

Summary
Addictive drugs induce a dopamine signal that contributes to the initiation of addiction, and the dopamine signal influences drug-associated memories that perpetuate drug use. The addiction process shares many commonalities with the synaptic plasticity mechanisms normally attributed to learning and memory. Environmental stimuli repeatedly linked to addictive drugs become learned associations, and those stimuli come to elicit memories or sensations that motivate continued drug use. Applying in vivo recording techniques to freely moving mice, we show that physiologically relevant concentrations of the addictive drug, nicotine, directly cause in vivo hippocampal synaptic potentiation of the kind that underlies learning and memory. The drug-induced long-term synaptic plasticity required a local hippocampal dopamine signal. Disrupting general dopamine signaling prevented the nicotine-induced synaptic plasticity and conditioned place preference. These results suggest that dopaminergic signaling serves as a functional label of salient events by enabling and scaling synaptic plasticity that underlies drug-induced associative memory.
Keywords: mesolimbic, addiction, dopaminergic, hippocampus, dentate gyrus, LTP
Introduction
Dopamine (DA) signaling has a vital role during addiction. When DA signaling is prevented, the reinforcing effects of drugs decrease, as indicated by prevention or attenuation of self-administration and place preference (Caine et al., 2007; Corrigall, 1999; Izzo et al., 2001; Stolerman and Shoaib, 1991). DA concentrations are not a direct indication of reward. Rather, DA likely participates in the ongoing associative learning of adaptive behaviors as an animal continually updates a construct of environmental saliency. That viewpoint is consistent with evidence indicating that addictive drugs act upon synaptic plasticity mechanisms that normally underlie learning and memory (Dani and Harris, 2005; Hyman et al., 2006; Jay, 2003; Jones and Bonci, 2005; Kauer, 2004; Kelley, 2004; Ungless et al., 2004; Winder et al., 2002).
The hippocampus, a key center of learning and memory, receives dopaminergic afferents from the ventral tegmental areas of the midbrain. Dopamine release in the hippocampus enhances long-term potentiation (LTP) and learning, which establishes a functional link between memory systems and reward centers (Lisman and Grace, 2005). Evidence also indicates that the hippocampus has an important role in the development of drug addiction. For example, transgenic mice with diminished hippocampal synaptic plasticity and deficits in long-term memory are no longer able to develop drug-induced place preference because the place of drug taking is hypothesized not to be associated with the drug-induced reward (Biala et al., 2005). One of the most important pathways for the formation of associative memory is the perforant path, which originates in the entorhinal cortex and relays convergent information from the neocortex to the hippocampus (Deadwyler et al., 1979; Lavenex and Amaral, 2000). In particular, we focused on the medial perforant path because it specifically carries place and spatial information (Hargreaves et al., 2005) that is important for drug associated memory. The accumulated evidence also supports that synaptic plasticity along this pathway is a substrate of memory (Lynch, 2004; Martinez and Derrick, 1996; McHugh et al., 2007; Rumpel et al., 2005).
There have been behavioral and in vitro advances showing that nicotine can influence the induction of synaptic potentiation (Davis et al., 2007; Ji et al., 2001; Matsuyama et al., 2000; Nashmi et al., 2007). The in vivo experiments, however, used deep urethane anesthesia that has been shown to alter the function of ligand-gated channels (Hara and Harris, 2002), and used doses of nicotine that would cause seizures in awake mice (Franceschini et al., 2002; Miner and Collins, 1989). There has been little or no research in freely moving animals that monitors the ongoing induction of in vivo synaptic plasticity by a biologically relevant dose of an addictive drug. Here we show that the addictive drug, nicotine, dose-dependently induces long-term synaptic potentiation of the kind that supports learning and memory. More importantly, the induction of the synaptic plasticity requires a local DA signal within the hippocampus, consistent with the view that DA enables memory for particular events (Schultz et al., 1997). The results also suggest that the magnitude of the DA signal influences the strength of the synaptic plasticity.
Results
Nicotine-induced Long-term In Vivo Synaptic Plasticity
Field potentials evoked by stimulation of the medial perforant path (Figure 1A) were recorded in the hilar region of the hippocampal dentate gyrus (Figure 1B) from freely moving C57BL/6 mice (Davis et al., 1997) (Figure 1C). We focused on the medial perforant path because it relays convergent information from the neocortex that is rich in contextual, place, and spatial content (Hargreaves et al., 2005), and evidence indicates that such information is associated with the drug experience (Biala et al., 2005; Kilts et al., 2001). Field recordings were made from the hilus to follow the field excitatory postsynaptic potential (fEPSP) and the population (pop) spike that is produced when a population of granule cells fire action potentials together. Synaptic transmission was quantified by measuring the pop spike amplitude (PS, angled arrow inset, Figure 1D) because the fEPSP is often obscured by an increase in the pop spike that occurs after synaptic potentiation induction in awake animals.
Figure 1. Nicotine-induced Synaptic Potentiation in the Dentate Gyrus (DG) of Freely Moving Mice.
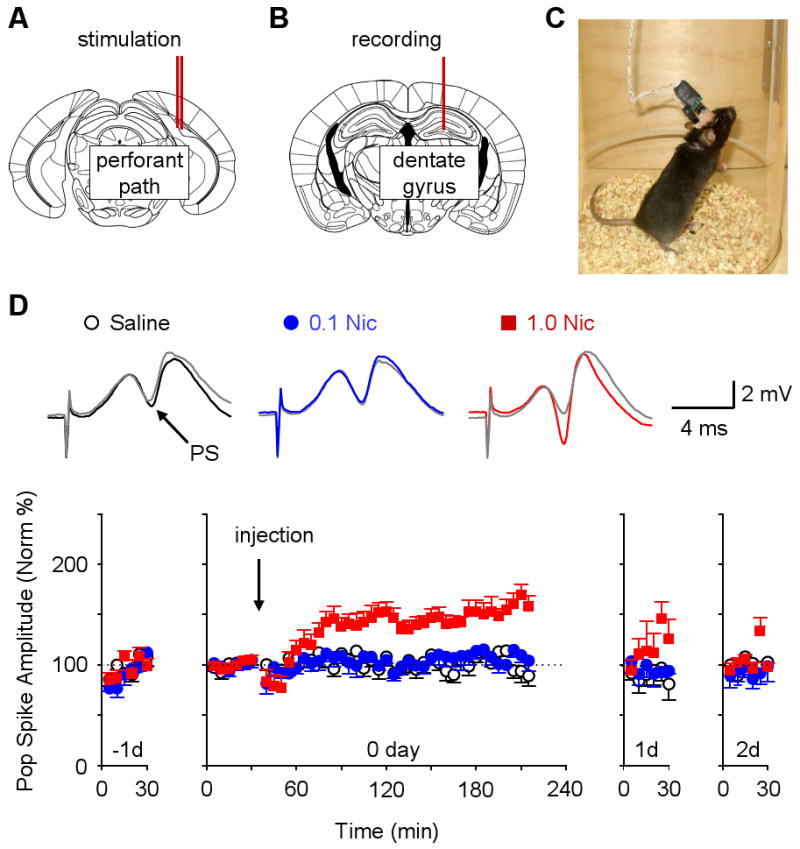
(A) Placement of the concentric bipolar stimulating electrodes (double red lines) into the medial perforant path.
(B) Placement of the recording electrode (red line) into the ipsilateral hilus of the dentate gyrus.
(C) Photograph of a freely moving mouse during a recording session.
(D) Nicotine-induced potentiation of the perforant path recorded in the DG. The time course of each procedure is across four days, from the day before (-1d) until two days after (2d) the nicotine administration. A 30 min baseline was obtained on -1d and confirmed to be stable for 30 min on day 0 (0d). Saline (open circles, n = 8), 0.1 mg/kg nicotine (blue circles, n = 7), or 1.0 mg/kg nicotine (red squares, n = 12) was injected (i.p.) at 30 min on 0d (downward arrow). The population spike (PS) amplitudes are plotted above to indicate potentiation of synaptic transmission compared to the baselines (gray traces). Two-way ANOVA with repeated measures on day 0 showed significant effects of groups (F2,24 = 20.36, P < 0.001) and group × time interactions (F82,984 = 2.96, P < 0.001). The PS amplitude increased significantly over baseline and stayed potentiated for at least 3 hrs after 1.0 mg/kg nicotine on 0d (F41,451 = 7.91, P < 0.001). The return to baseline is shown for 30 min on 1d and 2d.
Two weeks after surgical implantation of the electrodes and habituation to the recording situation, mice were treated with three 4-day counterbalanced sessions of systemic intraperitoneal injection (i.p.) of saline, 0.1, 0.5, or 1.0 mg/kg nicotine, respectively. Neither saline nor 0.1 mg/kg nicotine affected transmission, but 0.5 and 1.0 mg/kg nicotine induced long-term synaptic potentiation (Figure 1D, red data squares for 1.0 mg/kg, Supplemental Figure S1 for 0.5 mg/kg). Systemic administration of nicotine induced synaptic potentiation of the following amplitude measured 3 hours after administration: 124.1 ± 6.4%, n = 3, P < 0.05 for 0.5 mg/kg and 159 ± 10 %, n = 12, P < 0.05 for 1.0 mg/kg, paired t-test. Further tests indicated that the nicotine-induced synaptic enhancement lasted for more than 5 hours: 150.2 ± 9.6 % of baseline for 1.0 mg/kg, n = 3, P < 0.05, paired t-test, data not shown. Since the known ½ life of nicotine in mice is only 5–8 min (Petersen et al., 1984), the synaptic potentiation is not due to the continued presence of nicotine; rather, the nicotine-induced synaptic potentiation outlasts the presence of nicotine.
To test whether the nicotine-induced potentiation resulted from an increase in the number of contributing afferent axons, we measured the presynaptic fiber volley during the nicotine-induced potentiation by moving the recording electrode from the hilus to a position closer to the dendritic synaptic innervation. We found that the incoming afferent excitation (i.e. the presynaptic fiber volley) was the same before and after nicotine-induced potentiation of the pop spike (see Supplemental Figure S2): 102.6 ± 4.1%, n = 5, P > 0.05, paired t-test. Thus, nicotine-induced synaptic potentiation of the perforant–dentate pathway does not result from increased afferent fibers. This result suggests that the potentiation resides beyond the incoming action potential and likely postsynaptically as expected for long-term potentiation (LTP).
Next, we verified that nicotine acted via nicotinic acetylcholine receptors (nAChRs) to induce the synaptic potentiation. The nonselective neuronal nAChR inhibitor, mecamylamine (2.0 mg/kg, i.p.), was used (Gould and Wehner, 1999). We first showed that injections of mecamylamine alone, at the concentration (2.0 mg/kg, i.p.) we used, did not significantly alter baseline synaptic transmission (Figure 2A). Saline injections (i.p.) preceding the nicotine administration still showed synaptic potentiation (Figure 2B, gray data circles), but mecamylamine injection (2.0 mg/kg, i.p.) preceding the nicotine administration completely prevented synaptic potentiation (Figure 2B, black data circles): 95.8 ±11.4 % of baseline, n = 5, P > 0.05, paired t-test.
Figure 2. Nicotinic Receptor Antagonist Mecamylamine inhibits Nicotine-induced Potentiation.

(A) Mecamylamine (2.0 mg/kg, i.p., n = 3) alone did not significantly alter evoked synaptic transmission from the perforant path, as indicated by the stable amplitude of the population spike (F44,88 = 0.61, P > 0.05).
(B) Mecamylamine (2.0 mg/kg, i.p., black circles, n = 5) administered 15 min prior to nicotine inhibited the nicotine-induced synaptic potentiation when compared to saline injected 15 min before nicotine (gray circles, n = 3). ANOVA showed significant effects of groups (F1,6 = 35.28, P < 0.005) and groups × time (F44,264 = 2.76, P < 0.001).
Since the nicotine administrations are systemic, nicotine could be influencing hippocampal plasticity by acting at sites outside of the hippocampus. To determine whether nicotine is acting locally only within the hippocampal dentate gyrus, nicotine was ipsilaterally microinfused into the dentate gyrus 300–500 μm dorsal to the recording site (Figure 3A). Initially we microinfused nicotine (0.008 μg, 0.5 μl/min over 2 min) to achieve a concentration comparable to that attained with systemic i.p. injection of 1 mg/kg (as in Figure 1). When either nicotine (0.008 μg) or saline, as a control, was slowly microinfused, there was no significant change in synaptic transmission (Figure 3B). In a recent report, it was found that a much higher dose of nicotine (0.35 μg) infused into the dorsal hippocampus, altered contextual fear conditioning in mice (Davis et al., 2007). Therefore, we microinfused this higher dose into the dentate, but again there was no significant change in synaptic transmission (Figure 3C). Although these negative results are not conclusive, they suggest that nicotine has actions that extend beyond the hippocampal dentate gyrus during the induction of the in vivo synaptic potentiation.
Figure 3. Local Microinfusion of Nicotine into the Dentate Gyrus does not Induce Synaptic Plasticity of the Perforant Path.
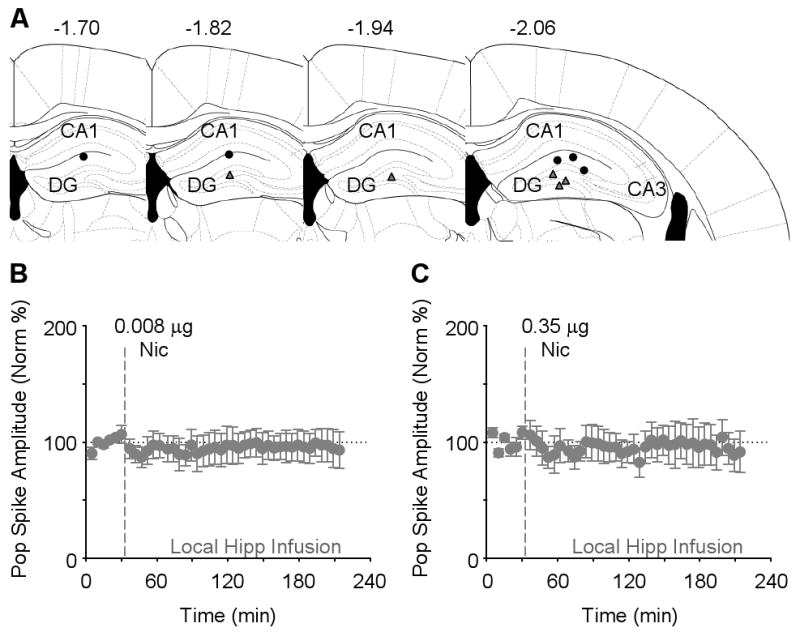
(A) Four different schematic representations of the rostral-caudal planes depict all 5 of the sites where nicotine was microinfused (black circles) and the field potential was recorded (gray triangles). The numbers represent the coordinate posterior from bregma.
(B) Nicotine at the infusion dose (0.008 μg, n = 4) comparable with systemic injection did not significantly alter evoked synaptic transmission from the perforant path, as indicated by the stable amplitude of the population spike (F41,123 = 0.45, P > 0.05).
(C) At an increased nicotine dose (0.35 μg, n = 5), local microinfusion still did not change the synaptic transmission of the perforant path (F41,164 = 0.62, P > 0.05).
Nicotine-induced Synaptic Potentiation Requires a Hippocampal Dopamine Signal Arriving from the Midbrain
An important feature of nicotine's addictive potential arises from nicotine evoked firing of midbrain DA neurons, which produces a potent DA signal (Nestler, 2004; Nisell et al., 1994; Pontieri et al., 1996; Zhang et al., 2009). To determine whether the DA signal induced by systemic injection of nicotine contributed to the nicotine-induced synaptic potentiation, we systemically inhibited D1 and D5 (D1-type) DA receptors using SCH23390 (Ushijima et al., 1995) because D1-type receptors are expressed in the hippocampus (Huang et al., 1992). Injections of SCH23390 (0.2 mg/kg, i.p.) alone did not significantly alter synaptic transmission (Figure 4A). Systemic injection of SCH23390 (0.2 mg/kg, i.p.) preceding the nicotine (1 mg/kg) administration, however, completely prevented nicotine-induced synaptic potentiation (Figure 4B, black data circles): 107.5 ±11.1 % of baseline, n = 7, P > 0.05. In control experiments where saline (i.p.) preceded the nicotine administration, nicotine induced a significant potentiation over the baseline (Figure 4B, gray data circles): 150.0 ± 7.7 % of baseline, n = 8, P < 0.001.
Figure 4. Systemic or Local Hippocampal Microinfusion of Dopamine D1-type Receptor Antagonist SCH23390 inhibits Nicotine-induced Potentiation.
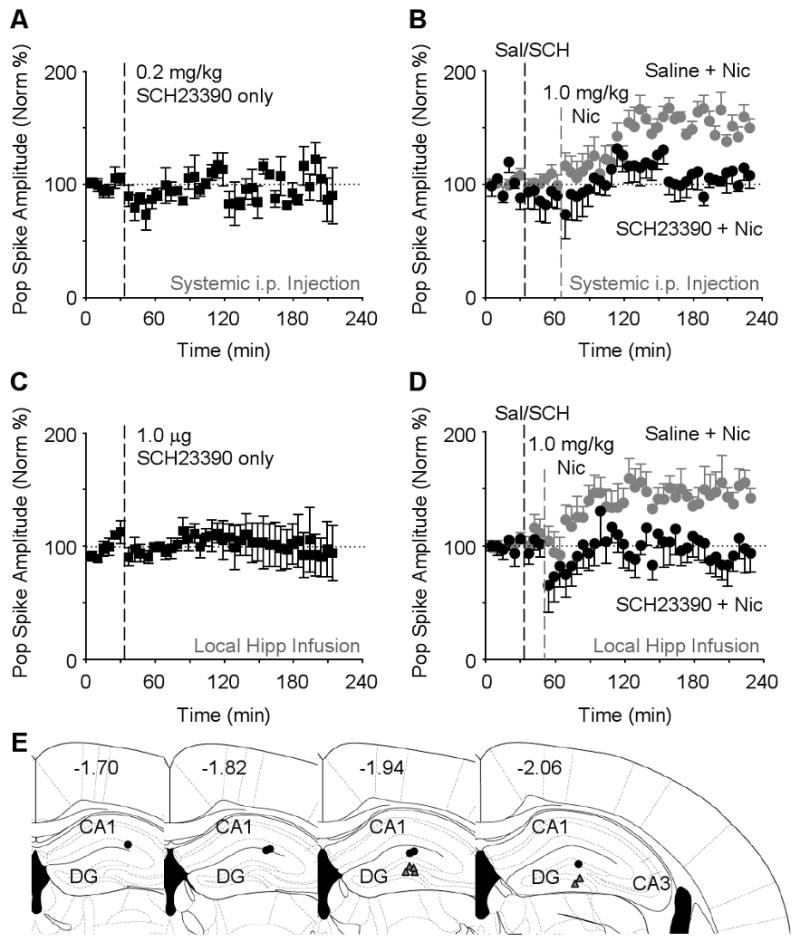
(A) Systemic administration of SCH23390 (0.2 mg/kg, i.p., n = 3) alone did not significantly alter evoked transmission of the perforant path (F41,82 = 1.06, P > 0.05).
(B) SCH23390 (0.2 mg/kg, i.p., black circles, n = 7) administered 30 min prior to nicotine inhibited the synaptic potentiation when compared to saline injected 30 min before nicotine (gray circles, n = 8). ANOVA showed significant effects of groups (F1,13 = 10.09, P < 0.01) and groups × time (F44,572 = 2.19, P < 0.001).
(C) Microinfusion of SCH23390 (1.0 μg, n = 5) directly into the DG had no effect on the basic synaptic transmission in the perforant path (F41,164 = 0.38, P > 0.05).
(D) Microinfusion of SCH23390 (1.0 μg, black circles, n = 6) directly into the DG 15 min before nicotine injection inhibited nicotine-induced potentiation. When saline (1.0 μl) was microinfused before nicotine (gray circles, n = 6), nicotine-induced potentiation was observed. ANOVA showed the significant differences (groups: F1,10 = 6.62, P < 0.05; groups × time: F44,440 = 1.48, P < 0.05).
(E) Four different schematic representations of the rostral-caudal planes depict all 6 of the sites where SCH23390 was microinfused (black circles) and the field potential was recorded (gray triangles). The numbers represent the coordinate posterior from bregma.
To determine whether the D1-type receptor antagonism was acting indirectly via distant targets or was acting locally within the hippocampal dentate gyrus, SCH23390 was ipsilaterally microinfused into the dentate gyrus (Figure 4E, black circles) 300–500 μm dorsal to the recording site (Figure 4E, gray triangles). When either SCH23390 (1.0 μg) or saline, as a control, was slowly microinfused alone into the dentate gyrus using an infusion pump (0.5 μl/min over 2 min), there was no significant change in synaptic transmission (Figure 4C). Microinfusion of SCH23390 (1.0 μg, 0.5 μl/min over 2 min) into the dentate gyrus preceding the nicotine administration, however, completely prevented nicotine-induced synaptic potentiation (Figure 4D, black data circles): 93.2 ± 16.6 % of baseline, n = 6, P > 0.05. In paired control experiments where saline microinfusion preceded the nicotine administration, nicotine induced a significant potentiation over the baseline (Figure 4D, gray data circles): 141.8 ± 8.7 %, n = 6, P < 0.001. The results indicate that D1-type receptors act locally within the hippocampal dentate gyrus to enable nicotine-induced long-term synaptic potentiation.
D2-type receptors regulate the firing of DA neurons by auto-inhibition at their midbrain source (Robinson et al., 2004; Zhang et al., 2009). Therefore, we used a D2-type agonist, quinpirole (0.2 mg/kg, i.p.), to inhibit DA neuron firing and, consequently, to decease the outgoing DA signal (Job et al., 2006; Robinson et al., 2004). Injections of quinpirole (0.2 mg/kg, i.p.) alone did not significantly alter perforant-path synaptic transmission (Figure 5A). Injection of quinpirole (0.2 mg/kg, i.p.) preceding the nicotine administration, however, prevented nicotine-induced synaptic potentiation (Figure 5B, black data circles): 119.9 ± 9.5 % of baseline, n = 5, P > 0.05. In control experiments where saline (i.p.) preceded the nicotine administration, nicotine induced a significant potentiation over the baseline (Figure 5B, gray data circles): 157.7 ± 8.1 %, n = 11, P < 0.001.
Figure 5. Systemic Dopamine D2-type Receptor Agonist Quinpirole inhibits Nicotine-induced Potentiation.

(A) Systemic administration of the D2-type receptor agonist quinpirole (0.2 mg/kg, i.p., n = 4) alone did not significantly alter evoked transmission of the perforant path (F42,126 = 0.93, P > 0.05).
(B) Quinpirole (0.2 mg/kg, i.p., black circles, n = 5) administered 30 min prior to nicotine (1.0 mg/kg, i.p.) inhibited the nicotine-induced potentiation when compared to saline injected 30 min before nicotine (gray circles, n = 11). ANOVA showed the significant differences (groups: F1,14 = 9.14, P < 0.01; groups × time: F43,602 = 2.06, P < 0.001).
To verify the DA signal was required and not just inhibition of D2-type receptors, we ipsilaterally microinfused tetrodotoxin (TTX) into the midbrain DA area to inhibit sodium channels and, consequently, inhibit action potentials from the DA neurons. When either TTX (2.0 μM in 0.5 μl/min over 1 min) or saline, as a control, was slowly microinfused alone into the midbrain DA area (Figure 6A), there was no significant change in hippocampal synaptic transmission (Figure 6B). Microinfusion of TTX (0.5 μl/min over 1 min) (Legault et al., 2000) preceding the nicotine administration, however, completely prevented nicotine-induced synaptic potentiation (Figure 6C, black data circles): 96.8 ± 6.7 % of baseline, n = 5, P > 0.05. In paired control experiments where saline microinfusion preceded the nicotine administration, nicotine induced a significant potentiation over the baseline (Figure 6C, gray data circles): 149.1 ± 12.6 %, n = 5, P < 0.05. The results indicate that a DA signal arising from the midbrain is required to enable nicotine-induced long-term synaptic potentiation within the hippocampal dentate gyrus.
Figure 6. Local Microinfusion of TTX inactivates Midbrain Neurons and abolishes Nicotine-induced Potentiation.
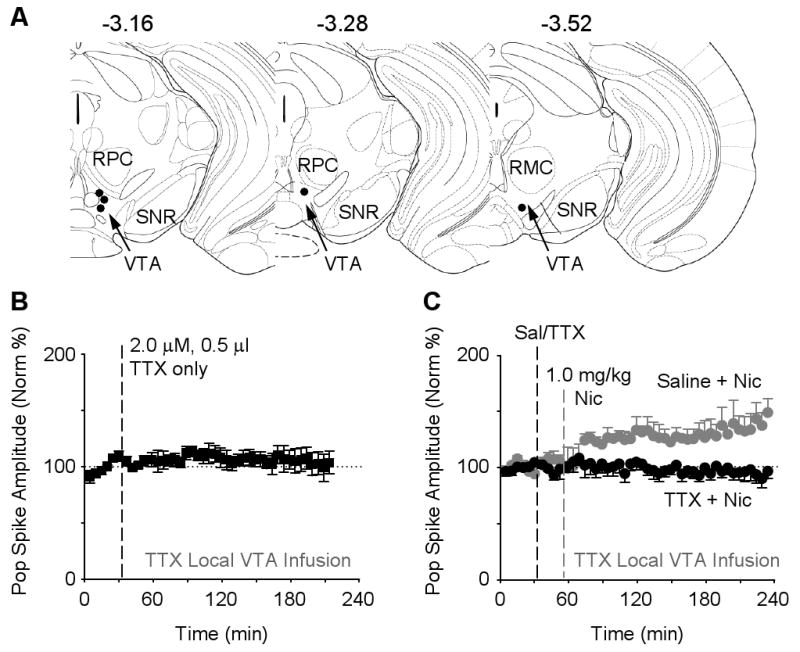
(A) Three different schematic representations of the rostral-caudal planes depict all 5 of the sites where TTX was microinfused (black circles) into the midbrain ventral tegmental area (VTA, arrow). The numbers represent the coordinate posterior from bregma, and the labels are parvicellular red nucleus (RPC), magnocellular red nucleus, (RMC), substantia nigra reticulata (SNR).
(B) Microinfusion of TTX (2.0 μM, 0.5 μl, n = 3) into the VTA had no effect on the basic synaptic transmission of the perforant path (F41,82 = 1.07, P > 0.05).
(C) Microinfusion of TTX (2.0 μM, 0.5 μl, black cycles, n = 5) into the VTA 20 min before nicotine injection inhibited nicotine-induced potentiation. When saline (0.5 μl) was microinfused before nicotine (gray circles, n = 5), nicotine-induced potentiation was observed. ANOVA showed the significant differences (groups: F1,8 = 11.02, P < 0.05; groups × time: F45,360 = 2.50, P < 0.01).
Rather than inhibiting DA signaling, the D2-type receptor antagonist, eticlopride, increases the firing rates of DA neurons at their midbrain source (Brady and O' Donnell, 2004; Robinson et al., 2004; Zhang et al., 2009). Injections of eticlopride (0.5 mg/kg, i.p.) alone did not significantly alter perforant-path synaptic transmission (Figure 7A). Injection of eticlopride (0.5 mg/kg, i.p.) preceding the nicotine administration, however, statistically enhanced nicotine-induced synaptic potentiation (Figure 7B, black data circles): 205.4 ± 17.5 % of baseline, n = 6, P < 0.001. In control experiments where saline (i.p.) preceded the nicotine administration, nicotine induced a significant (but statistically smaller) potentiation over the baseline (Figure 7B, gray data circles): 157.7 ± 8.0 %, n = 11, P < 0.001. The results suggest that the strength of the nicotine-induced synaptic potentiation is influenced by the magnitude of the DA signal.
Figure 7. Systemic Dopamine D2-type Receptor Antagonist Eticlopride increases Nicotine-induced Potentiation.

(A) Systemic administration of the D2-type receptor antagonist eticlopride (0.5 mg/kg, i.p., n = 3) alone did not significantly alter evoked transmission from the perforant path (F47,47 = 0.62, P > 0.05).
(B) Eticlopride (0.5 mg/kg, i.p., black circles, n = 6) administered 30 min prior to nicotine (1.0 mg/kg, i.p.) enhanced nicotine-induced potentiation when compared to saline injected 30 min before nicotine (gray circles, n = 11) (groups × time: F43,645= 1.91, P < 0.001).
Nicotine-induced Conditioned Placed Preference Requires Dopamine D1-type Signaling
The results to this point indicate that nicotine induces synaptic plasticity that requires D1-type receptor activity. To examine whether the electrophysiological results are consistent with memory-based behavioral outcomes, we tested nicotine-induced conditioned place preference (CPP). Others have shown that the hippocampus participates in CPP (Meyers et al., 2003; Meyers et al., 2006). In addition, nicotine-induced CPP requires nAChR activity, and there is an inverted U-shaped dose response with 0.5 mg/kg of nicotine causing the strongest CPP (Walters et al., 2006). We found that concentration of nicotine (0.5 mg/kg) also induces in vivo synaptic plasticity (Supplemental Figure S2).
Using procedures adapted from published methods (Walters et al., 2006), we found significant CPP (Figure 8) in response to nicotine (Nic, 0.5 mg/kg, i.p., P < 0.05, n = 10) compared to saline (Sal, n = 10). Then, we examined whether dopamine D1-type receptor activity, which is required for nicotine-induced synaptic plasticity, is also required for CPP. Although saline injection before conditioning still resulted in nicotine-induced CPP (Figure 8, Sal + Nic, n = 12), when the D1-type inhibitor SCH23390 (0.2 mg/kg, i.p.) was administered 20 min before conditioning, CPP was not induced (Figure 8, SCH + Nic, n = 12, P < 0.05).
Figure 8. Dopamine D1-type Receptor Antagonist SCH23390 inhibits Nicotine-induced Conditioned Place Preference.

Mice treated with 0.5 mg/kg nicotine (Nic, n = 10) exhibited significant CPP compared with saline-treated controls (Sal, n = 10, * P < 0.05). Saline injection before nicotine did not alter the CPP score (Sal + Nic, n = 12). In contrast, SCH23390 (0.2 mg/kg, i.p.) prior to nicotine abolished nicotine-induced CPP (SCH + Nic, n =12, * P < 0.05).
Discussion
Nicotine-induced In Vivo Synaptic Plasticity, Condition Place Preference, and Implications
In this study, in vivo synaptic transmission was directly measured in freely moving mice, and an addictive drug, nicotine, dose-dependently induced synaptic plasticity of the kind that underlies learning and memory (Kauer, 2004; Kelley, 2004; Whitlock et al., 2006; Winder et al., 2002). While saline or low doses of nicotine did not alter synaptic transmission, two higher and behaviorally relevant nicotine doses (Grabus et al., 2006) caused long-lasting synaptic potentiation that was stable for at least 5 hours. Biologically relevant nicotine doses in mice are larger than in rats because nicotine has a very short half-life of 5 to 8 minutes in mice (Petersen et al., 1984). The rapid turnover means that the nicotine concentration in the mice fell to less than 1% in an hour. Therefore, the nicotine-induced synaptic potentiation outlasted the presence of nicotine by hours.
More importantly, pharmacological manipulations indicated that the drug-induced synaptic plasticity required a DA signal broadcast from the midbrain that acted locally within the dentate gyrus of the hippocampus. Systemic or local inhibition of dopamine D1-type receptors within the dentate gyrus prevented the nicotine-induced potentiation. The impact of D2-type receptor manipulations is different. D2-type agonists activate negative feedback via D2 autoreceptors and decrease the DA signal to the hippocampus by suppressing DA neuron firing (Robinson et al., 2004; Zhang et al., 2009). Likewise, active D2 autoreceptors on DA terminals inhibit DA release in the hippocampus, which also decreases the DA signal. Therefore, systemic activation of D2-type receptors, which inhibits DA neuron firing and DA release, prevented nicotine-induced potentiation. This explanation was supported because inhibiting DA neuron action potentials by microinfusion of TTX into the midbrain also prevented nicotine-induced plasticity. On the other hand, systemic inhibition of D2 receptors, which boosts DA neuron firing (Robinson et al., 2004; Zhang et al., 2009), enhanced the amplitude of nicotine-induced potentiation. Overall, the results suggest that DA enables, at least, this form of synaptic plasticity, and the strength of the DA signal modulates the amplitude of the induced synaptic potentiation.
The nicotine-induced conditioned place preference (CPP) results help to link the synaptic measurements to a memory-based behavior. It has previously been shown that nicotine displays an inverted U-shaped dose response (Walters et al., 2006). Consistent with our nicotine-induced synaptic potentiation, nicotine induces CPP at 0.5 mg/kg but not at 0.1 mg/kg, which is likely too low a dose for either effect. Nicotine at 1 mg/kg induces synaptic potential, but it did not induce significant CPP (Walters et al., 2006). That result is not too surprising. At this higher concentration, nicotine is a slight sedative in mice, causing them to become relatively immobile. In fact, this property lowers the mouse core body temperature at 1 mg/kg nicotine (Marks et al., 1986), which likely causes the delayed onset of synaptic potentiation seen at 1 mg/kg nicotine (Figure 1D) but not see at 0.5 mg/kg, which does not cause sedation (compare Supplemental Figure S1). Furthermore, at 1 mg/kg nicotine is approaching doses that cause adverse autonomic effects. Therefore, the synaptic plasticity results lead us to hypothesis that the mice are learning in association with the 1 mg/kg nicotine injection, but they may be learning place avoidance nearly as often as place preference. Coupled with the complication of nicotine sedation, 1 mg/kg is a dose that is beyond the optimum for CPP.
It has been shown that drug-induced CPP requires the dorsal hippocampus, where we make our measurements. Excitotoxic lesion or inactivation of the dorsal hippocampus by muscimol prevented CPP (Meyers et al., 2003; Meyers et al., 2006). In a very recent study, the role of the dorsal hippocampus in CPP also was confirmed in C57 mice that we use (Tropea et al., 2008). Therefore, the dorsal hippocampus is contributor to CPP. Since the DA receptor antagonist was administered systemically in our CPP experiment, we can only correlate the nicotine-induced synaptic potentiation, the DA signaling, and the CPP. A study has shown, however, that dorsal hippocampal DA receptors play a role in morphine reward (Rezayof et al., 2003). Intrahippocampal injection of D1-type receptor agonist SKF38393 significantly potentiated morphine induced CPP, and this effect was reversed by D1-type receptor antagonist SCH23390 (Rezayof et al., 2003). Those results support a mechanistic connection between the hippocampal DA signal and the CPP results that we observed. Overall, our in vivo and behavioral results provide a link between an addictive drug and DA's influence over the memory circuits that create associations to the act of drug use (Dani and Montague, 2007; Kauer, 2004; Kelley, 2004).
The medial perforant path carries contextual and spacial information from the cortex to the hippocampus (Deadwyler et al., 1979; Hjorth-Simonsen and Jeune, 1972; Lavenex and Amaral, 2000). The hippocampus receives cholinergic innervation arising mainly from the medial septum and diagonal band (Woolf, 1991) and densely expresses nicotinic acetylcholine receptors (Dani and Bertrand, 2007). In addition, the hippocampus receives dopaminergic innervation from the ventral tegmental area, and nicotine increases the firing rates of those midbrain DA neurons (Grenhoff et al., 1986; Mameli-Engvall et al., 2006; Zhang et al., 2009). The results from this study indicate that the DA signal acts to enable synaptic potentiation by nicotine in the dentate gyrus. The results suggest that as information continually flows through the perforant path to the hippocampus, the DA signal provides a time stamp that labels particular events for storage as memories (Wise, 2004). In one view, the DA signal arises during unpredicted events in the environment that require learning to update the repertoire of adaptive behaviors (Schultz et al., 1997). We speculate that normally important events in the environment will produce the convergence of perforant path activity and a DA signal to produce synaptic potentiation. In our example, the nicotine administration produced the convergence of perforant path synaptic activity and the DA signal.
Drug Associated Memories Contribute to Continued Drug Use
This study was based on the hypothesis that addictive drugs produce synaptic plasticity that underlies drug-linked associative memory (Kauer, 2004; Kelley, 2004; Winder et al., 2002). This hypothesis is consistent with the emerging view that memories associated with addictive behaviors become internal motivational drives to continue drug use (Bonson et al., 2002; Dani and Montague, 2007; Everitt et al., 2001; Kenny and Markou, 2005; Kilts et al., 2001). That is, environmental cues previously associated with drug use elicit internal sensation-based memories that motivate the desire for the drug, making long-term abstinence difficult.
Normally, neural systems operating beneath our consciousness enable us to associate environmental situations with behavioral sequences that produce success (Everitt et al., 2001; Packard and Knowlton, 2002; Schultz et al., 1997). Eventually, those same environmental situations cue internal states (i.e., feelings or sensations) that motivate the successful behaviors. In that way, we continually learn how to exploit our environments successfully. Dopamine signals may serve as a label during this learning process, indicating when the environment presented unexpected events that needed to be updated in our mnemonic map of environmental saliency (Schultz, 2001; Schultz et al., 1997; Wise, 2004)
Addictive drugs usurp these normally adaptive systems, which include the dopaminergic system originating in the ventral tegmental area of the midbrain. When an addictive drug is used, there are associated behaviors and environmental events obligatorily linked to the usage. Addictive drug use can be considered a learned (conditioned) behavior reinforced by the drug. Those events associated with the drug also are subject to conditioning. A recent fMRI study of addicted smokers showed that cues related to smoking elicit neural activity in regions linked to attention, memory, emotion, and motivation (Smolka et al., 2005). In rats, presentation of nicotine-associated cues reinstated previously extinguished nicotine-seeking behavior, providing evidence that links nicotine-associated cues to relapse (Paterson et al., 2005).
Based on the background of information, the results provide interesting suggestions. As information continuously passes through the hippocampus, most information is not stored but particular salient events are labeled by a phasic DA signal that enables synaptic plasticity and, subsequently, memory. That is, an animal's construct or internal memories of the environment are updated when the DA signal labels the particular event as important, new, and salient. In the case presented in this study, those memories would be linked to the addictive drug. When specific environmental events are present (e.g., the location of drug use), they are capable of cuing drug-associated memories or feelings that motivate continued drug use or relapse (Paterson et al., 2005; Smolka et al., 2005).
Experimental Procedrues
Animal Care and Surgery
Adult male C57BL6/J mice (Jackson lab), 3-4 months, were used in all the experiments. For electrophysiology, they were housed individually with food and water ad libitum in a temperature-controlled room (23 ± 0.5 °C) with a 12-h artificial light-dark cycle (light on at 9 pm). All experiments took place during the dark phase of the cycle. The Institutional Animal Care and Use Committee approved all procedures in accordance with federal guidelines.
On Day 0, mice were anesthetized with sodium pentobarbital (80 mg/kg, i.p., supplemented with 15 mg/kg as necessary) and secured in stereotaxic apparatus. A concentric bipolar stimulating electrode consisting of a tungsten wire placed inside a stainless steel tube was placed at the medial part of the perforant path (0.2 mm posterior and 2.8–3.0 mm lateral of lambda, 1.0–1.3 mm below the dura). The recording electrode (Teflon-coated tungsten wire, bare diameter 50 μm) attached to a stainless steel guide tube (outer diameter 310 μm) was targeted ipsilaterally to the hilus of the dentate gyrus (DG) (1.8–2.0 mm posterior, 1.4–1.6 mm lateral of bregma, 2.2–2.3 mm below the skull) (Franklin and Paxinos, 1997). Final depths of the electrodes were determined by electrophysiological guidance. A cortical silver ball, placed contralaterally, served as a recording reference as well as ground. Surgical screws and dental cement were used to anchor the electrodes, a unity gain preamplifier, and the connecting device for chronic recordings. Mice were given at least two weeks to recover.
Handling, Habituation, Stimulation, and Recording Procedures
From Day 10 for a total of 5 days, each animal received 5-min handling followed by 0.3 ml saline (i.p.) in the home cage both in the morning and afternoon. Then the mice were individually habituated to the recording chamber (Plexiglas cylinder with bedding) and to the recording headstage system. Mice received 1 hour of habituation daily for 4 consecutive days starting from Day 11. On the days of habituation and testing, mice were slightly sedated with isoflurane for 3-5 s to connect the implanted electrode assembly to the recording headstage (Tang et al., 2001). Monophasic square pulses (100 μs) were delivered to the perforant path. Signals were amplified (× 100), filtered (bandpass 0.1 Hz – 5 kHz), digitized at 10 kHz and stored on disk for off-line analysis.
On Day 15, an input-output curve (I/O) was generated. Single test stimuli were then delivered at 30 s intervals at an intensity (35 – 100 μA) that evoked 40% of the maximal amplitude of the population spike. In the typical experiments examining evoked synaptic responses in the perforant path – dentate pathway, there were two to three counterbalanced sessions for every individual mouse: saline, 0.1 mg/kg, and 0.5 mg/kg or 1.0 mg/kg nicotine. Each session went across four days. On day minus one (-1d), the baseline amplitude of the population spike was examined for stability for 30 min. On 0d, after 30 min of baseline recording, saline, 0.1 mg/kg, 0.5 mg/kg, or 1.0 mg/kg nicotine was administered (i.p.) at a volume of 0.1 ml per 10 g body weight. The responses were usually monitored for another 3 hours following the injection, but in some cases the responses were monitored for 5 hours. The posttests were repeated for 30 min on 1d and 2d. I/O curves were recorded systematically between two sessions to verify the stability and the recovery after the treatment of each session.
Throughout the electrophysiological recording, the mouse's behavior was video monitered, and the EEG derived from the dentate gyrus recording also was displayed on an oscilloscope to ensure the absence of electrical after discharges and to ensure the mouse was in a “still-alert” state (Doyere et al., 2003).
Drug Administration and Histology
All the drugs were from Sigma (St. Louis, MO, USA). For intraperitoneal injection, (-)-nicotine hydrogen tartrate salt (calculated as the free base), mecamylamine hydrochloride, R(+)-SCH-23390 hydrochloride, S(−)-eticlopride hydrochloride, and (−)-quinpirole hydrochloride were dissolved in saline and adjusted for 0.1 ml per 10g body weight.
For intracranial microinfusion, solutions were adjusted to pH 7.4 with NaOH and back filled into a 30 gage injector. Nicotine (0.008 or 0.35 μg) or the dopamine D1 receptor antagonist SCH23390 or saline was injected slowly using an infusion pump into the dentate gyrus at 0.5 μl/min over either 1 or 2 min. Pilot tests with lidocaine in anesthetized and freely moving mice verified that this procedure worked well. To avoid the mechanical effect of the cannula insertion on the electrophysiological recording, the injector tip was located 0.3 – 0.5 mm above the recording sites in the DG. For VTA inactivation with TTX, a 24G guide cannula was implanted (3.2 mm posterior and 0.5 mm lateral of bregma, 3.0 mm below the skull). The tip of the injector was 1.2 mm below the guide tip. Saline or TTX (0.5 μl over 1 min) was infused into the VTA 20 min before nicotine administration (i.p.). Following drug infusion, cannulas were left in place for another minute to allow diffusion of the drug away from the cannula tip. At the end of the microinfusion experiments, 4% methylene blue in PBS (0.5 μl) was injected into the infusion site. Mice were later sacrificed with an overdose of isoflurane. An anodal current (30 μA, 10 s) was passed through the tungsten wire for identification of the electrode placements. Frozen 30-μm coronal sections were cut and stained with hematoxylin.
Behavioral Procedures
C57BL6/J mice (3.5-4 months) were used for the experiments of conditioned place preference (CPP). The procedure for handling and habituation of needle injection was the same as that used for the mice in physiological recordings. The procedures and apparatus for the nicotine-induced CPP was adapted from published methods (Walters et al., 2006).
Since most of our mice in the pilot test did not show initial bias to one of two sides of the conditioning apparatus, an unbiased protocol consisting of three phases was used: (1) Preconditioning was conduced on day 1. Each mouse was placed separately into the apparatus for 15 min, with free access to both compartments. The time spent in each compartment was recorded by ANY-maze software (Stoelting, 2008). (2) Conditioning took place on days 2-4. In experiment 1, mice received saline or 0.5 mg/kg nicotine (i.p.) and were immediately confined to the pairing compartment for 30 min in the morning. Five hours later in the afternoon, animals were injected with the alternate nicotine or saline and immediately confined to the opposite compartment for 30 min. Saline control groups received saline on both sides of the apparatus. This procedure was repeated on days 3-4. A counterbalanced design was used between the two compartments, morning and afternoon sessions and from day to day. In experiment 2, the conditioning protocol was exactly the same as Experiment 1 except for the pharmacological treatment. For this experiment, mice first received saline or 0.2 mg/kg SCH23390 (i.p.) and stayed in their home cages. Twenty minutes later all the animals were given 0.5 mg/kg nicotine (i.p.) before being confined to one of the two compartments. (3) Testing was done on day 5. The mice were able to freely explore the full apparatus for 15 min and time spent on each side was recorded. Preference score was expressed in seconds and is calculated by subtracting preconditioning day data from test day data.
Data Analysis
The amplitude of the population spike was measured for each evoked response. The values from every 10 consecutive recordings were averaged and normalized for each animal as a percentage of the mean baseline values obtained during the 30 min immediately before drug treatment. Data were expressed as mean ± SEM and analyzed with two-way repeated measures analysis of variance. Post hoc tests for time × group interactions were first assessed with another repeated measure analysis of variance for the time variable operating only on a single group. Significant time effects were then explored with paired t-tests by comparing the mean of all baseline points with each post-baseline time bin. Group effects in the time × group interaction were assessed with one-way analyses of variance for each time point. For experiments with more than two levels to the group variable (e.g., Figure 1A) a SNK posthoc was used.
Supplementary Material
Acknowledgments
The work was supported by Award Numbers and from the National Institute of Neurological Disorders and Stroke (R01 NS021229) and the National Institute on Drug Abuse (R01 DA009411) respectively. The content is solely the responsibility of the authors and does not necessarily represent the official views of NINDS, NIDA, or the National Institutes of Health.
We gratefully acknowledge the advice of Dr. Mariella De Biasi throughout this study and her student, Ms. Erica Perez, during the CPP experiments.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Biala G, Betancur C, Mansuy IM, Giros B. The reinforcing effects of chronic D-amphetamine and morphine are impaired in a line of memory-deficient mice overexpressing calcineurin. Eur J Neurosci. 2005;21:3089–3096. doi: 10.1111/j.1460-9568.2005.04132.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bonson KR, Grant SJ, Contoreggi CS, Links JM, Metcalfe J, Weyl HL, Kurian V, Ernst M, London ED. Neural systems and cue-induced cocaine craving. Neuropsychopharmacology. 2002;26:376–386. doi: 10.1016/S0893-133X(01)00371-2. [DOI] [PubMed] [Google Scholar]
- Brady AM, O'Donnell P. Dopaminergic modulation of prefrontal cortical input to nucleus accumbens neurons in vivo. J Neurosci. 2004;24:1040–1049. doi: 10.1523/JNEUROSCI.4178-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caine SB, Thomsen M, Gabriel KI, Berkowitz JS, Gold LH, Koob GF, Tonegawa S, Zhang J, Xu M. Lack of self-administration of cocaine in dopamine D1 receptor knock-out mice. J Neurosci. 2007;27:13140–13150. doi: 10.1523/JNEUROSCI.2284-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corrigall WA. Nicotine self-administration in animals as a dependence model. Nicotine Tob Res. 1999;1:11–20. doi: 10.1080/14622299050011121. [DOI] [PubMed] [Google Scholar]
- Dani JA, Bertrand D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharmacol Toxicol. 2007;47:699–729. doi: 10.1146/annurev.pharmtox.47.120505.105214. [DOI] [PubMed] [Google Scholar]
- Dani JA, Harris RA. Nicotine addiction and comorbidity with alcohol abuse and mental illness. Nat Neurosci. 2005;8:1465–1470. doi: 10.1038/nn1580. [DOI] [PubMed] [Google Scholar]
- Dani JA, Montague PR. Disrupting addiction through the loss of drug-associated internal states. Nat Neurosci. 2007;10:403–404. doi: 10.1038/nn0407-403. [DOI] [PubMed] [Google Scholar]
- Davis JA, Kenney JW, Gould TJ. Hippocampal alpha4beta2 nicotinic acetylcholine receptor involvement in the enhancing effect of acute nicotine on contextual fear conditioning. J Neurosci. 2007;27:10870–10877. doi: 10.1523/JNEUROSCI.3242-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis S, Bliss TV, Dutrieux G, Laroche S, Errington ML. Induction and duration of long-term potentiation in the hippocampus of the freely moving mouse. J Neurosci Methods. 1997;75:75–80. doi: 10.1016/s0165-0270(97)00053-8. [DOI] [PubMed] [Google Scholar]
- Deadwyler SA, West M, Lynch G. Activity of dentate granule cells during learning: differentiation of perforant path input. Brain Res. 1979;169:29–43. doi: 10.1016/0006-8993(79)90371-8. [DOI] [PubMed] [Google Scholar]
- Doyere V, Schafe GE, Sigurdsson T, LeDoux JE. Long-term potentiation in freely moving rats reveals asymmetries in thalamic and cortical inputs to the lateral amygdala. Eur J Neurosci. 2003;17:2703–2715. doi: 10.1046/j.1460-9568.2003.02707.x. [DOI] [PubMed] [Google Scholar]
- Everitt BJ, Dickinson A, Robbins TW. The neuropsychological basis of addictive behaviour. Brain Res Brain Res Rev. 2001;36:129–138. doi: 10.1016/s0165-0173(01)00088-1. [DOI] [PubMed] [Google Scholar]
- Franceschini D, Paylor R, Broide R, Salas R, Bassetto L, Gotti C, DeBiasi M. Absence of alpha7-containing neuronal nicotinic acetylcholine receptors does not prevent nicotine-induced seizures. Brain Res Mol Brain Res. 2002;98:29–40. doi: 10.1016/s0169-328x(01)00309-6. [DOI] [PubMed] [Google Scholar]
- Franklin KB, Paxinos G. The Mouse Brain in Stereotaxic Coordinates. London: Academic Press; 1997. [Google Scholar]
- Gould TJ, Wehner JM. Nicotine enhancement of contextual fear conditioning. Behav Brain Res. 1999;102:31–39. doi: 10.1016/s0166-4328(98)00157-0. [DOI] [PubMed] [Google Scholar]
- Grabus SD, Martin BR, Brown SE, Damaj MI. Nicotine place preference in the mouse: influences of prior handling, dose and strain and attenuation by nicotinic receptor antagonists. Psychopharmacology (Berl) 2006;184:456–463. doi: 10.1007/s00213-006-0305-7. [DOI] [PubMed] [Google Scholar]
- Grenhoff J, Aston-Jones G, Svensson TH. Nicotinic effects on the firing pattern of midbrain dopamine neurons. Acta Physiol Scand. 1986;128:351–358. doi: 10.1111/j.1748-1716.1986.tb07988.x. [DOI] [PubMed] [Google Scholar]
- Hara K, Harris RA. The anesthetic mechanism of urethane: the effects on neurotransmitter-gated ion channels. Anesth Analg. 2002;94:313–318. doi: 10.1097/00000539-200202000-00015. table of contents. [DOI] [PubMed] [Google Scholar]
- Hargreaves EL, Rao G, Lee I, Knierim JJ. Major dissociation between medial and lateral entorhinal input to dorsal hippocampus. Science. 2005;308:1792–1794. doi: 10.1126/science.1110449. [DOI] [PubMed] [Google Scholar]
- Hjorth-Simonsen A, Jeune B. Origin and termination of the hippocampal perforant path in the rat studied by silver impregnation. The Journal of comparative neurology. 1972;144:215–232. doi: 10.1002/cne.901440206. [DOI] [PubMed] [Google Scholar]
- Huang Q, Zhou D, Chase K, Gusella JF, Aronin N, DiFiglia M. Immunohistochemical localization of the D1 dopamine receptor in rat brain reveals its axonal transport, pre- and postsynaptic localization, and prevalence in the basal ganglia, limbic system, and thalamic reticular nucleus. Proc Natl Acad Sci U S A. 1992;89:11988–11992. doi: 10.1073/pnas.89.24.11988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyman SE, Malenka RC, Nestler EJ. Neural mechanisms of addiction: the role of reward-related learning and memory. Annu Rev Neurosci. 2006;29:565–598. doi: 10.1146/annurev.neuro.29.051605.113009. [DOI] [PubMed] [Google Scholar]
- Izzo E, Orsini C, Koob GF, Pulvirenti L. A dopamine partial agonist and antagonist block amphetamine self-administration in a progressive ratio schedule. Pharmacol Biochem Behav. 2001;68:701–708. doi: 10.1016/s0091-3057(01)00472-5. [DOI] [PubMed] [Google Scholar]
- Jay TM. Dopamine: a potential substrate for synaptic plasticity and memory mechanisms. Prog Neurobiol. 2003;69:375–390. doi: 10.1016/s0301-0082(03)00085-6. [DOI] [PubMed] [Google Scholar]
- Ji D, Lape R, Dani JA. Timing and location of nicotinic activity enhances or depresses hippocampal synaptic plasticity. Neuron. 2001;31:131–141. doi: 10.1016/s0896-6273(01)00332-4. [DOI] [PubMed] [Google Scholar]
- Job MO, Ramachandra V, Anders S, Low MJ, Gonzales RA. Reduced basal and ethanol stimulation of striatal extracellular dopamine concentrations in dopamine D2 receptor knockout mice. Synapse. 2006;60:158–164. doi: 10.1002/syn.20283. [DOI] [PubMed] [Google Scholar]
- Jones S, Bonci A. Synaptic plasticity and drug addiction. Curr Opin Pharmacol. 2005;5:20–25. doi: 10.1016/j.coph.2004.08.011. [DOI] [PubMed] [Google Scholar]
- Kauer JA. Learning mechanisms in addiction: synaptic plasticity in the ventral tegmental area as a result of exposure to drugs of abuse. Annu Rev Physiol. 2004;66:447–475. doi: 10.1146/annurev.physiol.66.032102.112534. [DOI] [PubMed] [Google Scholar]
- Kelley AE. Memory and addiction: shared neural circuitry and molecular mechanisms. Neuron. 2004;44:161–179. doi: 10.1016/j.neuron.2004.09.016. [DOI] [PubMed] [Google Scholar]
- Kenny PJ, Markou A. Conditioned nicotine withdrawal profoundly decreases the activity of brain reward systems. J Neurosci. 2005;25:6208–6212. doi: 10.1523/JNEUROSCI.4785-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kilts CD, Schweitzer JB, Quinn CK, Gross RE, Faber TL, Muhammad F, Ely TD, Hoffman JM, Drexler KP. Neural activity related to drug craving in cocaine addiction. Archives of general psychiatry. 2001;58:334–341. doi: 10.1001/archpsyc.58.4.334. [DOI] [PubMed] [Google Scholar]
- Lavenex P, Amaral DG. Hippocampal-neocortical interaction: a hierarchy of associativity. Hippocampus. 2000;10:420–430. doi: 10.1002/1098-1063(2000)10:4<420::AID-HIPO8>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- Legault M, Rompre PP, Wise RA. Chemical stimulation of the ventral hippocampus elevates nucleus accumbens dopamine by activating dopaminergic neurons of the ventral tegmental area. J Neurosci. 2000;20:1635–1642. doi: 10.1523/JNEUROSCI.20-04-01635.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisman JE, Grace AA. The hippocampal-VTA loop: controlling the entry of information into long-term memory. Neuron. 2005;46:703–713. doi: 10.1016/j.neuron.2005.05.002. [DOI] [PubMed] [Google Scholar]
- Lynch MA. Long-term potentiation and memory. Physiol Rev. 2004;84:87–136. doi: 10.1152/physrev.00014.2003. [DOI] [PubMed] [Google Scholar]
- Mameli-Engvall M, Evrard A, Pons S, Maskos U, Svensson TH, Changeux JP, Faure P. Hierarchical control of dopamine neuron-firing patterns by nicotinic receptors. Neuron. 2006;50:911–921. doi: 10.1016/j.neuron.2006.05.007. [DOI] [PubMed] [Google Scholar]
- Marks MJ, Romm E, Gaffney DK, Collins AC. Nicotine-induced tolerance and receptor changes in four mouse strains. J Pharmacol Exp Ther. 1986;237:809–819. [PubMed] [Google Scholar]
- Martinez JL, Jr, Derrick BE. Long-term potentiation and learning. Annu Rev Psychol. 1996;47:173–203. doi: 10.1146/annurev.psych.47.1.173. [DOI] [PubMed] [Google Scholar]
- Matsuyama S, Matsumoto A, Enomoto T, Nishizaki T. Activation of nicotinic acetylcholine receptors induces long-term potentiation in vivo in the intact mouse dentate gyrus. Eur J Neurosci. 2000;12:3741–3747. doi: 10.1046/j.1460-9568.2000.00259.x. [DOI] [PubMed] [Google Scholar]
- McHugh TJ, Jones MW, Quinn JJ, Balthasar N, Coppari R, Elmquist JK, Lowell BB, Fanselow MS, Wilson MA, Tonegawa S. Dentate gyrus NMDA receptors mediate rapid pattern separation in the hippocampal network. Science. 2007;317:94–99. doi: 10.1126/science.1140263. [DOI] [PubMed] [Google Scholar]
- Meyers RA, Zavala AR, Neisewander JL. Dorsal, but not ventral, hippocampal lesions disrupt cocaine place conditioning. Neuroreport. 2003;14:2127–2131. doi: 10.1097/00001756-200311140-00023. [DOI] [PubMed] [Google Scholar]
- Meyers RA, Zavala AR, Speer CM, Neisewander JL. Dorsal hippocampus inhibition disrupts acquisition and expression, but not consolidation, of cocaine conditioned place preference. Behav Neurosci. 2006;120:401–412. doi: 10.1037/0735-7044.120.2.401. [DOI] [PubMed] [Google Scholar]
- Miner LL, Collins AC. Strain comparison of nicotine-induced seizure sensitivity and nicotinic receptors. Pharmacol Biochem Behav. 1989;33:469–475. doi: 10.1016/0091-3057(89)90532-7. [DOI] [PubMed] [Google Scholar]
- Nashmi R, Xiao C, Deshpande P, McKinney S, Grady SR, Whiteaker P, Huang Q, McClure-Begley T, Lindstrom JM, Labarca C, et al. Chronic nicotine cell specifically upregulates functional alpha 4* nicotinic receptors: basis for both tolerance in midbrain and enhanced long-term potentiation in perforant path. J Neurosci. 2007;27:8202–8218. doi: 10.1523/JNEUROSCI.2199-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nestler EJ. Molecular mechanisms of drug addiction. Neuropharmacology. 2004;47 1:24–32. doi: 10.1016/j.neuropharm.2004.06.031. [DOI] [PubMed] [Google Scholar]
- Nisell M, Nomikos GG, Svensson TH. Infusion of nicotine in the ventral tegmental area or the nucleus accumbens of the rat differentially affects accumbal dopamine release. Pharmacol Toxicol. 1994;75:348–352. doi: 10.1111/j.1600-0773.1994.tb00373.x. [DOI] [PubMed] [Google Scholar]
- Packard MG, Knowlton BJ. Learning and memory functions of the Basal Ganglia. Annu Rev Neurosci. 2002;25:563–593. doi: 10.1146/annurev.neuro.25.112701.142937. [DOI] [PubMed] [Google Scholar]
- Paterson NE, Froestl W, Markou A. Repeated administration of the GABAB receptor agonist CGP44532 decreased nicotine self-administration, and acute administration decreased cue-induced reinstatement of nicotine-seeking in rats. Neuropsychopharmacology. 2005;30:119–128. doi: 10.1038/sj.npp.1300524. [DOI] [PubMed] [Google Scholar]
- Petersen DR, Norris KJ, Thompson JA. A comparative study of the disposition of nicotine and its metabolites in three inbred strains of mice. Drug Metab Dispos. 1984;12:725–731. [PubMed] [Google Scholar]
- Pontieri FE, Tanda G, Orzi F, DiChiara G. Effects of nicotine on the nucleus accumbens and similarity to those of addictive drugs. Nature. 1996;382:255–257. doi: 10.1038/382255a0. [DOI] [PubMed] [Google Scholar]
- Rezayof A, Zarrindast MR, Sahraei H, Haeri-Rohani A. Involvement of dopamine receptors of the dorsal hippocampus on the acquisition and expression of morphine-induced place preference in rats. J Psychopharmacol. 2003;17:415–423. doi: 10.1177/0269881103174005. [DOI] [PubMed] [Google Scholar]
- Robinson S, Smith DM, Mizumori SJ, Palmiter RD. Firing properties of dopamine neurons in freely moving dopamine-deficient mice: effects of dopamine receptor activation and anesthesia. Proc Natl Acad Sci U S A. 2004;101:13329–13334. doi: 10.1073/pnas.0405084101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rumpel S, LeDoux J, Zador A, Malinow R. Postsynaptic receptor trafficking underlying a form of associative learning. Science. 2005;308:83–88. doi: 10.1126/science.1103944. [DOI] [PubMed] [Google Scholar]
- Schultz W. Reward signaling by dopamine neurons. Neuroscientist. 2001;7:293–302. doi: 10.1177/107385840100700406. [DOI] [PubMed] [Google Scholar]
- Schultz W, Dayan P, Montague PR. A neural substrate of prediction and reward. Science. 1997;275:1593–1599. doi: 10.1126/science.275.5306.1593. [DOI] [PubMed] [Google Scholar]
- Smolka MN, Buhler M, Klein S, Zimmermann U, Mann K, Heinz A, Braus DF. Severity of nicotine dependence modulates cue-induced brain activity in regions involved in motor preparation and imagery. Psychopharmacology (Berl) 2005:1–12. doi: 10.1007/s00213-005-0080-x. [DOI] [PubMed] [Google Scholar]
- Stolerman IP, Shoaib M. The neurobiology of tobacco addiction. Trends Pharmacol Sci. 1991;12:467–473. doi: 10.1016/0165-6147(91)90638-9. [DOI] [PubMed] [Google Scholar]
- Tang J, Wotjak CT, Wagner S, Williams G, Schachner M, Dityatev A. Potentiated amygdaloid auditory-evoked potentials and freezing behavior after fear conditioning in mice. Brain Res. 2001;919:232–241. doi: 10.1016/s0006-8993(01)03020-7. [DOI] [PubMed] [Google Scholar]
- Tropea TF, Kosofsky BE, Rajadhyaksha AM. Enhanced CREB and DARPP-32 phosphorylation in the nucleus accumbens and CREB, ERK, and GluR1 phosphorylation in the dorsal hippocampus is associated with cocaine-conditioned place preference behavior. J Neurochem. 2008;106:1780–1790. doi: 10.1111/j.1471-4159.2008.05518.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ungless MA, Magill PJ, Bolam JP. Uniform inhibition of dopamine neurons in the ventral tegmental area by aversive stimuli. Science. 2004;303:2040–2042. doi: 10.1126/science.1093360. [DOI] [PubMed] [Google Scholar]
- Ushijima I, Mizuki Y, Yamada M. Alteration of cataleptic responses induced by dopamine receptor antagonists after chronic cocaine administration in mice. Eur J Pharmacol. 1995;285:55–59. doi: 10.1016/0014-2999(95)00382-u. [DOI] [PubMed] [Google Scholar]
- Walters CL, Brown S, Changeux JP, Martin B, Damaj MI. The beta2 but not alpha7 subunit of the nicotinic acetylcholine receptor is required for nicotine-conditioned place preference in mice. Psychopharmacology (Berl) 2006;184:339–344. doi: 10.1007/s00213-005-0295-x. [DOI] [PubMed] [Google Scholar]
- Whitlock JR, Heynen AJ, Shuler MG, Bear MF. Learning induces long-term potentiation in the hippocampus. Science. 2006;313:1093–1097. doi: 10.1126/science.1128134. [DOI] [PubMed] [Google Scholar]
- Winder DG, Egli RE, Schramm NL, Matthews RT. Synaptic plasticity in drug reward circuitry. Curr Mol Med. 2002;2:667–676. doi: 10.2174/1566524023361961. [DOI] [PubMed] [Google Scholar]
- Wise RA. Dopamine, learning and motivation. Nature reviews. 2004;5:483–494. doi: 10.1038/nrn1406. [DOI] [PubMed] [Google Scholar]
- Woolf NJ. Cholinergic systems in mammalian brain and spinal cord. Prog Neurobiol. 1991;37:475–524. doi: 10.1016/0301-0082(91)90006-m. [DOI] [PubMed] [Google Scholar]
- Zhang T, Zhang L, Liang Y, Siapas AG, Zhou FM, Dani JA. Dopamine signaling differences in the nucleus accumbens and dorsal striatum exploited by nicotine. J Neurosci. 2009;29:4035–4043. doi: 10.1523/JNEUROSCI.0261-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.