HIV mutant indicating that the effect was not dependent on targeting of Gag and Gag-Pol to the plasma membrane and that it occurs more efficiently in the cell cytoplasm. No decrease in viral particle release was observed with a HIV-1 mutant expressing the K103N RT mutation that confers efavirenz resistance or with a PR-defective HIV mutant. Furthermore, similar experiments performed with MoMLV demonstrated that efavirenz did not confer a non-specific effect on viral particle production. A model has been proposed to explain these data. In this model, potent NNRTIs bind to the RT embedded in Gag-Pol thereby promoting the interaction between individual Gag-Pol polyproteins. This leads to premature activation of the HIV-1 PR embedded within Gag-Pol, and the subsequent cleavage of the precursor polypeptides. As a consequence, the amount of full-length viral polyproteins available for assembly and budding from the host cell membrane decreases.
10. Conclusions and future perspectives
NNRTIs represent an important therapeutic class of inhibitors used in the treatment of HIV-1 infection. Although multiple studies have demonstrated that they primarily block HIV-1 replication by inhibiting the DNA polymerase active site of RT, recent work has suggested that their inhibition of reverse transcription might also be due to effects on RT RNase H activity and/or T/P binding. An in-depth understanding of the multiple mechanisms by which NNRTIs inhibit reverse transcription is essential because this information may be critical for the development of the next-generation of NNRTIs and for understanding drug resistance.
Some NNRTIs also inhibit the late stages of HIV-1 replication by interfering with HIV-1 Gag-Pol polyprotein processing. However, it should be noted that the concentration of NNRTI that is required to affect the late stage of HIV replication is three orders of magnitude greater than the concentration that blocks reverse transcription. Nevertheless, in the case of efavirenz, these drug concentrations are observed in the plasma of efavirenz treated individuals (Almond et al., 2005). The large differences in potency of the NNRTIs for the mature RT heterodimer and the proposed target for the late effect, the RT embedded within Gag-Pol, may be due to differences in the relative affinity of efavirenz for the two targets. In this regard, elucidation of the structure of RT embedded within Gag-Pol would contribute to our understanding of the difference between binding of NNRTIs to this target compared to the NNRTI-binding pocket of the mature RT, and might facilitate the development of more potent antiviral drugs that target Gag-Pol.
Acknowledgments
Research in the NSC laboratory is supported by a grant from the National Institutes of Health (R01 GM068406-01). GT was supported by NHMRC Career Development Award 235102 and NHMRC Project Grant 381705.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Almond LM, Hoggard PG, Edirisinghe D, Khoo SH, Back DJ. Intracellular and plasma pharmacokinetics of efavirenz in HIV-infected individuals. J Antimicrob Chemother. 2005;56:738–744. doi: 10.1093/jac/dki308. [DOI] [PubMed] [Google Scholar]
- Auwerx J, Stevens M, Van Rompay AR, Bird LE, Ren J, De Clercq E, Oberg B, Stammers DK, Karlsson A, Balzarini J. The phenylmethylthiazolylthiourea nonnucleoside reverse transcriptase (RT) inhibitor MSK-076 selects for a resistance mutation in the active site of human immunodeficiency virus type 2 RT. J Virol. 2004;78:7427–7437. doi: 10.1128/JVI.78.14.7427-7437.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzarini J, Perez-Perez MJ, San-Felix A, Schols D, Perno CF, Vandamme AM, Camarasa MJ, De Clercq E. 2′,5′-Bis-O-(tert-butyldimethylsilyl)-3′-spiro-5″-(4″-amino-1″,2″-oxathiole-2″,2′-dioxide)pyrimidine (TSAO) nucleoside analogues: highlyselective inhibitors of human immunodeficiency virus type 1 that are targeted at the viral reverse transcriptase. Proc Natl Acad Sci USA. 1992;15:4392–4396. doi: 10.1073/pnas.89.10.4392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balzarini J, Velazquez S, San-Felix A, Karlsson A, Perez-Perez MJ, Camarasa MJ, De Clercq E. Human immunodeficiency virus type 1-specific [2′,5′-bis-O-(tert-butyldimethylsilyl)-beta-D-ribofuranosyl]-3′-spiro-5″-(4″-amino-1″,2″- oxathiole-2″,2″-dioxide)-purine analogues show a resistance spectrum that is different from that of the human immunodeficiency virus type 1-specific non-nucleoside analogues. Mol Pharmacol. 1993;43:109–114. [PubMed] [Google Scholar]
- Buckheit RW, Jr, Hartman TL, Watson KM, Kwon HS, Lee SH, Lee JW, Kang DW, Chung SG, Cho EH. The structure-activity relationships of 2,4(1H,3H)-pyrimidinedione derivatives as potent HIV type 1 and type 2 inhibitors. Antivir Chem Chemother. 2007;18:259–275. doi: 10.1177/095632020701800502. [DOI] [PubMed] [Google Scholar]
- Buckheit RW, Jr, Watson K, Fliakas-Boltz V, Russell J, Loftus TL, Osterling MC, Turpin JA, Pallansch LA, White EL, Lee JW, Lee SH, Oh JW, Kwon HS, Chung SG, Cho EH. SJ-3366, a unique and highly potent nonnucleoside reverse transcriptase inhibitor of human immunodeficiency virus type 1 (HIV-1) that also inhibits HIV-2. Antimicrob Agents Chemother. 2001;45:393–400. doi: 10.1128/AAC.45.2.393-400.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camarasa MJ, San-Felix A, Velazquez S, Perez-Perez MJ, Gago F, Balzarini J. TSAO compounds: the comprehensive story of a unique family of HIV-1 specific inhibitors of reverse transcriptase. Curr Top Med Chem. 2004;4:945–963. doi: 10.2174/1568026043388600. [DOI] [PubMed] [Google Scholar]
- Clark SA, Shulman NS, Bosch RJ, Mellors JW. Reverse transcriptase mutations 118I, 208Y, and 215Y cause HIV-1 hypersusceptibility to non-nucleoside reverse transcriptase inhibitors. AIDS. 2006;20:981–984. doi: 10.1097/01.aids.0000222069.14878.44. [DOI] [PubMed] [Google Scholar]
- Condra JH, Emini EA, Gotlib L, Graham DJ, Schlabach AJ, Wolfgang JA, Colonno RJ, Sardana VV. Identification of the human immunodeficiency virus reverse transcriptase residues that contribute to the activity of diverse nonnucleoside inhibitors. Antimicrob Agents Chemother. 1992;36:1441–1446. doi: 10.1128/aac.36.7.1441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E. The role of non-nucleoside reverse transcriptase inhibitors (NNRTIs) in the therapy of HIV-1 infection. Antiviral Res. 1998;38:153–179. doi: 10.1016/s0166-3542(98)00025-4. [DOI] [PubMed] [Google Scholar]
- Ding J, Hughes SH, Arnold E. Protein-nucleic acid interactions and DNA conformation in a complex of human immunodeficiency virus type 1 reverse transcriptase with a double-stranded DNA template-primer. Biopolymers. 1997;44:125–138. doi: 10.1002/(SICI)1097-0282(1997)44:2<125::AID-BIP2>3.0.CO;2-X. [DOI] [PubMed] [Google Scholar]
- Divita G, Müller B, Immendörfer U, Gautel M, Rittinger K, Restle T, Goody RS. Structural mapping of catalytic site with respect to alpha-subunit and noncatalytic site in yeast mitochondrial F1-ATPase using fluorescence resonance energy transfer. J Biol Chem. 1993;268:13178–13186. [PubMed] [Google Scholar]
- Figueiredo A, Moore KL, Mak J, Sluis-Cremer N, De Bethune MP, Tachedjian G. Potent Nonnucleoside Reverse Transcriptase Inhibitors Target HIV-1 Gag-Pol. PLoS Pathog. 2006;2:e119. doi: 10.1371/journal.ppat.0020119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geitmann M, Unge T, Danielson UH. Biosensor-based kinetic characterization of the interaction between HIV-1 reverse transcriptase and non-nucleoside inhibitors. J Med Chem. 2006;49:2367–2374. doi: 10.1021/jm0504048. [DOI] [PubMed] [Google Scholar]
- Goody RS, Muller B, Restle T. Factors contributing to the inhibition of HIV reverse transcriptase by chain-terminating nucleotides in vitro and in vivo. FEBS Lett. 1991;7:1–5. doi: 10.1016/0014-5793(91)81089-q. [DOI] [PubMed] [Google Scholar]
- Gotte M, Kameoka M, McLellan N, Cellai L, Wainberg MA. Analysis of efficiency and fidelity of HIV-1 (+)-strand DNA synthesis reveals a novel rate-limiting step during retroviral reverse transcription. J Biol Chem. 2000;276:6711–6719. doi: 10.1074/jbc.M009097200. [DOI] [PubMed] [Google Scholar]
- Gotte M, Li X, Wainberg MA. HIV-1 reverse transcription: a brief overview focused on structure-function relationships among molecules involved in initiation of the reaction. Arch Biochem Biophys. 1999;365:199–210. doi: 10.1006/abbi.1999.1209. [DOI] [PubMed] [Google Scholar]
- Grobler JA, Dornadula G, Rice MR, Simcoe AL, Hazuda DJ, Miller MD. HIV-1 reverse transcriptase plus-strand initiation exhibits preferential sensitivity to non-nucleoside reverse transcriptase inhibitors in vitro. J Biol Chem. 2007;282:8005–8010. doi: 10.1074/jbc.M608274200. [DOI] [PubMed] [Google Scholar]
- Hang JQ, Li Y, Yang Y, Cammack N, Mirzadegan T, Klumpp K. Substrate-dependent inhibition or stimulation of HIV RNase H activity by non-nucleoside reverse transcriptase inhibitors. Biochem Biophys Res Commun. 2007;12:341–350. doi: 10.1016/j.bbrc.2006.11.018. [DOI] [PubMed] [Google Scholar]
- Hang JQ, Li Y, Yang Y, Zommer H, Mackenzie R, Mizadegan U, Reynen P, Cammack N, Klumpp K. Compound specific differences in the levels of resistance of HIV-RT catalyzed strand-transfer, DNA polymerase and RNase H activities to inhibition by NNRTIs. Antivir Ther. 2006;11:S145. [Google Scholar]
- Hazen RJ, Harvey RJ, St Clair MH, Ferris RG, Freeman GA, Tidwell JH, Schaller LT, Cowan JR, Short SA, Romines KR, Chan JH, Boone LR. Anti-human immunodeficiency virus type 1 activity of the nonnucleoside reverse transcriptase inhibitor GW678248 in combination with other antiretrovirals against clinical isolate viruses and in vitro selection for resistance. Antimicrob Agents Chemother. 2005;49:4465–4473. doi: 10.1128/AAC.49.11.4465-4473.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jochmans D, Deval J, Kesteleyn B, Van Marck H, Bettens E, De Baere I, Dehertogh P, Ivens T, Van Ginderen M, Van Schoubroeck B, Ehteshami M, Wigerinck P, Gotte M, Hertogs K. Indolopyridones inhibit human immunodeficiency virus reverse transcriptase with a novel mechanism of action. J Virol. 2006;80:12283–12292. doi: 10.1128/JVI.00889-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplan AH, Manchester M, Swanstrom R. The activity of the protease of human immunodeficiency virus type 1 is initiated at the membrane of infected cells before the release of viral proteins and is required for release to occur with maximum efficiency. J Virol. 1994;68:6782–6786. doi: 10.1128/jvi.68.10.6782-6786.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kati WM, Johnson KA, Jerva LF, Anderson KS. Mechanism and fidelity of HIV reverse transcriptase. J Biol Chem. 1992;267:25988–25997. [PubMed] [Google Scholar]
- Kohlstaedt LA, Wang J, Friedman JM, Rice PA, Steitz TA. Crystal structure at 3.5 A resolution of HIV-1 reverse transcriptase complexed with an inhibitor. Science. 1992;26:1783–1790. doi: 10.1126/science.1377403. [DOI] [PubMed] [Google Scholar]
- Motakis D, Parniak MA. A tight-binding mode of inhibition is essential for anti-human immunodeficiency virus type 1 virucidal activity of nonnucleoside reverse transcriptase inhibitors. Antimicrob Agents Chemother. 2002;46:1851–1856. doi: 10.1128/AAC.46.6.1851-1856.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nissley DV, Radzio J, Ambrose Z, Sheen CW, Hamamouch N, Moore KL, Tachedjian G, Sluis-Cremer N. Characterization of novel non-nucleoside reverse transcriptase (RT) inhibitor resistance mutations at residues 132 and 135 in the 51 kDa subunit of HIV-1 RT. Biochem J. 2007;404:151–157. doi: 10.1042/BJ20061814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palaniappan C, Kim JK, Wisniewski M, Fay PJ, Bambara RA. Control of initiation of viral plus strand DNA synthesis by HIV reverse transcriptase. J Biol Chem. 1998;273:3808–3816. doi: 10.1074/jbc.273.7.3808. [DOI] [PubMed] [Google Scholar]
- Parniak MA, Sluis-Cremer N. Inhibitors of HIV-1 reverse transcriptase. Adv Pharmacol. 2000;49:67–109. doi: 10.1016/s1054-3589(00)49024-1. [DOI] [PubMed] [Google Scholar]
- Park J, Morrow CD. The nonmyristylated Pr160gag-pol polyprotein of human immunodeficiency virus type 1 interacts with Pr55gag and is incorporated into viruslike particles. J Virol. 1992;66:6304–6313. doi: 10.1128/jvi.66.11.6304-6313.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pettit SC, Sheng N, Tritch R, Erickson-Viitanen S, Swanstrom R. The regulation of sequential processing of HIV-1 Gag by the viral protease. Adv Exp Med Biol. 1998;436:15–25. doi: 10.1007/978-1-4615-5373-1_2. [DOI] [PubMed] [Google Scholar]
- Quan Y, Liang C, Inouye P, Wainberg MA. Enhanced impairment of chain elongation by inhibitors of HIV reverse transcriptase in cell-free reactions yielding longer DNA products. Nucleic Acids Res. 1998;15:5692–5698. doi: 10.1093/nar/26.24.5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quan Y, Rong L, Liang C, Wainberg MA. Reverse transcriptase inhibitors can selectively block the synthesis of differently sized viral DNA transcripts in cells acutely infected with human immunodeficiency virus type 1. J Virol. 1999;73:6700–6707. doi: 10.1128/jvi.73.8.6700-6707.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabson AB, Graves BJ. Synthesis and processing of viral RNA. In: Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. Cold Spring Harbor Laboratory Press; Plainview, NY: 1997. pp. 205–261. [PubMed] [Google Scholar]
- Radzio J, Sluis-Cremer N. Efavirenz Accelerates HIV-1 Reverse Transcriptase Ribonuclease H Cleavage Leading to Diminished Zidovudine Excision. Mol Pharmacol. 2007 doi: 10.1124/mol.107.038596. In Press. [DOI] [PubMed] [Google Scholar]
- Ren J, Diprose J, Warren J, Esnouf RM, Bird LE, Ikemizu S, Slater M, Milton J, Balzarini J, Stuart DI, Stammers DK. Phenylethylthiazolylthiourea (PETT) non-nucleoside inhibitors of HIV-1 and HIV-2 reverse transcriptases. Structural and biochemical analyses. J Biol Chem. 2000;275:5633–5639. doi: 10.1074/jbc.275.8.5633. [DOI] [PubMed] [Google Scholar]
- Rittinger K, Divita G, Goody RS. Human immunodeficiency virus reverse transcriptase substrate-induced conformational changes and the mechanism of inhibition by nonnucleoside inhibitors. Proc Natl Acad Sci USA. 1995;92:8046–8049. doi: 10.1073/pnas.92.17.8046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato A, Hammond J, Alexander TN, Graham JP, Binford S, Sugita K, Sugimoto H, Fujiwara T, Patick AK. In vitro selection of mutations in human immunodeficiency virus type 1 reverse transcriptase that confer resistance to capravirine, a novel nonnucleoside reverse transcriptase inhibitor. Antiviral Res. 2006;70:66–74. doi: 10.1016/j.antiviral.2006.01.001. [DOI] [PubMed] [Google Scholar]
- Shaw-Reid CA, Feuston B, Munshi V, Getty K, Krueger J, Hazuda DJ, Parniak MA, Miller MD, Lewis D. Dissecting the effects of DNA polymerase and ribonuclease H inhibitor combinations on HIV-1 reverse-transcriptase activities. Biochemistry. 2005;8:1595–1606. doi: 10.1021/bi0486740. [DOI] [PubMed] [Google Scholar]
- Sluis-Cremer N, Arion D, Abram ME, Parniak MA. Proteolytic processing of an HIV-1 pol polyprotein precursor: insights into the mechanism of reverse transcriptase p66/p51 heterodimer formation. Int J Biochem Cell Biol. 2004;36:1836–1847. doi: 10.1016/j.biocel.2004.02.020. [DOI] [PubMed] [Google Scholar]
- Sluis-Cremer N, Dmitrienko GI, Balzarini J, Camarasa MJ, Parniak MA. Human immunodeficiency virus type 1 reverse transcriptase dimer destabilization by 1-[Spiro[4″-amino-2″,2″ -dioxo-1″,2″ -oxathiole-5″,3′-[2′, 5′-bis-O-(tert-butyldimethylsilyl)-beta-D-ribofuranosyl]]]-3-ethylthy mine. Biochemistry. 2000;15:427–1433. doi: 10.1021/bi991682+. [DOI] [PubMed] [Google Scholar]
- Sluis-Cremer N, Hamamouch N, San Felix A, Velazquez S, Balzarini J, Camarasa MJ. Structure-activity relationships of [2′,5′-bis-O-(tert-butyldimethylsilyl)-beta-D-ribofuranosyl]- 3′-spiro-5″-(4″-amino-1″,2″-oxathiole-2″,2″-dioxide)thymine derivatives as inhibitors of HIV-1 reverse transcriptase dimerization. J Med Chem. 2006;10:4834–4841. doi: 10.1021/jm0604575. [DOI] [PubMed] [Google Scholar]
- Smith AJ, Srinivasakumar N, Hammarskjold ML, Rekosh D. Requirements for incorporation of Pr160gag-pol from human immunodeficiency virus type 1 into virus-like particles. J Virol. 1993;67:2266–2275. doi: 10.1128/jvi.67.4.2266-2275.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spence RA, Kati WM, Anderson KS, Johnson KA. Mechanism of inhibition of HIV-1 reverse transcriptase by nonnucleoside inhibitors. Science. 1995;267:988–993. doi: 10.1126/science.7532321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swanstrom R, Wills JW. Synthesis, assembly, and processing of viral proteins. In: Coffin JM, Hughes SH, Varmus HE, editors. Retroviruses. Cold Spring Harbor Laboratory Press; Plainview, NY: 1997. pp. 205–261. [PubMed] [Google Scholar]
- Tachedjian G, Moore KL, Goff SP, Sluis-Cremer N. Efavirenz enhances the proteolytic processing of an HIV-1 pol polyprotein precursor and reverse transcriptase homodimer formation. FEBS Lett. 2005;579:379–384. doi: 10.1016/j.febslet.2004.11.099. [DOI] [PubMed] [Google Scholar]
- Tachedjian G, Orlova M, Sarafianos SG, Arnold E, Goff SP. Nonnucleoside reverse transcriptase inhibitors are chemical enhancers of dimerization of the HIV type 1 reverse transcriptase. Proc Natl Acad Sci USA. 2001;98:7188–7193. doi: 10.1073/pnas.121055998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venezia CF, Howard KJ, Ignatov ME, Holladay LA, Barkley MD. Effects of efavirenz binding on the subunit equilibria of HIV-1 reverse transcriptase. Biochemistry. 2006;45:2779–2789. doi: 10.1021/bi051915z. [DOI] [PubMed] [Google Scholar]
- Wang LZ, Kenyon GL, Johnson KA. Novel mechanism of inhibition of HIV-1 reverse transcriptase by a new non-nucleoside analog, KM-1. J Biol Chem. 2004;79:38424–38432. doi: 10.1074/jbc.M406241200. [DOI] [PubMed] [Google Scholar]
- Xia Q, Radzio J, Anderson KS, Sluis-Cremer N. Probing nonnucleoside inhibitor-induced active-site distortion in HIV-1 reverse transcriptase by transient kinetic analyses. Protein Sci. 2007;16:1728–1737. doi: 10.1110/ps.072829007. [DOI] [PMC free article] [PubMed] [Google Scholar]