

Abstract
This study determined a) the association between stages of Alzheimer's disease (AD) and overall gene expression change, and b) brain regions of greatest vulnerability to transcriptional change as the disease progressed. Fifteen cerebrocortical sites and the hippocampus were examined in persons with either no cognitive impairment or neuropathology, or with only AD-associated lesions. Cases were stratified into groups of 7−19 based on the degree of cognitive impairment (clinical dementia rating scale; CDR); neurofibrillary tangle distribution and severity (Braak staging) or density of cerebrocortical neuritic plaque (NP; grouping by NP density). Transcriptional change was assessed by Affymetrix U133 mRNA microarray analysis. The results suggested that a) gene expression changes in the temporal and prefrontal cortices are more closely related to disease severity than other regions examined; b) more genes are down-regulated at any given disease severity stage than up-regulated; c) the degree of gene expression change in a given regions depends on the disease severity classification scheme used; and d) the classification of cases by CDR provides a more orderly gradient of gene expression change in most brain regions than Braak staging or NP grouping.
Keywords: Gene expression, Microarray, Alzheimer's disease, Dementia, Cerebral cortex, Hippocampus
Introduction
Significant progress has been made in understanding neuropathological, neuroanatomical, neurochemical and molecular biological abnormalities in the brains of persons with Alzheimer's disease (AD) (Hardy 2006;Roberson and Mucke 2006;Terry 2006). An inherent feature of many neurobiological studies has been the identification of brain regions, cortical laminae and specific cell types that are particularly vulnerable in AD and in dementia(Bussiere et al. 2003;Morrison et al. 2005;Von Gunten et al. 2005). For example, landmark studies by Braak and Braak(Braak et al. 2002;Braak and Braak 1991;Braak and Braak 1997) have outlined the progression of neuritic plaque (NP) and neurofibrillary tangle (NFT) pathology; neuroanatomical studies have identified the susceptibility of specific brain regions and neurons in specific cerebral cortical laminae(Braak et al. 2002;Giannakopoulos et al. 2003;Von Gunten et al. 2005), studies of neurochemical abnormalities have pointed to the vulnerability of neurotransmitter and neuropeptide systems(Haroutunian and Davis 2003), while other studies have attempted to place neuropathological, molecular biological and neurochemical systems within the framework of the progression of the dementia symptoms of AD(Davis et al. 1999b;Davis et al. 1999c;Haroutunian et al. 1998;Haroutunian et al. 1999;Haroutunian et al. 2006;Haroutunian and Davis 2003;Masliah et al. 1993;Morris et al. 2001;Naslund et al. 2000;Parvathy et al. 2001;Price et al. 2001;Terry 2006;Uboga and Price 2000).
The advent of high throughput gene expression technologies has raised our capacities by permitting the study of the expression level of multiple genes simultaneously and defining AD-associated changes in numerous molecular biological systems(Blalock et al. 2004;Blalock et al. 2005;Emilsson et al. 2006;Ginsberg et al. 2000;Ginsberg et al. 2004;Ginsberg et al. 2006;Loring et al. 2001;Parachikova et al. 2006;Pasinetti 2001;Ricciarelli et al. 2004;Xu et al. 2006;Yao et al. 2003). In addition, microarray techniques have been combined with microdissection capabilities to hone in on gene expression abnormalities in discrete neurons and in brain regions known to be affected by AD neuropathology(Ginsberg et al. 2000;Ginsberg et al. 2004;Ginsberg et al. 2006). Despite the breadth of our current technical abilities and understanding of transcriptional abnormalities in AD, most of the studies cited above have approached the question of abnormal gene expression in AD by examining gene expression in a limited number of brain regions and/or at specific neuropathologically or cognitively distinct stages of the disease. Thus, an understanding of the extent and distribution of gene expression abnormalities free from preconceived expectations based on the extent and distribution of specific neuropathologic features remains elusive.
The classical neuropathological studies of AD and studies of dementia defined the progression of disease in terms of selected neuropathological lesions in different brain region or the progressive deterioration of different cognitive functions and domains such as memory and executive function(Rapp et al. 2005;Stern et al. 1996). The molecular biological and gene expression counterparts of these progressive changes are obscure, however. It is of interest to learn how the progression of dementia or each of the different hallmark neuropathological lesions of AD such as NPs or NFTs is associated with gene expression within the brain. In addition, it is important to our understanding of the disease process to ascertain how faithfully gene expression changes in different brain regions reflect the different stages of AD, whether disease stage is defined by different neuropathological criteria or by functional and cognitive criteria.
The current study aimed to determine the topography of transcriptional abnormalities in the brain of persons at different stages of AD and dementia. The approach was based on large-scale gene expression profiling in 15 different brain regions of a large and varied enough cohort of cases (N=53) as to permit case stratification using different schemas for neuropathological and cognitive staging of AD. Transcriptional vulnerability was operationally defined as the number of genes that were abnormally expressed in different brain regions at different stages of AD. The study cohort was specifically selected to represent persons who died at different stages of dementia and with different densities of NFTs and NPs in diagnostically relevant (CERAD-defined(Mirra et al. 1991)) brain regions. The gene expression data was then assembled to permit stratification of cases along a) the dementia dimension as defined by clinical dementia rating (CDR)(Dooneief et al. 1996;Morris 1993) scores at the time of death; b) by emphasis on the distribution of NFT pathology using Braak neuropathology staging(Braak and Braak 1991); and c) by grouping of cases based on mean density of neuritic plaques in the cerebral cortex. Confounds associated with comorbid neuropathology such as significant cerebrovascular disease or Lewy body lesions were avoided by the selection of cases who had either no significant neuropathology or only those neuropathological lesions that are associated with AD. Additionally, close control was exercised over key variables such as age at death and brain tissue pH by maintaining them within a narrow range so that they were not statistically different from each other irrespective of the 3 group stratification strategies employed in the data analyses.
Methods
General
Brain tissue specimens were derived from the Mount Sinai School of Medicine Alzheimer's disease and Dementia Brain Bank. The precise tissue handling procedures have been described in detail(Davis et al. 1999a;Haroutunian et al. 1998;Haroutunian et al. 1999;Haroutunian et al. 2006).Tissue donors were subjects who had been residents of the Jewish Home and Hospital in Manhattan and the Bronx, NY and were participants in a longitudinal study of aging and dementia. All neuropsychological, diagnostic and autopsy protocols were approved by the Mount Sinai and the Jewish Home and Hospital Institutional Review Boards. Following brain tissue donation each brain specimen was divided midsagittally. The right half of the brain was fixed in 4% paraformaldehyde for neuropathological assessment. Approximately 0.8 cm coronal slabs of the entire left half of the brain were flash frozen and kept at −80°C until later dissection at −20°C. Cerebral cortical regions (approximately 1 cm3) from Brodmann areas (Bm)-8 (superior frontal gyrus), Bm-10 (frontal pole), Bm-44 (inferior frontal gyrus), Bm-4 (precentral gyrus), Bm-46 (dorsolateral prefrontal cortex), Bm- 24/32 (anterior cingulate – at the level of the genu of the corpus callosum), Bm-23/31 (posterior cingulate cortex – at the level of the pulvinar), Bm-7 (superior parietal lobule), Bm-20 (inferior temporal gyrus) , Bm-21 (middle temporal gyrus), Bm-22 (superior temporal gyrus), Bm-38 (temporal pole), Bm-36/28 (parahippocampal gyrus/entorhinal cortex), Bm-17 (occipital cortex primary visual cortex) regions and from the hippocampus (at the level of the red nucleus) were dissected from flash frozen coronal sections, pulverized at −80°C and aliquoted. Fifty to 100 mg aliquots from each region were used for microarray gene expression analysis.
Not all brain regions for all subjects were available for analysis and some aliquots from some brain regions did not yield RNA of sufficient quality or quantity for microarray analysis. Table 1 summarizes the demographic characteristics of the studied cases based on the different disease staging stratification schemes used (please see below). This table shows the characteristics of the subject groups based on the maximum and minimum number of specimens included in each group and brain region combination. More detailed demographic information on a brain region by brain region and group stratification basis is provided in the Supplementary Table. All subjects died of natural causes. The predominant causes of death were cardiovascular disease and myocardial infarction, cancer, septicemia and bronchopneumonia. Brain specimens from subjects who were comatose for more than 6 hours prior to death were excluded from the current study.
Table 1.
Demographic characteristics of the study cohort in different stratification groupings. Values represent mean ± SEM. The first line of each row represents the scores from the maximum number of subjects in each group, the values in the second line represent the values from the minimum number of subjects in each group. A more detailed table of demographic characteristics is provided in the Supplementary Table.
| |
N |
PMI (min.) |
CDR |
Age (yr.) |
Sex |
pH |
|---|---|---|---|---|---|---|
| Clinical Dementia Rating score (CDR) Groups | ||||||
| CDR 0.0 | 17 | 540 ± 105 | 0.0 | 82 ± 2.9 | 5 M, 12 F | 6.52 ± 0.07 |
| |
9 |
403 ± 94 |
0.0 |
87 ± 3.3 |
2 M, 7 F |
6.53 ± 0.10 |
| CDR 0.5 | 14 | 321 ± 55 | 0.5 | 84 ± 2.7 | 5 M, 9 F | 6.41 ± 0.07 |
| |
8 |
259 ± 19 |
0.5 |
82 ± 3.7 |
4 M, 4 F |
6.42 ± 0.09 |
| CDR 1.0 | 13 | 285 ± 37 | 1.0 | 85 ± 2.8 | 5 M, 8 F | 6.35 ± 0.07 |
| |
7 |
274 ± 57 |
1.0 |
87 ± 2.6 |
4 M, 3 F |
6.31 ± 0.11 |
| CDR 2.0 | 13 | 313 ± 69 | 2.0 | 89 ± 0.7 | 1 M, 12 F | 6.37 ± 0.09 |
| |
7 |
306 ± 95 |
2.0 |
87 ± 1.6 |
0 M, 7F |
6.28 ± 0.12 |
| CDR ≥ 3.0 | 26 | 266 ± 50 | 3.7 ± 0.05 | 87 ± 1.8 | 9 M, 17 F | 6.40 ± 0.05 |
| |
16 |
281 ± 64 |
3.9 ± 0.06 |
89 ± 1.7 |
6 M, 10 F |
6.45 ± 0.06 |
| Braak Staging Groups | ||||||
| Braak 0&I | 13 | 387 ± 65 | 0.7 ± 0.1 | 79 ± 2.8 | 3 M, 10 F | 6.38 ± 0.10 |
| |
6 |
290 ± 54 |
0.9 ± 0.2 |
81 ± 2.7 |
1 M, 5 F |
6.20 ± 0.12 |
| Braak II | 15 | 240 ± 22 | 1.2 ± 0.2 | 85 ± 2.7 | 6 M, 9 F | 6.50 ± 0.06 |
| |
9 |
217 ± 18 |
1.2 ± 0.2 |
87 ± 2.8 |
3 M, 6 F |
6.61 ± 0.07 |
| Braak III | 19 | 335 ± 54 | 1.5 ± 0.1 | 88 ± 1.9 | 4M, 15 F | 6.38 ± 0.05 |
| |
13 |
321 ± 67 |
1.2 ± 0.1 |
87 ± 1.6 |
4 M, 9 F |
6.38 ± 0.06 |
| Braak IV | 12 | 203 ± 20 | 2.3 ± 0.2 | 90 ± 0.6 | 1 M, 11 F | 6.41 ± 0.08 |
| |
8 |
210 ± 24 |
2.4 ± 0.2 |
88 ± 1.7 |
1 M, 7 F |
6.48 ± 0.10 |
| Braak V&VI | 23 | 312 ± 62 | 2.9 ± 0.1 | 88 ± 1.6 | 7 M, 16 F | 6.36 ± 0.06 |
| |
13 |
367 ± 79 |
2.7 ± 0.1 |
88 ± 2.8 |
4 M, 9 F |
6.43 ± 0.09 |
| Cortical Neuritic Plaque density( mm2) Groups | ||||||
| Plaque Grp. 0 | 18 | 459 ± 96 | 0.5 ± 0.3 | 78 ± 2.7 | 9 M, 8 F | 6.57 ± 0.06 |
| |
9 |
238 ± 21 |
0.3 ± 0.4 |
83 ± 2.6 |
3 M, 6 F |
6.42 ± 0.12 |
| Plaque Grp. 1 | 19 | 278 ± 46 | 1.4 ± 0.1 | 90 ± 0.6 | 3 M, 16 F | 6.46 ± 0.06 |
| 1−6 |
15 |
302 ± 60 |
2.0 ± 0.1 |
89 ± 1.6 |
2 M, 13 F |
6.42 ± 0.07 |
| Plaque Grp. 2 | 25 | 290 ± 51 | 2.3 ± 0.1 | 87 ± 0.3 | 6 M, 19 F | 6.28 ± 0.05 |
| 7−12 |
19 |
212 ± 16 |
2.3 ± 0.1 |
88 ± 1.6 |
5 M, 14 F |
6.32 ± 0.06 |
| Plaque Grp. 3 | 15 | 347 ± 85 | 2.9 ± 0.1 | 85 ± 1.6 | 5 M, 10 F | 6.36 ± 0.08 |
| 13+ | 8 | 308 ± 83 | 2.4 ± 0.2 | 84 ± 2.7 | 3 M, 5 F | 6.41 ± 0.10 |
Subject selection, cognitive assessment, neuropathological assessment and group stratification
The cohort of subjects included in the present analysis was part of a larger clinical and neuropsychological study of early AD and had been evaluated in detail for cognitive status during the last 6 months of life as previously described(Davis et al. 1999a;Haroutunian et al. 1998;Haroutunian et al. 1999;Haroutunian et al. 2006). One hundred fifteen subjects were included in this postmortem study. Exclusion criteria were: presence of neuropathological lesions not associated with AD (including, but not limited to, Pick's disease, Lewy body inclusions, Parkinson's disease, stroke, multi-infarct dementia, and severe cerebrovascular disease judged to be sufficient to affect cognitive function(Blessed et al. 1968;Tomlinson et al. 1970)), coma > 6 hours prior to death, seizures or fever (>39°C) during the 24 hours prior to death, unnatural cause of death; and comorbid psychiatric disease such as schizophrenia. The Clinical Dementia Rating scale (CDR)(Dooneief et al. 1996;Morris 1993) was used as the primary measure of dementia severity for the 6 months preceding death. Subjects were rated by the CDR using a multi-step consensus-dependent approach(Haroutunian et al. 1998;Haroutunian et al. 1999) to have no cognitive deficits (CDR=0), questionable dementia (CDR=0.5), mild dementia (CDR=1.0), moderate dementia (CDR=2.0), and severe to terminal dementia (CDR=3.0−5.0). When available, longitudinal neuropsychological assessment results were also considered in deriving the final consensus CDR score.
The neuropathological assessment procedures used have been described in detail(Davis et al. 1999a;Haroutunian et al. 1998;Haroutunian et al. 2006). Neuropathological assessments were performed on the right hemisphere and consisted of examining representative blocks from the superior and middle frontal gyrus, orbital cortex, basal ganglia with basal forebrain, amygdala, hippocampus (rostral and caudal levels with adjacent parahippocampal and inferior temporal cortex), superior temporal gyrus, parietal cortex (angular gyrus), calcarine cortex, hypothalamus with mammillary bodies, thalamus, midbrain, pons, medulla, cerebellar vermis, and lateral cerebellar hemisphere according to the Consortium to Establish a Registry for Alzheimer's Disease (CERAD) protocol(Mirra et al. 1991). Sections from paraffin embedded blocks were variably stained with hematoxylin and eosin, modified Bielschowski, modified thioflavin S, and anti-β amyloid (4G8, gift of Dr. N. Robakis, The Mount Sinai School of Medicine), anti-tau (AD2, gift of Dr. A. Delacourte, Lille, France) and anti-ubiquitin (Daka Corp. Carpinteria, CA staining of representative) as required. All neuropathology data regarding the extent and distribution of neuropathologic lesions were collected in a blinded fashion relative to the subject's dementia status. Each case was assigned a Braak AD-staging score for progression of neurofibrillary neuropathology(Braak et al. 2006;Braak and Braak 1991). In addition, quantitative data regarding the density of neuritic plaques in the middle frontal gyrus (Brodmann area 9), orbital frontal cortex (Brodmann area 45/47), superior temporal gyrus (Brodmann area 21/22), inferior parietal cortex (Brodmann area 39) and calcarine cortex (Brodmann area 17) were collected as described(Haroutunian et al. 1998). The average of these NP density measures was used for the stratification of subjects into groups based on the severity of NP neuropathology. Previous studies have indicated that this composite measure of plaque density corresponds closely to the average plaque densities in most cortical regions examined(Haroutunian et al. 1998).
Microarray procedures
The procedures used for microarray analysis of gene expression in human postmortem specimens have been published previously(Katsel et al. 2005a;Katsel et al. 2005b;Katsel et al. 2005c). RNA quality was assessed using a combination of a 260/280 ratio derived from a high resolution electrophoresis system (LabChip™, Agilent Technologies, Palo Alto, CA) and 3’ to 5’ hybridization ratios for glyceraldehide-3 phosphate (GAPDH) probes. No correlation was observed between RNA quality and the relatively narrow range of PMIs in this study. Microarray analysis was performed by Gene Logic Inc. (Gaithersburg, MD) using Affymetrix (Santa Clara, CA) HG-U133 A&B Human genome GeneChip® sets (44928 probe sets) as described in the standard protocol outlined in the GeneChip® Expression Analysis Technical Manual (Affymetrix). GeneChips were scanned under low PMT. Image quality was subjected to strict quality control protocols using proprietary Gene Logic algorithms and visual inspection.
Data from 992 samples (1984 individual GeneChips) were normalized using MAS 5.0 algorithms and GXTM Explorer v.2.0 (expression, comparative and contrast analysis; Gene Logic Inc. http://www.genelogic.com/solutions/other/data.cfm)) as described previously(Katsel et al. 2005a;Katsel et al. 2005b;Katsel et al. 2005c). To reduce the false discovery (Type I error) error rate a change in gene expression was considered to be significant if it met the relatively stringently criteria of p<0.05 relative to the expression level in the designated control group; fold change ≥ 1.7, present calls ≥80%. These criteria were applied on a brain region by region basis after subject stratification (see below). To validate the results obtained by MAS 5.0 analysis the raw microarray data from 1984 HG-U133A and B individual chips were transformed and analyzed using GeneSpring GX 7.3.1 (Agilent Technologies/Silicon Genetics, Santa Clara, CA). Expression data were pre-normalized with Robust Multi-Chip Average (RMA)(Bolstad et al. 2003;Irizarry et al. 2003) with subsequent log transformation, normalization to 50th percentile of all values per chip and median-centered per gene using the GeneSpring normalization options. Statistical comparisons were done using the GeneSpring's Cross Gene Error Model (CGEM), based on the deviation from 1.0 algorithm. To minimize the false positive/negative error rate in these GeneSpring/RMA/CGEM analyses, we used a combination of confidence (P≤0.05) and fold change (≥1.6) with Benjamini and Hochberg multiple testing correction(Benjamini and Hochberg 1995).
Stratification
Gene expression changes were calculated after stratification of the subjects based on 3 primary disease severity measures. The first schema was contingent on stratification based on functional behavioral/cognitive status as defined by the CDR. Subjects with no significant cognitive deficits (CDR score = 0) were compared to those with questionable (CDR = 0.5), mild (CDR = 1.0), moderate (CDR = 2.0) or severe to terminal (CDR ≥ 3) dementia (Table 1). The second schema was based on Braak neurofibrillary pathology staging where subjects with Braak scores of 0 and I (no neurofibrillary pathology or very mild neurofibrillary pathology confined to the transentoshinal region) were compared to those classified as meeting criteria for stage II (numerous neurofibrillary tangles in the transentorhinal region and modest NFT pathology in the CA1 sector of the hippocampus), stage III (transentorhinal region NFTs involving the pre-α layer, presence of “ghost tangles” and involvement of the pyramidal cells of the subiculum), stage IV (severe pre-β layer involvement of the transentorhinal and entorhinal cortex, numerous NFTS in the hippocampus, mild NFT changes in the isocortex and no or only mild NFT pathology in the primary motor and sensory cortices); and stages V plus VI (severe pathology of the pre-α layers with numerous “ghost tangles”, extensive involvement of the hippocampus and destruction of isocortical association areas). Persons meeting criteria for stage 0 and I and those meeting criteria for stage V and VI were grouped together to ensure a sample size of at least 8 cases per group in the majority of the studied brain region. Stratification based on neuritic plaque involvement was more arbitrary and relied loosely on tertile-split of the distribution of cases and the neuritic plaque-based diagnostic schema of Khachaturian(Khachaturian 1985) where cases with mean neocortical neuritic plaque density score of 0 were compared to NP-group 1 (1−6 mean neocortical NPs), NP-group 2 (7−12 mean neocortical NPs) and NP-group 3 (≥ 13 mean neocortical NPs). For each stratification schema, the subjects falling into the first (least severe) strata were used as the “control” comparison group for each of the other strata within that stratification scheme.
Statistical analysis of data
Multiple statistical procedures were employed for different aspects of the study using SPSS (release 14.0.0) and Statistica (release 6.0). To determine overall transcriptional vulnerability (i.e., total numbers of genes expressed at higher or lower levels than controls) based on CDR, Braak and NP classification of the cases, the total number of genes expressed at higher or lower levels than the designated control group (p<0.05 relative to the expression level in the designated control group; fold change ≥ 1.7, present calls ≥80% as defined by MAS 5.0) was calculated for each brain regions examined in each subject. These regional gene expression changes relative to controls were then summed for each subject. Within group differences between up and down regulated gene changes, and pair-wise between group differences in up, down, and total gene expression changes were compared using the binomial distribution. Group differences in the relative numbers of up or down regulated genes were assessed by a χ2 test. Pearson correlations were used to test associations including the comparison of MAS 5.0 based vs. RMA based expression level normalization and the correlation of the number of abnormally expressed genes with each level of group stratification. As detailed below, Benjamini and Hochberg (Benjamini and Hochberg 1995) false discovery multiple comparison corrections were applied to RMA normalized gene expression results and Bonferroni corrections were applied to the χ2 and binomial distribution tests.
Results
Comparison of the MAS 5.0 normalization with GXTM-based differential gene expression discovery vs. RMA normalization with Benjamini and Hochberg corrected-CGEM-based differential gene expression discovery showed that although the two procedures differed with respect to the absolute number of differentially expressed genes identified (e.g., the total number of genes that were found to be up or down regulated in the CDR 0.5 group was 127 and 175 using MAS 5.0 and RMA normalization respectively), the relative ranking of brain regions and the relationships of change with changes in CDR staging remained constant resulting in very robust correlations between the two methods for the 15 brain regions examined (r=0.85, p=0.000001; r=0.76, p=0.000001 and r=0.81, p=0.000001 for numbers of total, up- and down-regulated genes, respectively). Therefore, the results reported here are based on the slightly more conservative MAS 5.0 normalized results.
Figures 1-3 provide a visual representation of the numbers of abnormally expressed genes relative to the designated control group in each brain region studied when subjects were grouped on the basis of the severity of cognitive impairment (Figure 1, CDR), Braak NFT pathology staging (Figure 2, Braak staging) or the mean density of NPs within the 5 cortical regions where NP density was measured (Figure 3, NP). Although each of these figures shows a general increase in the number of abnormally expressed genes in different brain regions as a function of increasing disease severity, the pattern of change from one level of severity to the next differs for each classification scheme. Figure 1 shows a pattern of gene expression dysregulation in persons with questionable dementia (CDR 0.5) that predominantly involves the middle and inferior temporal gyri and the superior parietal lobule. This pattern of brain region involvement shifts noticeably in persons with mild, but definite dementia (CDR 1.0) where the regions of greatest gene expression abnormalities involve the superior temporal gyrus, the frontal pole and the anterior cingulate gyrus. Progression from mild dementia to moderate and severe dementia is marked by a general increase in the numbers of abnormally expressed genes in previously affected brain regions and the recruitment of the dorsolateral prefrontal cortex, the middle frontal gyrus, the motor cortex, the parahippocampal gyrus and the hippocampus.
Figure 1.
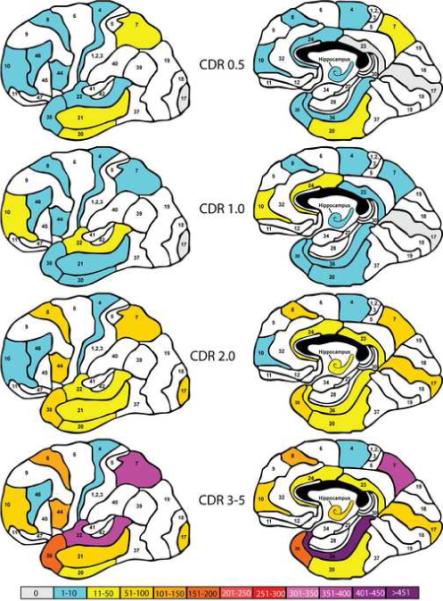
Representation of the total number of aberrantly expressed genes (p<0.05 relative to the CDR=0.0 group; fold change ≥ 1.7, present calls ≥80%) in each of the brain regions examined as a function of increasing dementia severity. The left and right panels represent lateral and medial views of the brain, respectively. Different numbers of aberrantly expressed genes are represented by different color codes according to the scale shown at the bottom of the figure. White regions were not sampled for study. Numbers within each demarcated region represent the corresponding Brodmann regions and the studied regions correspond to Brodmann areas (Bm)-8 (superior frontal gyrus), Bm-10 (frontal pole), Bm-44 (inferior frontal gyrus), Bm-4 (precentral gyrus), Bm-46 (dorsolateral prefrontal cortex), Bm- 24/32 (anterior cingulate – at the level of the genu of the corpus callosum), Bm-23/31 (posterior cingulate cortex – at the level of the pulvinar), Bm-7 (superior parietal lobule), Bm-20 (inferior temporal gyrus) , Bm-21 (middle temporal gyrus), Bm-22 (superior temporal gyrus), Bm-38 (temporal pole), Bm-36/28 (parahippocampal gyrus/entorhinal cortex), Bm-17 (occipital cortex primary visual cortex)
Figure 3.
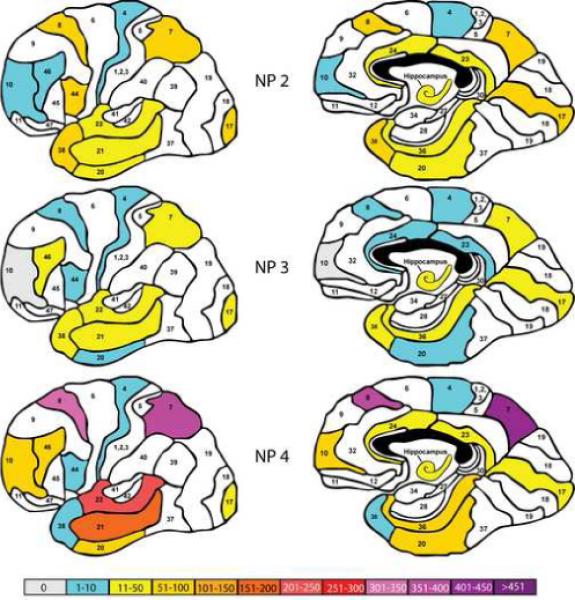
Representation of the total number of aberrantly expressed genes (p<0.05 relative to the mean NP density =0/mm2 group; fold change ≥ 1.7, present calls ≥80%) in each of the brain regions examined as a function of increasing NP density. The color coding, panel organization and Brodmann area numbers are identical to Figure 1.
Figure 2.
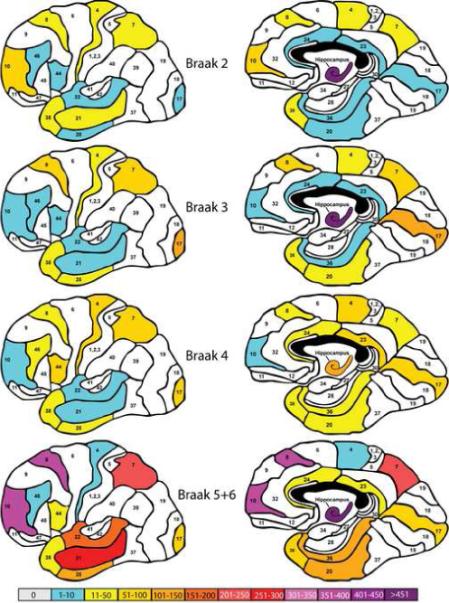
Representation of the total number of aberrantly expressed genes (p<0.05 relative to Braak stage 0+I comparison group; fold change ≥ 1.7, present calls ≥80%) in each of the brain regions examined as a function of increasing Braak disease progression stage. The color coding, panel organization and Brodmann area numbers are identical to Figure 1.
A somewhat different pattern of gene expression change is revealed when the subjects are classified on the basis of Braak staging of NFTs. Figure 2 shows that by Braak stage II many brain regions are already significantly involved. The most severely affected region is the hippocampus, but portions of the temporal cortex, the frontal pole, middle frontal gyrus and the motor cortex and superior parietal lobule are also affected. This pattern of cortical and hippocampal involvement is more or less maintained in Braak stages III and IV. However it is interesting that at Braak stage IV the absolute number of abnormally expressed genes is generally reduced in many regions, including the hippocampus. By Braak stage V and VI, many of the brain regions examined are maximally or near maximally involved with the greatest gene expression abnormalities detected in the middle temporal gyrus, the frontal pole, the middle frontal gyrus, the superior temporal lobule and the hippocampus.
Similar to early Braak stages, classification by NP density (Figure 3) reveals a predominant involvement of the temporal cortex, hippocampus and middle and dorsolateral frontal cortex, superior parietal lobule and somewhat surprisingly the primary visual cortex in cases with mild (≤6/mm2) NP pathology. As was the case with Braak staging, this pattern persists, albeit with somewhat reduced numbers of affected genes, in the moderate NP density grouping (>6, ≤12/mm2) category. As expected from the pattern of gene expression abnormalities found with CDR and Braak grouping, the numbers of abnormally expressed genes increases dramatically in subjects with NP density scores of ≥13/mm2. In this group, the numbers of affected genes is particularly high in the superior and middle temporal gyri, the middle frontal gyrus and in the superior parietal lobule.
Figures 4 provides a graphic representation of the cumulative number of abnormally expressed genes relative to the designated control groups when subjects are stratified on the basis of dementia severity (CDR, Figure 4, upper panel), Braak NFT pathology staging (Figure 4, middle panel) or the mean density of NPs within the 5 cortical regions where NP density was measured (Figure 4, lower panel). The gestalt of differences in the numbers of up-regulated and down-regulated genes as a function of disease severity, irrespective of the classification system, gained from figures 1-3 are confirmed by these representations of the data and the results of the statistical analyses shown in Figure 4. Because the results shown in this figure were derived by assessing the numbers of genes that were significantly up or down-regulated relative to the relevant control group, by definition differences between the groups shown in the figures were statistically significant compared to the control group. In general, differences between the two least severe disease categories, irrespective of the severity classification system used were not statistically significant, or were statistically significant in the case of NP density but in the opposite to the expected direction. All other group comparisons between the severity conditions were highly significant and remained so, even after conservative (Bonferroni) correction for multiple comparisons.
Figure 4.

Numbers of aberrantly expressed genes (number down-regulated, number up-regulated) relative to the control comparison group as a function of increasing disease severity (upper panel: CDR; middle panel: Braak stage; lower panel: mean cortical NP density).
Figure 4 also show that irrespective of the classification scheme used for subject stratification, the number of genes that were down-regulated at any stage of disease was higher compared to the number of genes that were upregulated at that disease severity stage (χ2 test; all ps<0.00001).
These figures also suggest that the proportion of down-regulated genes increased as a function of increasing disease severity for all severity classification systems (overall χ2 tests; all ps<0.0000001). However, the differences between proportions were most pronounced when subjects were classified by Braak staging and by NP density scores (χ2 tests; all ps<0.01). The change in the proportion of up- vs. down-regulated genes in the CDR classification system as a function of increasing disease severity became statistically significant only when subjects with moderate dementia (CDR 2.0) were compared to the group of subjects with severe or terminal dementia (CDR ≥3.0; p=0.03).
The correlation between each measure of disease severity with the total number of significantly altered genes in each brain region is shown in Figure 5. This Figure shows that the extent and pattern of correlation between the numbers of abnormally expressed genes with disease severity is dependent, to some extent, on the measure of disease severity used. In general, gene expression changes in the temporal and frontal cortex correlate highly and positively with the total number of differentially expressed genes, however, there are differences. The most dramatic differences between the classification schemes are apparent in the temporal pole and the cingulate gyrus where the correlations with disease severity are positive and high when subjects are classified on the basis of their CDR and Braak scores, but these relationships are inversed when subjects are classified on the basis of the density of NPs in the cerebral cortex. In addition, gene expression changes in the hippocampus correlate better with increases in dementia severity than they do with increasing Braak stage or increasing densities of NPs in the neocortex.
Figure 5.
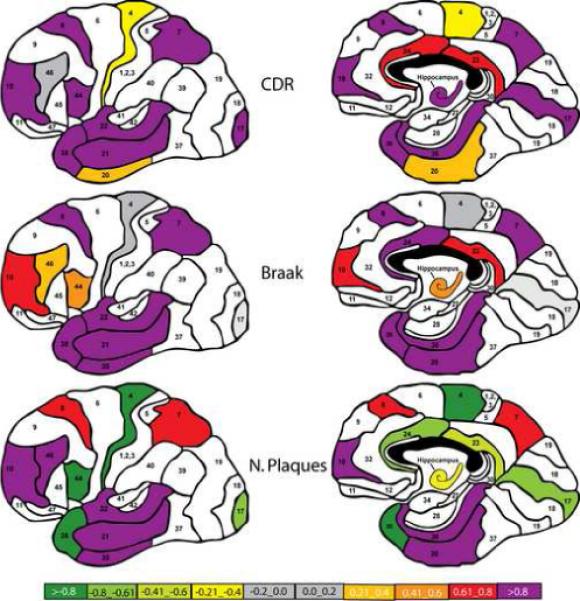
Correlation of the total number of aberrantly expressed genes in each studied brain region with the severity of disease (upper panel: CDR; middle panel: Braak staging; lower panel: NP density). Positive correlations imply that the number of aberrantly expressed genes increases as the index of disease severity increases. Negative correlations suggest that the number of aberrantly expressed genes decrease as a function of increasing disease severity. The panel organization and Brodmann area numbers are identical to Figure 1.
Discussion
Microarray analyses of gene expression in the human brain generate enormous data sets even when the analyses are limited to a single brain region and to comparisons between two groups. The wealth of information generated by multiregional comparisons of multiple groups varying across multiple classification dimensions becomes extraordinarily complex and requires analysis of discreet questions and tests of well-defined hypotheses. The results reported here are of course subject to all the limitations of microarray-based approaches (e.g., false discovery resulting from multiple statistical tests of significance even though similar results were obtained following two different analytical methods) which are compounded by the simultaneous multiregional analysis approach. However, despite these obvious caveats, the results reported provide significant insights into the global molecular changes that are associated with disease progression and to their correspondence with the different classifications of disease progression commonly used in AD and dementia research. Other studies(Blalock et al. 2004;Emilsson et al. 2006;Loring et al. 2001;Parachikova et al. 2006) have reported on microarray findings in AD. However, none have examined as many different brain regions as the current study and none have categorized the cases and controls based on different disease severity categorization approaches with relatively large numbers of cases in each demographically well-balanced disease category. For example, in one study that objectively assessed antemortem cognitive status and directly tested hypotheses to discover changes in gene expression at the earliest stages of disease(Parachikova et al. 2006), only 2 brain regions (hippocampus and dorsolateral prefrontal cortex) were assessed and the study was limited to the analysis of the brains of persons with mild dementia and mild neuropathology as assessed by Braak staging. The study that assessed the largest study cohort of 114 cases(Emilsson et al. 2006), measured gene expression in the superior frontal gyrus and did not stratify the studied cases based on disease severity.
The CDR measures the severity of dementia symptoms and as such is perhaps the most valid measure employed in this study from the perspective of impact on the wellbeing of persons with AD. Examination of Figure 1 suggests that the temporal and parietal cortices undergo the greatest number of gene expression changes at the time of mild cognitive impairment and dementia onset (e.g., CDR=0.5). Although the hippocampus and the frontal cortex are also affected, these regions suffer fewer gene expression changes early in the course of dementia. The cingulate cortex, especially the posterior cingulate gyrus, and the primary visual cortex are affected least. As dementia progresses to the moderate and severe stages (CDR=2 and 3−5) this general pattern is reinforced such that more and more genes become abnormally expressed in the brain regions affected during the initial stages of dementia, and brain regions such as the visual cortex and posterior cingulate gyrus that were unaffected at the mildest stages become involved. Interestingly, the precentral gyrus (Brodmann 4) and the dorsolateral prefrontal cortex (Brodmann 46) evidence no greater number of abnormally expressed genes at the end stages of dementia than they did at the earliest measured stage. It is also interesting, and perhaps surprising, that the numbers of abnormally affected genes in some brain regions such as the inferior and middle temporal gyri and the inferior parietal lobule declines as dementia progresses from questionable (CDR=0.5) to mild (CDR=1) before returning to CDR0.5 levels and assuming an upward trajectory as the disease progresses. As discussed below, this pattern of reductions in abnormally expressed genes in the mild to moderate stages of disease is not unique to the progression of transcriptional abnormalities when using the CDR staging system, but is observed to an even greater extent with Braak and NP density staging.
Figure 4 shows that the general trend toward increasing numbers of abnormally expressed genes with increasing dementia severity is characterized primarily by increasing numbers of down-regulated genes. Although there is an increase in the numbers of up-regulated genes with increasing dementia severity, they represent a much smaller proportion of dysregulated genes. This figure illustrates also that the most dramatic increase in the number of abnormally expressed genes occurs as dementia severity progresses from mild but definite dementia (CDR=1) to moderate dementia (CDR=2) with a significantly smaller increment as dementia progresses to the severe and terminal stage. One implication of this finding is support of current approaches to clinical trials that target treatment strategies to persons in the mild to moderate stages of dementia severity. The data reported here suggest that once the disease process has begun, therapeutic approaches should aim to prevent the dramatic increase in dysregulated genes that normally occurs following the onset of mild dementia symptoms.
The pattern of gene expression change in the cerebral cortex and hippocampus is somewhat different from that observed following subject grouping by dementia severity when the study subjects are stratified on the basis of severity of NFT pathology (Braak staging). Among the most notable differences is the near maximal dysregulation of genes in the hippocampus in persons at the earliest Braak stages (Braak stage II and III) with progressive recruitment of the parahippocampal gyrus, inferior temporal gyrus and the middle temporal gyrus and prefrontal regions at terminal stages (IV and V+VI). Also of note is the less dramatic involvement of several brain regions even at the final stages of the disease. These regions include the modest changes apparent in the cingulate cortex, the temporal pole and the precentral gyrus (Bm 4). Comparison of the cartoons of the most severe disease state in Figures 1 (CDR stratification) and 2 (Braak stratification) also suggests a more moderate involvement of the temporal cortex when the study cohort was stratified on the basis of Braak staging. Since the subjects represented in the different stratification schemes are the same, the differences between the gene expression changes shown in Figures 1 and 2 imply differences in group membership. The most parsimonious explanation for the differences in gene expression after stratification by these two methods is that under the conditions of Braak staging defined in the current study, the “disease free” group (Braak stage 0 and I) included subjects who were found to be demented clinically already. This inference is supported by the fact that the CDR scores of persons grouped into the Braak 0 and I stage approached mild dementia (CDR 1; Table 1) and were significantly different from the CDR 0 group (p=0.03). In addition, as shown in Figure 4, the absolute number of genes that were abnormally expressed relative to the control group in Braak stage II was significantly higher than the numbers of abnormally expressed genes in CDR stages 0.5 and 1.0 and higher even than the number of abnormally expressed genes in CDR stages 0.5 and 1.0 combined. These observations suggest that significant gene expression abnormalities are present before the onset of NFTs and neuropathological lesions sufficient in magnitude to meet criteria for Braak stage II.
Stratification of subjects by Braak stage, like stratification by CDR stage, showed that the proportion of down regulated genes was significantly higher at every stage of disease than up-regulated gene (Figure 4; all ps<0.001 except Braak stage IV). This disparity between up- and down-regulated genes became more pronounced in the advanced stages of disease (Braak stage V+VI). Figure 4 also shows an unexpected result of a general reduction in the numbers of abnormally expressed genes in Braak stage IV relative to the other stages studied (ps<0.001). The reasons for this generalized reduction in abnormally expressed genes are not obvious. It could be argued that the reduction in the numbers of abnormally expressed genes may represent the induction of compensatory mechanisms; however, a simplistic view would expect compensatory changes to occur earlier than Braak stage IV. Although a number of hypotheses, such as cell loss, can be invoked as an explanation for this observation, none can be supported or refuted empirically by the current data set. However, it is unlikely that this observation is spurious, since a generally similar trend is observed when subjects are stratified on the basis of the density neuritic plaques in the cerebral cortex (see below) and as discussed above a similar, albeit statistically non-significant trend was observed following stratification by CDR staging also.
The overall pattern of gene expression change when subjects were grouped on the basis of cerebrocortical plaque density (Figure 3) was more similar to that observed following Braak staging than CDR staging (Figures 1-3). Even subjects with 1−6/cm2 mean cerebrocortical NPs, an insufficient number of NPs to meet neuropathological diagnostic criteria for AD(Khachaturian 1985;Mirra et al. 1991), evidenced significant involvement of the hippocampus, temporal cortex, cingulate cortex, inferior and superior frontal gyri, inferior parietal lobule and the primary visual cortex.
The similarity in the patterns of gene expression change between groups stratified on the basis of NP density and Braak staging is also evident in Figure 4 with respect to the total numbers of abnormally expressed genes, as well as down-and up-regulated genes. As was the case with the CDR and Braak staging group stratification schemes, the numbers of down-regulated genes exceeded the numbers of up-regulated genes and the numbers of affected genes in the moderate NP group was significantly lower than the numbers of dysregulated genes in the mildly and severely affected groups. It is also interesting that the numbers of dysregulated genes in the least affected group (NP group 2), like the Braak staging group II, was significantly higher than that observed in the CDR 0.5 group. This is perhaps not surprising given that the mean CDR for this group was more similar to the CDR 2 group than to the CDR 0.5 group (Table 1) as reported previously(Haroutunian et al. 1998). As was the case with Braak staging, one parsimonious explanation for these findings is that neuropathologically evident changes lag behind changes in cognition and in abnormal gene expression.
The “correlation map” in Figure 5 shows that altered gene expression in some brain regions correlate better with cognitive and neuropathological indices of disease than transcriptional changes in other brain regions. High positive correlations between altered gene expression and measures of disease severity are observed in most regions of the temporal cortex and in the prefrontal cortex (Bm10 and Bm8) irrespective of the disease severity classification scheme. The most consistently positive and highly correlated brain region examined with all 3 classification methods were the superior and middle temporal gyri and the parahippocampal gyrus. Conversely, altered gene expression in other brain regions (e.g., precentral gyrus, Bm4) does not correlate significantly or correlates negatively with measures of disease severity regardless of the disease severity classification method used. It is noteworthy that the negative correlations shown in Figure 5 were not attributable to artifactual confounds such as reductions in the number of expressed genes. The relationship between gene expression in other brain regions such as the inferior frontal gyrus (Bm 44) and the hippocampus and indices of disease severity depends very much on the classification system used. Altered gene expression in these two brain regions is highly correlated with cognitive status (CDR), moderately correlated with Braak stage, and negatively correlated with the density of NPs.
There are a number of qualifications that must be kept in mind in considering the results described above. Foremost among these is that lack of independence of the 3 staging schemes from each other. Table 1 shows that in general persons with high CDR scores evidence generally greater numbers of NPs and NFTs than persons with low CDR scores. Similarly, persons meeting criteria for Braak stages V and VI have a higher density of NPs than persons in lower Braak stages. It is therefore not surprising that the general pattern of transcriptional change in different brain regions is similar between the different stratification schemes used. On the other hand, the different patterns of transcriptional change between the three grouping strategies suggests that the correspondence between these different categorization systems is not absolute and significant information regarding transcriptional vulnerability can be gleaned despite this lack of independence. From a different perspective, it can be argued that comparing different CDR, Braak or NP stages to each other is like comparing apples to oranges. CDR is a global measure of dementia severity whereas Braak staging is based on the specific progression of NFT pathology in the course of the disease and the NP stages constructed were based roughly on neuropathological categorization(Khachaturian 1985;Mirra et al. 1991) and frequency tertiles. Nevertheless, one would expect that there would be some positive relationship between dementia progression with a general increase in the degree of neuropathology(Haroutunian et al. 1998;Haroutunian et al. 1999) and that these general associations would be reflected in the amount of abnormally expressed genes. Indeed the results described above show such a general relationship, but it is the correspondence of transcriptional change between the different stages that is less robust. Another caveat and cautionary note derives from the fact that to permit comparison of groups with reasonable sample sizes, Braak stages 0 and 1 had to be combined to constitute the baseline comparator for the other Braak stages. It is possible that a somewhat different pattern of gene expression results would have emerged if the baseline comparison group was comprised of cases with no NFT pathology at all. It is important to note, however, that it is not uncommon for elderly persons with no discernable cognitive impairment to evidence some NPs and adequate NFT neuropathology to meet criteria for Braak stage 1(Haroutunian et al. 1998;Haroutunian et al. 1999;Haroutunian et al. 2006;Price and Morris 1999) and the grouping of Braak stages 0 and 1 into a single category of neuropathologically normal or mildly affected is not unique to the current study(Parachikova et al. 2006).
In this study we made no attempt to analyze the expression of specific genes, gene ontology groups or clusters. The aim of the current set of analyses was to determine the association between different disease staging methods and overall gene expression change in different brain regions and to identify brain regions of greatest vulnerability to transcriptional change as the disease process progressed. Even with the caveats mentioned above, the results suggest some tentative general conclusion regarding these two basic questions; a) gene expression changes in the temporal and prefrontal cortices are more closely related to disease severity than some of the other brain regions examined; b) in general, more genes are down-regulated at any given disease severity stage than genes that are up-regulated, irrespective of the methods used for the classification of disease severity; c) the degree of gene expression change in most brain regions examined depends to a significant degree on the disease severity classification scheme used; and d) the classification of cases by CDR provides a more orderly gradient of gene expression change in most brain regions than Braak staging or NP grouping.
This later observation suggests that there are gene expression changes that are closely associated with the earliest cognitive changes in AD. That the expression of only a relatively few genes is altered at the early stages of dementia relative to cognitively intact persons suggests that the cognitive deficits identified by the CDR represent some of the earliest events in the disease process. That significantly more genes are affected in early Braak stages or when only a few NPs can be detected indicates that multiple neurobiological systems must be affected and engaged before these neuropathological lesions become manifest.
Supplementary Material
Acknowledgements
Supported by AG02219 (VH)
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Disclosure Statement: The authors have no actual or potential conflicts of interest.
Although we are unable at this time to publish the entire array dataset, we are committed to sharing the gene expression data broadly with our academic colleagues. We will be happy to collaborate with interested investigators and to interrogate the data set for the expression levels of genes of interest.
Reference List
- Benjamini Y, Hochberg Y. Controlling the false discovery rate — a practical and powerful approach to multiple testing. Journal of the Royal Society Series B (Methodology) 1995;57:289–300. [Google Scholar]
- Blalock EM, Chen KC, Stromberg AJ, Norris CM, Kadish I, Kraner SD, Porter NM, Landfield PW. Harnessing the power of gene microarrays for the study of brain aging and Alzheimer's disease: statistical reliability and functional correlation. Ageing Res Rev. 2005;4:481–512. doi: 10.1016/j.arr.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Blalock EM, Geddes JW, Chen KC, Porter NM, Markesbery WR, Landfield PW. Incipient Alzheimer's disease: microarray correlation analyses reveal major transcriptional and tumor suppressor responses. Proc Natl Acad Sci U S A. 2004;101:2173–2178. doi: 10.1073/pnas.0308512100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blessed G, Tomlinson BE, Roth M. The association between quantitative measures of dementia and of senile change in the cerebral grey matter of elderly subjects. Br J Psychiatry. 1968;114:797–811. doi: 10.1192/bjp.114.512.797. [DOI] [PubMed] [Google Scholar]
- Bolstad BM, Irizarry RA, Astrand M, Speed TP. A comparison of normalization methods for high density oligonucleotide array data based on variance and bias. Bioinformatics. 2003;19:185–93. doi: 10.1093/bioinformatics/19.2.185. [DOI] [PubMed] [Google Scholar]
- Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease-associated neurofibrillarypathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112:389–404. doi: 10.1007/s00401-006-0127-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braak H, Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol (Berl) 1991;82:239–259. doi: 10.1007/BF00308809. [DOI] [PubMed] [Google Scholar]
- Braak H, Braak E. Staging of Alzheimer-related cortical destruction. Int Psychogeriatr. 1997;9(Suppl 1):257–261. [PubMed] [Google Scholar]
- Braak H, Tredici KD, Schultz C, Braak E. Vulnerability of select neuronal types to Alzheimer's disease. Ann N Y Acad Sci. 2002;924:53–61. doi: 10.1111/j.1749-6632.2000.tb05560.x. [DOI] [PubMed] [Google Scholar]
- Bussiere T, Gold G, Kovari E, Giannakopoulos P, Bouras C, Perl DP, Morrison JH, Hof PR. Stereologic analysis of neurofibrillary tangle formation in prefrontal cortex area 9 in aging and Alzheimer's disease. Neuroscience. 2003;117:577–592. doi: 10.1016/s0306-4522(02)00942-9. [DOI] [PubMed] [Google Scholar]
- Davis KL, Mohs RC, Marin D, Purohit DP, Perl DP, Lantz M, Austin G, Haroutunian V. Cholinergic markers in elderly patients with early signs of Alzheimer disease. JAMA. 1999a;281:1401–6. doi: 10.1001/jama.281.15.1401. [DOI] [PubMed] [Google Scholar]
- Davis KL, Mohs RC, Marin DB, Purohit DP, Perl DP, Lantz M, Austin G, Haroutunian V. Cholinergic markers are not decreased in early Alzheimer's disease. JAMA. 1999b;281:1401–1406. doi: 10.1001/jama.281.15.1401. [DOI] [PubMed] [Google Scholar]
- Davis KL, Mohs RC, Marin DB, Purohit DP, Perl DP, Lantz M, Austin G, Haroutunian V. Neuropeptide abnormalities in patients with early Alzheimer disease. Arch Gen Psychiatry. 1999c;56:981–987. doi: 10.1001/archpsyc.56.11.981. [DOI] [PubMed] [Google Scholar]
- Dooneief G, Marder K, Tang MX, Stern Y. The Clinical Dementia Rating scale: community-based validation of “profound’ and “terminal’ stages. Neurology. 1996;46:1746–1749. doi: 10.1212/wnl.46.6.1746. [DOI] [PubMed] [Google Scholar]
- Emilsson L, Saetre P, Jazin E. Alzheimer's disease: mRNA expression profiles of multiple patients show alterations of genes involved with calcium signaling. Neurobiol Dis. 2006;21:618–625. doi: 10.1016/j.nbd.2005.09.004. [DOI] [PubMed] [Google Scholar]
- Giannakopoulos P, Herrmann FR, Bussiere T, Bouras C, Kovari E, Perl DP, Morrison JH, Gold G, Hof PR. Tangle and neuron numbers, but not amyloid load, predict cognitive status in Alzheimer's disease. Neurology. 2003;60:1495–1500. doi: 10.1212/01.wnl.0000063311.58879.01. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Che S, Counts SE, Mufson EJ. Single cell gene expression profiling in Alzheimer's disease. NeuroRx. 2006;3:302–318. doi: 10.1016/j.nurx.2006.05.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg SD, Elarova I, Ruben M, Tan F, Counts SE, Eberwine JH, Trojanowski JQ, Hemby SE, Mufson EJ, Che S. Single-cell gene expression analysis: implications for neurodegenerative and neuropsychiatric disorders. Neurochem Res. 2004;29:1053–64. doi: 10.1023/b:nere.0000023593.77052.f7. [DOI] [PubMed] [Google Scholar]
- Ginsberg SD, Hemby SE, Lee VM, Eberwine JH, Trojanowski JQ. Expression profile of transcripts in Alzheimer's disease tangle-bearing CA1 neurons. Ann Neurol. 2000;48:77–87. [PubMed] [Google Scholar]
- Hardy J. A hundred years of Alzheimer's disease research. Neuron. 2006;52:3–13. doi: 10.1016/j.neuron.2006.09.016. [DOI] [PubMed] [Google Scholar]
- Haroutunian V, Davies PJ, Vianna C, Buxbaum JD, Purohit DP. Tau protein abnormalities associated with the progression of Alzheimer disease type dementia. Neuro-biology of Aging. 2006;28:1–7. doi: 10.1016/j.neurobiolaging.2005.11.001. [DOI] [PubMed] [Google Scholar]
- Haroutunian V, Davis KL. Pathophysiology of Dementia. In: Tasman A, Kay J, Lieberman J, editors. Psychiatry. Vol. 1. John Wiley & Sons; Chichester, England: 2003. pp. 338–349. [Google Scholar]
- Haroutunian V, Perl DP, Purohit DP, Marin D, Khan K, Lantz M, Davis KL, Mohs RC. Regional distribution of neuritic plaques in the nondemented elderly and subjects with very mild Alzheimer disease. Arch Neurol. 1998;55:1185–91. doi: 10.1001/archneur.55.9.1185. [DOI] [PubMed] [Google Scholar]
- Haroutunian V, Purohit DP, Perl DP, Marin D, Khan K, Lantz M, Davis KL, Mohs RC. Neurofibrillary tangles in nondemented elderly subjects and mild Alzheimer disease. Arch Neurol. 1999;56:713–8. doi: 10.1001/archneur.56.6.713. [DOI] [PubMed] [Google Scholar]
- Irizarry RA, Hobbs B, Collin F, Beazer-Barclay YD, Antonellis KJ, Scherf U, Speed TP. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–64. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Katsel P, Davis KL, Gorman JM, Haroutunian V. Variations in differential gene expression patterns across multiple brain regions in schizophrenia. Schizo Res. 2005a;79:157–173. doi: 10.1016/j.schres.2005.03.020. [DOI] [PubMed] [Google Scholar]
- Katsel P, Davis KL, Gorman JM, Haroutunian V. Variations in myelin and oligodendrocyte-related gene expression across multiple brain regions: A gene ontology study. Schizo Res. 2005b;17:241–252. [Google Scholar]
- Katsel PL, Davis KL, Haroutunian V. Large-scale microarray studies of gene expression in multiple regions of the brain in schizophrenia and Alzheimer's disease. Int Rev Neurobiol. 2005c;63:41–82. doi: 10.1016/S0074-7742(05)63003-6. [DOI] [PubMed] [Google Scholar]
- Khachaturian ZS. Diagnosis of Alzheimer's disease. Arch Neurol. 1985;42:1097–1105. doi: 10.1001/archneur.1985.04060100083029. [DOI] [PubMed] [Google Scholar]
- Loring JF, Wen X, Lee JM, Seilhamer J, Somogyi R. A gene expression profile of Alzheimer's disease. DNA Cell Biol. 2001;20:683–695. doi: 10.1089/10445490152717541. [DOI] [PubMed] [Google Scholar]
- Masliah E, Miller A, Terry RD. The synaptic organization of the neocortex in Alzheimer's disease. Med Hypotheses. 1993;41:334–340. doi: 10.1016/0306-9877(93)90078-5. [DOI] [PubMed] [Google Scholar]
- Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM, Vogel FS, Hughes JP, van Belle G, Berg L. The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology. 1991;41:479–486. doi: 10.1212/wnl.41.4.479. [DOI] [PubMed] [Google Scholar]
- Morris JC. The clinical dementia rating (CDR): current version and scoring rules. Neurology. 1993;43:2412–2414. doi: 10.1212/wnl.43.11.2412-a. [DOI] [PubMed] [Google Scholar]
- Morris JC, Storandt M, Miller JP, McKeel DW, Price JL, Rubin EH, Berg L. Mild cognitive impairment represents early-stage Alzheimer disease. Arch Neurol. 2001;58:397–405. doi: 10.1001/archneur.58.3.397. [DOI] [PubMed] [Google Scholar]
- Morrison JH, Hof PR, Rapp PR. Neuropathology of normal aging in cerebral cortex. In: Beal MF, Lang AE, Ludolph A, editors. Neurodegenerative Diseases. Cambridge University Press; Cambridge: 2005. pp. 396–406. [Google Scholar]
- Naslund J, Haroutunian V, Mohs R, Davis KL, Davies P, Greengard P, Buxbaum JD. Correlation between elevated levels of amyloid beta-peptide in the brain and cognitive decline. JAMA. 2000;283:1571–1577. doi: 10.1001/jama.283.12.1571. [DOI] [PubMed] [Google Scholar]
- Parachikova A, Agadjanyan MG, Cribbs DH, Blurton-Jones M, Perreau V, Rogers J, Beach TG, Cotman CW. Inflammatory changes parallel the early stages of Alzheimer disease. Neurobiol Aging. 2006 doi: 10.1016/j.neurobiolaging.2006.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parvathy S, Davies P, Haroutunian V, Purohit DP, Davis KL, Mohs RC, Park H, Moran TM, Chan JY, Buxbaum JD. Correlation between Abetax-40-, Abetax-42-, and Abetax-43-containing amyloid plaques and cognitive decline. Arch Neurol. 2001;58:2025–2032. doi: 10.1001/archneur.58.12.2025. [DOI] [PubMed] [Google Scholar]
- Pasinetti GM. Use of cDNA microarray in the search for molecular markers involved in the onset of Alzheimer's disease dementia. J Neurosci Res. 2001;65:471–6. doi: 10.1002/jnr.1176. [DOI] [PubMed] [Google Scholar]
- Price JL, Ko AI, Wade MJ, Tsou SK, McKeel DW, Morris JC. Neuron number in the entorhinal cortex and CA1 in preclinical Alzheimer disease. Arch Neurol. 2001;58:1395–1402. doi: 10.1001/archneur.58.9.1395. [DOI] [PubMed] [Google Scholar]
- Price JL, Morris JC. Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Ann Neurol. 1999;45:358–368. doi: 10.1002/1531-8249(199903)45:3<358::aid-ana12>3.0.co;2-x. [DOI] [PubMed] [Google Scholar]
- Rapp MA, Schnaider-Beeri M, Sano M, Silverman J, Haroutunian V. The relationship of neuropsychological performance to functional status in old age: a comparison of nursing home residents and community dwellers. American Journal of Geriatric Psychiatry. 2005;13:450–459. doi: 10.1176/appi.ajgp.13.6.450. [DOI] [PubMed] [Google Scholar]
- Ricciarelli R, d'Abramo C, Massone S, Marinari U, Pronzato M, Tabaton M. Microarray analysis in Alzheimer's disease and normal aging. IUBMB Life. 2004;56:349–354. doi: 10.1080/15216540412331286002. [DOI] [PubMed] [Google Scholar]
- Roberson ED, Mucke L. 100 years and counting: prospects for defeating Alzheimer's disease. Science. 2006;314:781–784. doi: 10.1126/science.1132813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern Y, Liu X, Albert M, Brandt J, Jacobs DM, Castillo-Castaneda C, Marder K, Bell K, Sano M, Bylsma F, Lafleche G, Tsai WY. Application of a growth curve approach to modeling the progression of Alzheimer's disease. J Gerontol A Biol Sci Med Sci. 1996;51:M179–M184. doi: 10.1093/gerona/51a.4.m179. [DOI] [PubMed] [Google Scholar]
- Terry RD. Alzheimer's disease and the aging brain. J Geriatr Psychiatry Neurol. 2006;19:125–128. doi: 10.1177/0891988706291079. [DOI] [PubMed] [Google Scholar]
- Tomlinson BE, Blessed G, Roth M. Observations on the brains of demented old people. Journal of the Neurological Sciences. 1970;11:205–242. doi: 10.1016/0022-510x(70)90063-8. [DOI] [PubMed] [Google Scholar]
- Uboga NV, Price JL. Formation of diffuse and fibrillar tangles in aging and early Alzheimer's disease. Neurobiol Aging. 2000;21:1–10. doi: 10.1016/s0197-4580(00)00091-9. [DOI] [PubMed] [Google Scholar]
- Von Gunten A, Kovari E, Rivara CB, Bouras C, Hof PR, Giannakopoulos P. Stereologic analysis of hippocampal Alzheimer's disease pathology in the oldest-old: Evidence for sparing of the entorhinal cortex and CA1 field. Experimental Neurology. 2005;193:198–206. doi: 10.1016/j.expneurol.2004.12.005. [DOI] [PubMed] [Google Scholar]
- Xu PT, Li YJ, Qin XJ, Scherzer CR, Xu H, Schmechel DE, Hulette CM, Ervin J, Gullans SR, Haines J, Pericak-Vance MA, Gilbert JR. Differences in apolipo-protein E3/3 and E4/4 allele-specific gene expression in hippocampus in Alzheimer disease. Neurobiol Dis. 2006;21:256–275. doi: 10.1016/j.nbd.2005.07.004. [DOI] [PubMed] [Google Scholar]
- Yao PJ, Zhu M, Pyun EI, Brooks AI, Therianos S, Meyers VE, Coleman PD. Defects in expression of genes related to synaptic vesicle trafficking in frontal cortex of Alzheimer's disease. Neurobiol Dis. 2003;12:97–109. doi: 10.1016/s0969-9961(02)00009-8. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.