

Abstract
Recently, mass spectrometry has been employed in many studies to provide unbiased, reproducible, and quantitative protein abundance information on a proteome-wide scale. However, how instruments’ limited dynamic ranges impact the accuracy of such measurements has remained largely unexplored, especially in the context of complex mixtures. Here, we examined the distribution of peptide signal versus background noise (S/N) and its correlation with quantitative accuracy. With the use of metabolically labeled Jurkat cell lysate, over half of all confidently identified peptides had S/N ratios less than 10 when examined using both hybrid linear ion trap–Fourier transform ion cyclotron resonance and Orbitrap mass spectrometers. Quantification accuracy was also highly correlated with S/N. We developed a mass precision algorithm that significantly reduced measurement variance at low S/N beyond the use of highly accurate mass information alone and expanded it into a new software suite, Vista. We also evaluated the interplay between mass measurement accuracy and S/N; finding a balance between both parameters produced the greatest identification and quantification rates. Finally, we demonstrate that S/N can be a useful surrogate for relative abundance ratios when only a single species is detected.
Keywords: mass spectrometry, quantitation, dynamic range, computational algorithms
Introduction
Understanding the molecular regulation of a cell or tissue requires both the identification of the proteins involved and an appraisal of their respective activities. Quantitative proteomic studies gather both types of information, on a global scale, through direct assessment of protein identities, modification states, and relative abundances. In particular, mass-spectrometry-based approaches have been shown to provide fast, sensitive, and accurate quantification of thousands of peptides from a single liquid chromatography/tandem mass spectrometry (LC-MS/MS) analysis.1–3
Most stable-isotope-based LC-MS/MS methods currently in use measure relative differences in abundance between two distinct sample pools of the same protein species, where one pool was exposed to a stimulus of interest. Through the incorporation of stable isotopes into peptides derived from one sample,4–6 these pools are easily distinguished by mass within a full survey (MS) scan while being chemically identical and possessing equal ionization efficiencies. Peptide ions from either the labeled (‘heavy’) or unlabeled (‘light’) pools are then fragmented along the peptide backbone through a tandem (MS/MS) scan, yielding primary amino acid sequence information via the use of database searching algorithms.7,8
Quantitative information is typically gathered from consecutive MS scans in the form of chromatographic peak distributions for each peptide. These peak distributions, known as isotopic envelopes, consist of the multiple spectral peaks generated from a given peptide species and collectively represent the total signal from that species. These peaks are traced across time to produce an extracted ion chromatogram (XIC). The chromatograms detail the signal intensity of the pair of heavy and light species as both coelute from online liquid chromatography into the mass spectrometer. Comparing the area under each XIC curve yields a ratio that directly correlates with the relative abundances of the labeled and unlabeled peptides (Figure 1a).
Figure 1.
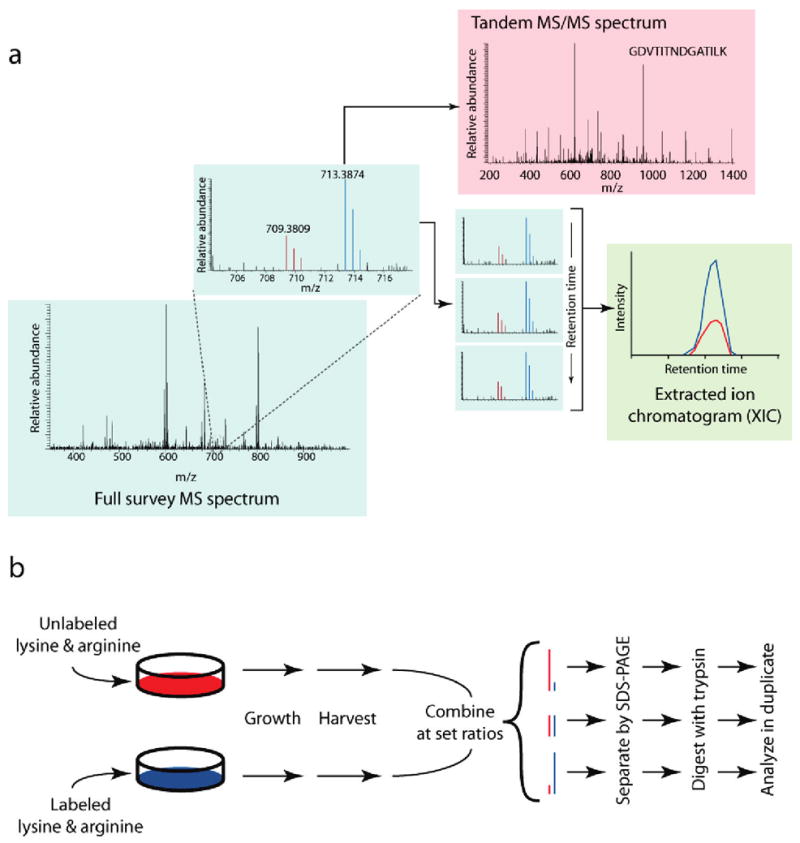
Experimental design. (a) Typical workflow for quantitative proteomic methods using stable isotopes. Stable isotope labeling produces two chemically identical peptide pools which differ only in their masses. This difference in mass is easily resolved within the full (MS) survey spectrum (blue box) into separate isotopic envelopes composed of all spectral peaks for the labeled and unlabeled species (red and blue peaks; see inset). To identify the eluting peptide, the mass spectrometer isolates and fragments the peptide ion of either the labeled (blue) or unlabeled (red) species to produce a tandem MS/MS spectrum (red box). The relative spectral peak intensities of both species are culled from successive MS spectra into an extracted ion chromatogram (green box). These chromatographic peaks, when compared across time, correlate with the change in abundance between the two peptide species. (b) Stable-isotope-labeled protein mixtures for testing signal-to-noise and quantitative accuracy. Jurkat lymphoblastic T-cell lines were grown using the SILAC method in two separate cultures differing in their growth media: one culture contained 13C615N2-lysine and 13C615N4-arginine, while the other contained natural forms of these amino acids. Cells were harvested, lysed, and combined at set protein concentration ratios of 5:1, 2.5:1, 1:1, 1:2.5, and 1:5. Sample mixtures were then gel-separated, trypsin-digested and analyzed in duplicate by LC-MS/MS techniques.
To cope with the large amounts of data generated in proteome-scale analyses, these quantitative calculations and peptide identification are usually performed in an automated fashion. However, beyond bioinformatic considerations, the wide range of protein concentration found in a given cell or tissue further confounds analysis, as it challenges the detection limits and dynamic range of the instrumentation. While recent advances in instrument design have improved mass measurement accuracy, enhanced sensitivity, and increased scanning speeds,9–13 the detection of low-abundance proteins and the separation of signal from noise remain major obstacles in proteome-scale studies by mass spectrometry.
These challenges affect the gathering of quantitative information in particular, as abundance data is commonly extracted from the MS scan. Consequently, quantitation does not benefit from the enrichment of specific peptide ions that often boost signal levels in MS/MS fragmentation. While it is likely that low signal levels affect the accuracy of quantitative results, most large-scale studies in the literature provide no information on the signal-to-noise (S/N) levels for measured peptide ratios; in fact, since most measurements are aggregated at the whole protein level, few include any assessment of peptide quantitative accuracy at all.
In this study, we investigated the S/N characteristics of complex mixtures analyzed by high-throughput, mass-spectrometry-based methods and the ramifications of low S/N on quantitative analysis. We found that a vast majority of peptides were identified at low S/N levels, and a strong correlation existed between S/N and the accuracy of calculated abundance ratios. To improve the accuracy at low S/N, we developed an algorithm that leverages the high precision of mass measurement in addition to commonly used mass accuracy information. We further found that signal-to-noise levels can be employed to establish minimum abundance differences in instances where just one member of the light/heavy pair was exclusively detected. This estimation provided valuable, quantitative information about minimum ratio changes where none had previously been reported.
Methods
Jurkat Cell Lysate Preparation, SDS-PAGE, and In-Gel Proteolysis
Jurkat cells were cultured in RPMI 1640 media lacking L-lysine and L-arginine (Invitrogen, Carlsbad, CA) with a supplement of 10% dialyzed fetal bovine serum (Calbiochem, San Diego, CA), antibiotics, and 12C614N2 L-lysine and 12C614N4 L-arginine (light) or 13C615N2 L-lysine and 13C6 15N4 L-arginine (heavy; Cambridge Isotope Laboratories, Andover, MA; 98% purity).
Approximately 5 × 107 cells each were harvested from both labeled and unlabeled conditions. Cells were lysed in 2 mL of buffer containing 50 mM Tris (pH 7.6), 150 mM NaCl, 4 mM MgCl2, 0.5% Triton X-100, 10 mM Na2P4O7, 5 mM NaF, 1 mM β-glycerophosphate, 1 mM Na3VO4, and a protease inhibitor cocktail (Complete Mini, Roche, Indianapolis, IN). Cells were thrice sonicated for 30 s with a 3 min rest on ice between each round of sonication, and centrifuged for 10 min at 15 000 rpm to remove cell debris and insoluble material.
Protein concentrations were determined by BCA assay. Disulfide bonds were reduced with DTT (3 mM, 50 °C, 30 min), and free sulfhydryl groups were alkylated with iodoacetamide (~8 mM, 25 °C, 1 h in dark).
Labeled and unlabeled lysate were mixed at ratios as indicated in the text and separated in a preparative SDS-PAGE bis-tris (4–12%) gel (Invitrogen). Electrophoresis was stopped when the buffer front had migrated ~5 cm into the gel. Gels were stained with Coomassie, and a single gel region containing proteins between 50 and 100 kDa was excised, and prepared for trypsin digestion as previously described.9
Samples were subjected to C18 solid-phase extraction (Seppak, Waters, Milford, MA), dried under vacuum, and resuspended in 20 μL of buffer containing 7.5% acetonitrile and 5% formic acid in water.
Mass Spectrometry and Peptide Identification
LC-MS/MS was performed on a hybrid linear ion trap-Fourier ion cyclotron resonance (FT-ICR) mass spectrometer12 (LTQ FT; ThermoFisher, San Jose, CA) and a hybrid linear ion trap–Orbitrap mass spectrometer13 (LTQ Orbitrap), as indicated in the text. Peptide mixtures were loaded onto a 125-μm i.d. fused-silica microcapillary column packed in-house with C18 resin (Michrom Bioresources, Inc., Auburn, CA) and separated using an 80-min gradient from 5% to 28% solvent B (0.15% HCOOH, 97.5% CH3CN). For the LTQ FT analyses, 10 MS/MS spectra were acquired in a data-dependent fashion from a preceding FTMS master (MS) spectrum (375–1800 m/z at a resolution setting of 1 × 105) with an automatic gain control (AGC) target of 1 × 106, unless otherwise specified in the text. LTQ Orbitrap analyses were identical to those in the LTQ FT, except master (MS) spectra were acquired from the Orbitrap (375–1800 m/z at a resolution setting of 6 × 104) instead of the FT-ICR cell. Charge-state screening was employed to reject singly charged peptides, and a threshold of 4000 counts was required to trigger an MS/MS. All data were collected in centroided mode. When possible, the LTQ and FT-ICR or Orbitrap were operated in parallel processing mode.
Database Searching and Data Processing
In-house software was employed to verify the precursor charge-state and monoisotopic mass from the isotopic envelope information in high mass accuracy FT-ICR or Orbitrap MS spectra, as previously described.28
MS/MS spectra were searched using the SEQUEST algorithm7 against a composite database containing the human IPI protein database (ftp.ebi.ac.uk/pub/databases/IPI/current) and its reversed complement. Search parameters included fully tryptic specificity, a static modification of 57.0214 on cysteine, dynamic modifications of 8.01420 on lysine, 10.00827 on arginine, and 15.9949 on methionine, and a mass tolerance of ±50 ppm.
XCorr, ppm, and dCn score cutoffs were empirically determined for each instrument analysis to maximize the number of peptide spectral matches while maintaining an estimated false discovery rate of ~1% at maximum sensitivity.15
Software
Instrument-specific RAW files were converted to the mzXML file format27 using ReAdW.exe (http://sashimi.sourceforge.net) and imported into a relational database using MySQL (http://www.mysql.com). Data analysis was performed using the in-house GFY analysis suite.
Peptide quantification and S/N determination were performed using Vista, an automated software suite developed in-house. The software functioned as follows: the theoretical masses of both labeled and unlabeled species were determined from the sequence composition of each peptide. These masses were then used to extract ion chromatogram intensities separately for each species from high resolution MS spectra within 20 MS survey scans and within a user-defined mass window. The mass window was separately defined for each run at ±5σ of the total mass accuracy distribution of all estimated confidently assigned peptides. Spectral peaks for each species were separated from surrounding noise using an iterative mass precision algorithm described in the text. Chromatographic peak boundaries were determined by extending each peak from the location of the data-dependent MS/MS scan within which the peptide was identified to a dynamically determined noise baseline, incorporating regions where the MP z-score was greater than zero. This noise baseline was calculated from all peaks observed within a ±25 m/z window around the theoretical species masses and within the adjacent ±20 MS spectra. The S/N ratio for each species was determined as the ratio of the maximum chromatographic peak intensity observed to the noise baseline, as calculated in the text. The area under the curve was then separately determined for each species, and compared to generate a relative abundance ratio. The monoisotopic peak for each species only was used to generate the area measurements; we note that, while multiple peaks may be used in theory, we found that such an approach is problematic at low signal levels with extreme quantitative ratios due to issues with the limit of detection of the mass spectrometer.
Quantified peptide species were individually scored for quality using both the Random Forest classifier16 and a heuristic score. This score represents a weighted average of a variety of empirically determined Boolean predictors, including signal-to-noise ratios, number of observations across the chromatographic peak, mass accuracy statistics, unlabeled/labeled pair coelution, distance from the tandem MS scan, split peak signature, and encapsulation of the surrounding data. Except where otherwise noted, all successfully quantified peptides within 5σ of the mean log2 ratio were included in all analyses, regardless of score. Vista also includes additional enhancements to improve quantification, including rescaling of the heavy peptide ion series, deconvolution to normalize for label impurity, and compensation for the interconversion of arginine to proline, as is sometimes seen in studies using SILAC.29
The software supports data supplied in the pepXML and mzXML formats,27 from a wide variety of high-mass-accuracy-capable instrumentation, analyzed using any of a variety of search algorithms. The algorithm also supports most differential labeling methods, including SILAC,6 cleavable ICAT,30 and CAR.24 It is further extensible to deuterium-based approaches.
All analyses in this study were performed on a dual-processor, dual-core 3.00 GHz Xeon system running Fedora Core 7. On average, Vista currently performs approximately one analysis per second; a typical analysis of 3445 peptides from the 1:1 labeled sample using LTQ FT data took 57 min.
More information on the Vista software suite, including information on obtaining the software, can be found at http://vista.hms.harvard.edu/.
Results and Discussion
Signal-to-Noise Characteristics of Complex Proteome Mixtures
To produce a test set of peptides typical of a large-scale experiment, we collected whole cell lysate from Jurkat T-cell lymphoblastic human cells differentially labeled using the SILAC method:6 one pool was grown in the presence of isotopically substituted 13C615N2-lysine and 13C615N4-arginine (heavy), while the other was grown in the presence of naturally occurring lysine and arginine (light; Figure 1b). These pools were mixed in set proportions (5:1, 2.5:1, 1:1:, 1:2.5, 5:1) based on total protein concentration and gel-digested, and the fractions corresponding to proteins with molecular weights between 50 and 100 kDa were analyzed in duplicate on a hybrid linear ion trap/Fourier transform ion cyclotron resonance (FT-ICR) mass spectrometer12 (LTQ FT, ThermoFisher Scientific) and a hybrid linear ion trap/Orbitrap mass spectrometer13 (LTQ Orbitrap, ThermoFisher Scientific).
To determine chromatographic peak S/N, we first developed a method for background noise estimation in full scan (MS) spectra. Noise may arise from a number of factors including atmospheric sources, electrical interference, and chemical contaminants. These sources combine to produce noise peaks, which are observed alongside peaks generated from the detection of peptide ions. However, the intensities of most noise peaks are characteristically different from peptide signal: noise peak intensities are typically lower and more similar than those from peptide signal. The intensity distribution of all peaks measured within several m/z and retention time windows of varying sizes indicated that many noise peaks reproducibly fell within a narrow intensity range, and each window had a similar median intensity (Figure 2a). We defined this median intensity observed in the local vicinity of the chromatographic peak as the noise level for this study, providing a suitable estimation of the background noise. The S/N of a particular peptide’s chromatographic peak can then be calculated by dividing the maximum peak intensity by the noise level.
Figure 2.
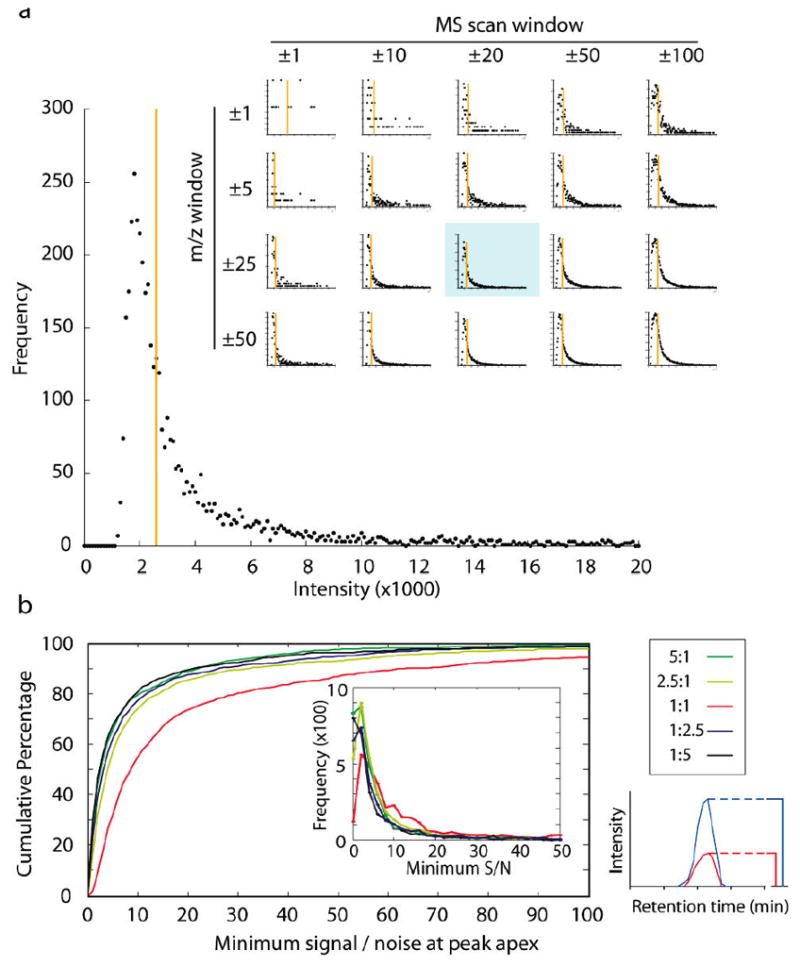
Signal-to-noise ratios in an FT-ICR instrument. (a) Spectral peak intensity distribution in FT-ICR MS survey scan data. A histogram of all spectral peak intensity information recorded across a defined retention time (±20 MS scans; 46.3–48.7 min) and mass range (±25 m/z; 675–725 m/z) from an 80-min analysis illustrates the distribution of peaks arising from both signal and noise, binned at 100 intensity unit increments. Noise peaks tend to appear with similar intensities, producing a large spike in frequency at the lower end of the intensity range. While the definition of the graph can be controlled by varying the time and mass windows examined (inset), the median value (orange bar) remains similar. (The blue-shaded graph in the inset is the same as the larger graph within the figure.) (b) Most complex-mixture peptide identifications from an FT-ICR instrument are at low signal-to-noise. A single 80-min LC-MS/MS analysis of a 1:1 mixture (Figure 1b) generated more than 3000 confident peptide identifications. However, the majority of these peptides were detected with S/N levels of less than 10 in the MS scan, and the median S/N value was only 8.6. Other mixing ratios produced still lower overall S/N distributions with similar numbers of peptides identified. Signal-to-noise values were measured at the chromatographic peak apex; the smaller S/N ratio from either the heavy or light peak was then chosen and plotted (bottom left). Any point along the graph indicates the percentage of peptides identified at or below the given S/N threshold. Graph inset shows a histogram of the S/N distribution of the same 3000 peptide identifications.
Using this system for estimating noise, we generated the S/N distribution for all peptides from our defined test mixtures with FT-ICR instrumentation (Figure 2b). For the 1:1 sample, the median S/N value was 8.6, and 28% of peptides had a S/N ratio of less than 5. Similar results were found at other mixing ratios. Within all test mixtures, most peptides were identified at S/N levels less than 10.
The Role of High Mass Accuracy in Separating Signal from Noise
Although it was clear most peptides in our test mixture were analyzed at low S/N, the consequences of low signal levels on quantitative accuracy remained to be tested. As mass spectrometers capable of highly accurate mass measurement are quickly becoming the norm in high-throughput quantitative analyses, we investigated the impact of highly accurate mass information (HMA) on the accuracy and precision of quantitative measurements. We reasoned that HMA should improve quantification accuracy over previous generations of instrumentation by requiring a tolerance window of only a few parts-per-million (the “mass window”) when defining the XIC, thus reducing the number of interfering, nonsignal peaks observed. Furthermore, HMA should enable the isolation of each spectral peak present in the peptide ion isotopic envelope separately from the others. Checking for these additional peaks provided additional discriminatory power to distinguish signal from noise, and to separate overlapping signals from other, unrelated peptides (data not shown).
With the use of HMA (±20 ppm), quantification accuracy in the 1:1 mixture was highest at the largest S/N levels (Figure 3a): at S/N levels above 10, the standard deviation in reported ratios was 0.22 (n = 1604). However, below S/N of 10, the standard deviation of ratio measurement expanded to 0.83 (n = 1738). These findings demonstrate that the standard deviation of ratio measurement was highly negatively correlated with S/N (r = −0.67). Similar correlations were found with mixtures of varying proportions (data not shown).
Figure 3.

A mass precision algorithm improves quantitation accuracy. (a) Standard deviation in ratio measurement as a function of S/N. A moving window (100 samples, centered) of the mean standard deviation from over 3000 observed abundance ratios illustrates the variance in ratio measurements obtained using only a high mass accuracy filter (red line), or through use of a mass precision algorithm (blue line; see text). Relative abundance ratios from the same 1:1 mixture were plotted against the signal-to-noise ratio of the less intense of the heavy or light peptide species. Individual ratio observations are shown as light red (mass accuracy filter) and light blue points (mass precision algorithm). While both approaches performed similarly at higher signal levels, the mass precision score significantly improved accuracy (Ansari-Bradley test: p = 0.0019) when compared to the simple mass window filter at lower signal levels. Blue and red stars indicate S/N and ratio values for the example peptide illustrated in panel c. (b) Mass precision algorithm methodology. The algorithm assesses the similarity in mass accuracy between the light and heavy peptide species. Theoretical masses of the light and heavy peptide peaks are indicated by red and blue triangles, respectively; noise peaks are shown in gray, while peptide peaks are shown in black. Although the peptide peak mass accuracy varies from scan to scan, both species deviate in a similar manner (as indicated by their mass difference from the theoretical mass). Conversely, noise peaks are randomly distributed, distinguishing them from peptide signal. (c) Mass precision algorithm resolves interfering peaks. To calculate the mass precision (MP) score, the mass accuracies of the light and heavy peaks (bottom panels; red and blue lines) were employed to discriminate against noise peaks and to determine chromatographic peak boundaries (see text). Using high mass accuracy as the only filter (top left) permitted a confounding peptide eluting earlier than the target heavy peptide to interfere with quantitation. The mass precision score (bottom right; green line) successfully discriminated between the two sets of spectral peaks so that only the correct peak data was chosen (top right).
An Algorithm To Improve Ratio Accuracy at Low Signal-to-Noise
A distinct approach was necessary to improve quantitation accuracy at low S/N levels, beyond the reduction in mass window size allowed by HMA. While the individual mass accuracy of a particular peptide species may drift from scan to scan, we observed that both the labeled and unlabeled versions of the same peptide exhibited coordinated variation: although the observed m/z of both species deviated from their theoretical m/z in varying amounts throughout sequential scans, both species tend to drift in a similar fashion (Figure 3b). In comparison, across the same scans, interfering peptide peaks are not perfectly coeluting and can have deviations that are much less correlated. We reasoned that this property could be used to discriminate specific peptide signal from interference and assist in peak boundary determination.
To validate this approach, we assessed the mass accuracy of all confidently identified labeled and unlabeled peptide species in the 1:1 test mixture, and then compared their measurement precision by calculating the similarity of mass accuracy between both species. The difference between observed light and heavy peptide peaks was more consistent than those same species’ deviation from their respective theoretical masses (Supplementary Figure 1 in Supporting Information).
We developed these observations into a mass precision algorithm (MP) to distinguish signal from noise and other interfering peaks in an automated manner (see Methods). For each spectral peak within the mass window, the mass deviations gauging the precision of a particular peak with respect to its labeled (or unlabeled) counterpart were summed, both in that spectrum and adjacent spectra. This score was recursively determined for all spectral peak combinations in the mass window. Scores were normalized to generate a z-score in which higher values corresponded to peak sets with similar mass precision, suggesting that these peaks were likely of peptide origin; the algorithm also assisted in chromatographic peak edge definition.
We applied the MP algorithm to quantification of the 1:1 test mixture (Figure 3a; blue line). The algorithm performed similarly to HMA alone at S/N levels above 10 (relative abundance ratio σ = 0.20 vs 0.22; n = 1604; respectively). However, for peptides with a maximum S/N of less than 10, the mass precision algorithm significantly reduced the standard deviation of the reported ratios (0.47 vs 0.83; n = 1738; Ansari-Bradley test: p = 0.0019). While the algorithm was designed to reject noise, it also performed well in deconvoluting signals from two different peptides, with similar masses, that eluted concurrently. In one example, the MP algorithm filtered out an unrelated peptide with a mass similar to the labeled peptide, yielding the correct result, while the HMA approach incorrectly combined both peptides (Figure 3c). The MP algorithm similarly improved accuracy when applied to other mixing ratios (5:1, 2.5:1, etc.) of the same sample (data not shown).
To determine if measurement accuracy was affected by the overall mixing ratio, we examined data from five samples mixed at varying proportions (5:1 to 1:5) and found the standard deviation of the reported ratio linearly scaled with the mixing ratio (Figure 4a).
Figure 4.

Assessing accuracy and reproducibility of quantitation. (a) Measurement accuracy scales linearly over a 10-fold ratio difference. Identical, labeled peptide samples were mixed at five different proportions (5:1 to 1:5) and analyzed in duplicate, producing an average of 2900 successfully quantified peptides per analysis. The variance in each mixture is presented as a box and whisker plot, where the box contains lines denoting the lower quartile, median, and upper quartile values. Whiskers extend to 1.5 times the interquartile range. (b) Distribution of Random Forest classifications as a function of abundance ratio and S/N. Color denotes the Random Forest true class probability (RF Score; see text) that a particular quantitation event from the 1:1 mixture illustrated in Figure 3a is reproducible. The classifier indicates the expectation that a replicate analysis of the same peptide would yield a ratio within 5% of this observation. (c) Distribution of Vista heuristic score classifications. Color denotes the Vista heuristic score (see text) for a particular quantitation event from the same 1:1 mixture illustrated in panel b. The heuristic score is a weighted sample of a series of Boolean predictors to predict reproducibility. (d) Replicate analyses and source of variation in measurement. Replicate analyses from the same biological sample were acquired in two separate instrument runs, and the relative abundance ratios of 1558 peptides identified and quantified in both analyses were plotted against each other on the x- and y-axes. Color indicates the average S/N level of peptides quantified at a particular ratio; ratios were binned into 50 equal increments (0.07 ratio units) and the average S/N level of all binned species is displayed. The spread of peptide quantitation events along a 1:1 identity axis (bottom left to top right) suggests a majority of the variation in ratio measurement was due to factors external to the algorithm or instrument (e.g., biological).
Assessing Quantitative Accuracy
Although the MP algorithm significantly improved the accuracy of ratio determination, especially at low S/N, it did not completely reduce the variation in ratio measurement. A method to assess the credibility of any single quantitative result was therefore necessary. While scoring systems for peptide identification are fairly mature,14,15 comparable systems for quantitative data were lacking. We trained a number of commonly employed machine learning strategies and a simple, Boolean scoring heuristic (see Methods) on 47 features gathered from the raw data, ranging from peptide charge state to S/N (Supplementary Figure 2 in Supporting Information). The Random Forest algorithm16 (Figure 4b) and the heuristic score (Figure 4c) yielded the greatest predictive power across all mixtures, and both scoring systems have proven useful in validation of concurrent studies.1,17,18 Each score provides a measure of the credibility of each individual quantitation event.
Sources of Variation in Quantitative Mass-Spectrometry-Based Methods
Variation in quantitative data may be biological (e.g., variation in protein abundance inside the cells or tissue examined), technical (caused by differences in sample handling), or instrument-based (dependent upon random or systematic biases of the mass spectrometer). To better understand the variation within our samples, we tested the reproducibility of our algorithm on separate instrument analyses of the same 2.5:1 peptide mixture. Measurement precision of the same peptide in different data acquisitions was much higher than the ratio accuracy between different peptides within the same sample (Figure 4d). These findings suggest the majority of variation in our test mixture was due to bias within the underlying biological samples and not affected by the instrument. Similar results were found within samples mixed at other proportions (data not shown).
Mass Accuracy versus Signal-to-Noise Levels
Signal intensity in ion-trap instrumentation is directly proportional to the number of ions analyzed. While increasing the number of ions analyzed improves S/N levels, more ions decrease the accuracy of mass measurement due to space charging effects,19 and lengthen collection time, reducing the number of spectra obtained per hour. We investigated the interplay between these two properties by varying the automatic gain control setting (roughly equivalent to the maximum number of ions allowed in the trap for any MS scan) from 300 000 to 10 million ion counts. While the 300 000-count method produced the best mass accuracy (Supplementary Figure 3a in Supporting Information), it possessed the lowest overall S/N (Supplementary Figure 3b in Supporting Information). The converse was true for the 10M-count method. To examine overall throughput and the method which yielded the highest number of peptides identified and quantified, we determined the number of MS/MS collected, the number of peptide identifications deemed correct at an estimated 1% false discovery rate, and the number of successful quantitation events (Supplementary Figure 3c in Supporting Information). The 1 and 3 M methods were most efficient in generating both peptide identification and quantitative information.
Using Signal-to-Noise Levels To Estimate Relative Abundance Ratios
Both the labeled and unlabeled peptide species must reach above the instrument detection threshold in order for their relative abundances to be established in most commonly used algorithms. In cases where one species reaches above the threshold while the other does not, it is not possible to perform a direct comparison (based upon area or spectral peak height) to determine the relative ratio. However, the minimum ratio difference between the two peptide species can be estimated by employing the signal-to-noise ratio of the observed peptide chromatographic peak, even when one species is below the detection limit of the instrument. For example, given that up-regulation of a protein causes it to appear in 100-fold excess of its basal state, if the up-regulated protein is detected at a S/N level of only 50, the signal generated from its basal counterpart will be below the detection limit of the mass spectrometer. Nevertheless, although it is impossible to determine the exact ratio, the abundance ratio between the species must be at least 50-fold. We compared the ratio of S/N levels for a 1:5 text mixture to the area-based method and found that S/N provided a reasonable approximation of our area-based results (Figure 5a).
Figure 5.

Signal-to-noise measurements provided useful estimations of relative ratio abundance when only one peptide species was present. (a) Comparison of area-based and signal-to-noise-based ratios for peptides where both species were found. For 1522 peptides with high S/N ratios (>15 S/N) from a 1:5 (labeled/unlabeled) mixture where both the labeled and unlabeled species were present, ratios were calculated based on the areas under each extracted ion chromatogram (gray), and by dividing the signal-to-noise ratio of the unlabeled species by that of the labeled species (blue). S/N-based ratios approximated those calculated by the area-based method. (b) Exclusive quantification events reflect minimum changes in abundance. In the same 1:5 mixture as above, signal from the unlabeled peptide species was exclusively detected for 866 peptides. Although the labeled species was not observed, the S/N ratio of the unlabeled species was used as a surrogate for an area-based calculation, allowing the minimum change in abundance to be calculated for these species (green bars). Gray bars depict the same distribution of area-based quantification events from panel a.
We analyzed 866 exclusive quantification events where only the unlabeled peptide species was detected from a 1:5 test mixture (Figure 5b). As predicted, the reported ratios were similar to or less than the ratios calculated for 1522 confidently quantified peptides where both species were found. To our knowledge, all previously described algorithms do not return quantitative measurements when only one species is detected. Assigning values to these one-species-only occurrences allowed identification of large changes in peptide concentration–changes which often denote key proteins of interest.
Comparison to Orbitrap Results
The Orbitrap mass spectrometer has recently become a popular platform for high-throughput proteomic analyses. Because of its frequent use in the study of complex mixtures20–23 and to confirm our findings from the FT-ICR platform, we examined the Orbitrap’s S/N characteristics and quantitative performance. As Orbitrap MS spectra generally contain fewer noise peaks, the intensity distribution of the Orbitrap mass analyzer was broader (Supplementary Figure 4a in Supporting Information) than the distribution of FT-ICR MS spectra. Nonetheless, the S/N distributions of the test mixtures were similar: the median S/N for the 1:1 mixture was 9.2 (FT-ICR median S/N: 8.6), while the median S/N for a 5:1 mixture was only 3.03 (Supplementary Figure 4b in Supporting Information). In terms of quantitative performance, we observed the same strong negative correlation between S/N and variance in ratio measurement in a 1:1 mixture as with the FT-ICR (Orbitrap r = −0.69; FT-ICR r = −0.67). Furthermore, while ratio accuracy in Orbitrap data was similar between HMA and the MP algorithm at S/N greater than 10 (σ = 0.29 and σ = 0.26, n = 2077, respectively), the MP score significantly improved accuracy at low S/N (<10) over the use of HMA alone (HMA σ = 0.80 vs MP algorithm σ = 0.76; n = 2107; Ansari-Bradley test: p = 6.1 × 10−4). This improvement in ratio accuracy (Supplementary Figure 4c in Supporting Information) was similar to results obtained with the mass precision algorithm on FT-ICR data.
A Software Platform To Automate Peptide Quantification
We integrated our findings into Vista, a software platform designed for efficient, high-throughput quantitative analysis of mass spectrometric data (Supplementary Figure 5 in Supporting Information). Automation of the quantification process is essential for its application to large-scale data sets, and integrates the MP algorithm into a comprehensive software package while also providing objective quality assessment and a number of other features. This software has been applied in a number of concurrent studies;1,17,18,24–26 more information about the software and its availablity can be found at http://vista.hms.harvard.edu/.
Conclusions
The sheer number of proteins, modification states, and the vast concentration range between the most and least abundant proteins in the cell provide significant challenges to global protein analysis via mass spectrometry. While much attention has been focused on improving the depth of analysis in high-throughput studies, understanding the data quality of such methodologies is equally important. Here, we found that, even with the introduction of instrumentation capable of highly accurate mass measurement, a majority of peptides within all samples analyzed in this study were identified at S/N values of less than 10, with over a quarter of all peptides identified at a S/N less than 5. While these low signal levels do not necessarily affect the quality of fragmentation spectra and subsequent peptide identification due to a separate selection event during fragmentation (data not shown), they nonetheless present a substantial challenge to the use of full MS spectra to determine relative abundance ratios. Not surprisingly, we present clear evidence that the accuracy of ratio measurement is highly correlated with S/N, a finding which should apply regardless of the quantitative method chosen.
Through the development of the mass precision algorithm outlined here, we were able to significantly improve ratio measurement accuracy for peptides identified at low S/N. By exploiting the relative stability of peptide ion masses, the algorithm was able to distinguish signal from noise, improving quantitation. It was also particularly effective at distinguishing peptide peaks of interest from other coeluting peptide peaks with similar masses (shown in Figure 3c), a frequent occurrence in the analysis of complex mixtures.
Although the MP algorithm was able to improve accuracy, it is still essential that quantitative data include an assessment of their overall precision and accuracy when published. Such measurements have been infrequently reported in the literature and are often not presented with sufficient detail for assessment at the individual peptide level. We therefore developed two unbiased scores, based upon the Random Forest classifier and a heuristic score, to aid in this process. In lieu of such scores, other assessors may be supplied, such as ratio standard deviations from multiple analyses, or, if replicate studies are impractical or impossible, the S/N of each measurement. S/N levels likely provide the simplest calculated metric which correlates with abundance measurement accuracy.
Of course, our findings with an FT-ICR mass analyzer may not apply to other instrument configurations or other detector types. Thus, we examined the performance of the Orbitrap mass analyzer, which has been used in a variety of recent high-throughput studies. Here, we found a similar correlation between signal levels and ratio measurement accuracy. However, the spectral peak intensity profile of the Orbitrap data was less dense than that of the FT-ICR and exhibited a broader distribution. This disparity is likely due to different signal processing algorithms for data thresholding from each detector. Nonetheless, we demonstrated that ratio measurement accuracy and S/N levels were very similar between both ion detector types.
Beyond the assessment of data quality, perhaps one of the most intriguing roles S/N determination can play is in supplying abundance information where only one species in a heavy-light pair is detected. These events are arguably the most significant for follow-up study, as they frequently represent widely varying abundances between two states. In these cases, the S/N ratio signifies the minimum possible change in abundance, providing crucial clues to the investigator concerning these one-species-only occurrences. Nevertheless, as many incorrectly identified peptides and common contaminants may masquerade as one-species-only events, great care must be taken, both in careful experimental design (to reduce the chance of contaminants being mistaken for real changes) and in data validation, to verify peptide identities before relying upon S/N information to deduce changes in abundance.
This work uncovered significant relationships affecting the interplay between the wide range of protein abundance, the dynamic range of the mass spectrometer, and the ability of large-scale experiments to accurately provide relative abundance information. Becasue of the large range of protein concentration within the cell, even as technologies improve the depth at which the proteome can be sampled, the efficient separation of peptide signal from background noise remains paramount. In this study, we present not only an assessment of how low signal levels affect quantitation, but also describe methods to improve ratio measurement accuracy and mitigate the effects of low signal levels in the analysis of complex mixtures.
Supplementary Material
Acknowledgments
This work was supported in part by National Institutes of Health Grants GM67945 and HG3456 (to S.P.G.). We thank F. McKeon and A. Kettenbach for supplying the Jurkat cells used in this study. We also thank J. Mintseris, S. Beausoleil, and B. Faherty for helpful discussions.
Footnotes
Supporting Information Available: Supporting Information includes a demonstration that mass precision is a more consistent indicator of signal peaks than mass accuracy (Supplementary Figure 1), receiver operating characteristic (ROC) curves for the machine learning algorithms applied to assess ratio reproducibility (Supplementary Figure 2), and the relationship between instrument gain control settings, mass accuracy, and signal-to-noise ratios (Supplementary Figure 3). An evaluation of S/N ratios and quantitative accuracy in Orbitrap data (Supplementary Figure 4) and an overview of the Vista quantitation algorithm (Supplementary Figure 5) are also provided. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.Matsuoka S, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science. 2007;316:1160–1166. doi: 10.1126/science.1140321. [DOI] [PubMed] [Google Scholar]
- 2.Dengjel J, et al. Quantitative proteomic assessment of very early cellular signaling events. Nat Biotechnol. 2007;25:566–568. doi: 10.1038/nbt1301. [DOI] [PubMed] [Google Scholar]
- 3.Dong MQ, et al. Quantitative mass spectrometry identifies insulin signaling targets in C. elegans. Science. 2007;317:660–663. doi: 10.1126/science.1139952. [DOI] [PubMed] [Google Scholar]
- 4.Ong SE, Foster LJ, Mann M. Mass spectrometric-based approaches in quantitative proteomics. Methods. 2003;29:124–130. doi: 10.1016/s1046-2023(02)00303-1. [DOI] [PubMed] [Google Scholar]
- 5.Gygi SP, et al. Quantitative analysis of complex protein mixtures using isotope-coded affinity tags. Nat Biotechnol. 1999;17:994–999. doi: 10.1038/13690. [DOI] [PubMed] [Google Scholar]
- 6.Ong SE, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1:376–386. doi: 10.1074/mcp.m200025-mcp200. [DOI] [PubMed] [Google Scholar]
- 7.Eng JK, McCormack AL, Yates I, John R. An approach to correlate tandem mass spectral data of peptides with amino acid sequences in a protein database. J Am Soc Mass Spectrom. 1994;5:976–989. doi: 10.1016/1044-0305(94)80016-2. [DOI] [PubMed] [Google Scholar]
- 8.Perkins DN, Pappin DJ, Creasy DM, Cottrell JS. Probability-based protein identification by searching sequence databases using mass spectrometry data. Electrophoresis. 1999;20:3551–3567. doi: 10.1002/(SICI)1522-2683(19991201)20:18<3551::AID-ELPS3551>3.0.CO;2-2. [DOI] [PubMed] [Google Scholar]
- 9.Bakalarski CE, Haas W, Dephoure NE, Gygi SP. The effects of mass accuracy, data acquisition speed, and search algorithm choice on peptide identification rates in phosphoproteomics. Anal Bioanal Chem. 2007;389:1409–1419. doi: 10.1007/s00216-007-1563-x. [DOI] [PubMed] [Google Scholar]
- 10.Mayya V, Rezaul K, Cong YS, Han D. Systematic comparison of a two-dimensional ion trap and a three-dimensional ion trap mass spectrometer in proteomics. Mol Cell Proteomics. 2005;4:214–223. doi: 10.1074/mcp.T400015-MCP200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Schwartz JC, Senko MW, Syka JE. A two-dimensional quadrupole ion trap mass spectrometer. J Am Soc Mass Spectrom. 2002;13:659–669. doi: 10.1016/S1044-0305(02)00384-7. [DOI] [PubMed] [Google Scholar]
- 12.Syka JE, et al. Novel linear quadrupole ion trap/FT mass spectrometer: performance characterization and use in the comparative analysis of histone H3 post-translational modifications. J Proteome Res. 2004;3:621–626. doi: 10.1021/pr0499794. [DOI] [PubMed] [Google Scholar]
- 13.Hu Q, et al. The Orbitrap: a new mass spectrometer. J Mass Spectrom. 2005;40:430–443. doi: 10.1002/jms.856. [DOI] [PubMed] [Google Scholar]
- 14.Keller A, Nesvizhskii AI, Kolker E, Aebersold R. Empirical statistical model to estimate the accuracy of peptide identifications made by MS/MS and database search. Anal Chem. 2002;74:5383–5392. doi: 10.1021/ac025747h. [DOI] [PubMed] [Google Scholar]
- 15.Elias JE, Gygi SP. Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods. 2007;4:207–214. doi: 10.1038/nmeth1019. [DOI] [PubMed] [Google Scholar]
- 16.Breiman L. Random Forest. Machine Learning. 2001;45:5–32. [Google Scholar]
- 17.Everley PA, et al. Enhanced analysis of metastatic prostate cancer using stable isotopes and high mass accuracy instrumentation. J Proteome Res. 2006;5:1224–1231. doi: 10.1021/pr0504891. [DOI] [PubMed] [Google Scholar]
- 18.Neher SB, et al. Proteomic profiling of ClpXP substrates after DNA damage reveals extensive instability within SOS regulon. Mol Cell. 2006;22:193–204. doi: 10.1016/j.molcel.2006.03.007. [DOI] [PubMed] [Google Scholar]
- 19.Zhang LK, Rempel D, Pramanik BN, Gross ML. Accurate mass measurements by Fourier transform mass spectrometry. Mass Spectrom Rev. 2005;24:286–309. [Google Scholar]
- 20.Li X, et al. Large-scale phosphorylation analysis of alpha-factor-arrested Saccharomyces cerevisiae. J Proteome Res. 2007;6:1190–1197. doi: 10.1021/pr060559j. [DOI] [PubMed] [Google Scholar]
- 21.Olsen JV, et al. Global, in vivo, and site-specific phosphorylation dynamics in signaling networks. Cell. 2006;127:635–648. doi: 10.1016/j.cell.2006.09.026. [DOI] [PubMed] [Google Scholar]
- 22.Villen J, Beausoleil SA, Gerber SA, Gygi SP. Large-scale phosphorylation analysis of mouse liver. Proc Natl Acad Sci USA. 2007;104:1488–1493. doi: 10.1073/pnas.0609836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yates JR, Cociorva D, Liao L, Zabrouskov V. Performance of a linear ion trap-Orbitrap hybrid for peptide analysis. Anal Chem. 2006;78:493–500. doi: 10.1021/ac0514624. [DOI] [PubMed] [Google Scholar]
- 24.Gartner CA, Elias JE, Bakalarski CE, Gygi SP. Catch-and-release reagents for broadscale quantitative proteomics analyses. J Proteome Res. 2007;6:1482–1491. doi: 10.1021/pr060605f. [DOI] [PubMed] [Google Scholar]
- 25.Haas W, et al. Optimization and use of peptide mass measurement accuracy in shotgun proteomics. Mol Cell Proteomics. 2006;5:1326–1337. doi: 10.1074/mcp.M500339-MCP200. [DOI] [PubMed] [Google Scholar]
- 26.Denison C, et al. A proteomic strategy for gaining insights into protein sumoylation in yeast. Mol Cell Proteomics. 2005;4:246–254. doi: 10.1074/mcp.M400154-MCP200. [DOI] [PubMed] [Google Scholar]
- 27.Pedrioli PG, et al. A common open representation of mass spectrometry data and its application to proteomics research. Nat Biotechnol. 2004;22:1459–1466. doi: 10.1038/nbt1031. [DOI] [PubMed] [Google Scholar]
- 28.Beausoleil SA, Villen J, Gerber SA, Rush J, Gygi SP. A probability-based approach for high-throughput protein phosphorylation analysis and site localization. Nat Biotechnol. 2006;24:1285–1292. doi: 10.1038/nbt1240. [DOI] [PubMed] [Google Scholar]
- 29.Van Hoof D, et al. An experimental correction for arginine-to-proline conversion artifacts in SILAC-based quantitative proteomics. Nat Methods. 2007;4:677–678. doi: 10.1038/nmeth0907-677. [DOI] [PubMed] [Google Scholar]
- 30.Li J, Steen H, Gygi SP. Protein profiling with cleavable isotope-coded affinity tag (cICAT) reagents: the yeast salinity stress response. Mol Cell Proteomics. 2003;2:1198–1204. doi: 10.1074/mcp.M300070-MCP200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.