

Abstract
Forest trees exhibit remarkable adaptations to their environments. The genetic basis for phenotypic adaptation to climatic gradients has been established through a long history of common garden, provenance, and genecological studies. The identities of genes underlying these traits, however, have remained elusive and thus so have the patterns of adaptive molecular diversity in forest tree genomes. Here, we report an analysis of diversity and divergence for a set of 121 cold-hardiness candidate genes in coastal Douglas fir (Pseudotsuga menziesii var. menziesii). Application of several different tests for neutrality, including those that incorporated demographic models, revealed signatures of selection consistent with selective sweeps at three to eight loci, depending upon the severity of a bottleneck event and the method used to detect selection. Given the high levels of recombination, these candidate genes are likely to be closely linked to the target of selection if not the genes themselves. Putative homologs in Arabidopsis act primarily to stabilize the plasma membrane and protect against denaturation of proteins at freezing temperatures. These results indicate that surveys of nucleotide diversity and divergence, when framed within the context of further association mapping experiments, will come full circle with respect to their utility in the dissection of complex phenotypic traits into their genetic components.
A long history of common garden experiments has shown that forest trees are adapted phenotypically to local and regional climates (Morgenstern 1996). Latitudinal clines for phenological traits have been documented across a diverse set of temperate forest tree species ranging from Norway spruce [Picea abies (L.) Karst.] to European aspen (Populus tremula L.). The frequent nature of these patterns, as well as the high heritabilities and polygenic nature for many of these traits (Howe et al. 2003), suggest that forest tree genomes should be rife with signatures of positive natural selection.
Early efforts to detect these signatures using molecular markers were largely unsuccessful (McKay and Latta 2002). One of the limiting factors for these efforts was the genomic distribution, availability, and nonfunctional nature of the genetic markers that were utilized (Merkle and Adams 1987). The development of high-throughput allele-specific marker detection for large sets of functional genes has revolutionized the search for adaptive genetic variation within conifers (González-Martínez et al. 2006). Surveys of nucleotide diversity across a diverse set of conifer species have highlighted common genomic attributes, such as the rapid decay of linkage disequilibrium (LD), and have documented the first tangible evidence of natural selection in forest tree genomes (Savolainen and Pyhäjärvi 2007; Eveno et al. 2008; Palmé et al. 2008, 2009).
Here, we take a candidate gene approach to the identification of genes underlying cold-hardiness phenotypes in coastal Douglas fir [Pseudotsuga menziesii (Mirb.) Franco var. menziesii]. A 40-year history of common garden experiments, provenance trials, and genecological studies has documented the genetic basis of growth, germination, and bud phenology traits, as well as the steep clines in genetic variability for those traits along environmental gradients (Campbell and Sorensen 1973; Campbell 1979; Aitken and Adams 1996, 1997; St. Clair et al. 2005; St. Clair 2006). The structure of these clines is consistent with the hypothesis that diversifying selection (McKay and Latta 2002) or local selective sweeps (Slatkin and Wiehe 1998; Santiago and Caballero 2005) should characterize most of the signatures of positive natural selection in the genome of coastal Douglas fir. If this is the case, we expect to observe signatures of elevated levels of intermediate-frequency polymorphisms in the site frequency spectrum for those genes that underlie these clinal traits.
While the phenotypes involved with adaptation to cold temperatures are well known, the identities of the genes underlying these traits have remained elusive. Several quantitative trait loci (QTL) have been identified (Jermstad et al. 2001a,b, 2003), and a handful of candidate genes have been shown to collocate with these QTL (Wheeler et al. 2005). The genes underlying physiological responses to cold, however, are well known in Arabidopsis thaliana L. (Thomashow 1999). Similar types of genes are thought to underlie cold-hardiness related traits in conifers (Yakovlev et al. 2006; Holliday et al. 2008). An initial survey of nucleotide diversity at 18 candidate genes in coastal Douglas fir, however, found only limited departures from neutrality (Krutovsky and Neale 2005). The vast majority of candidate genes, as well as inference of demographic influences upon genomic patterns of nucleotide diversity, thus remain to be investigated.
Mounting evidence indicates that patterns of positive selection are pervasive throughout the genomes of Drosophila (Begun et al. 2007), Arabidopsis (Clark et al. 2007), and humans (Voight et al. 2006). Evidence on a genome scale is only beginning to emerge for conifers (Namroud et al. 2008). We used an expanded set of candidate genes to address three goals: (i) describe patterns of polymorphism and divergence within a set of 121 candidate genes for cold hardiness, (ii) identify those genes with patterns of polymorphism and divergence inconsistent with neutral expectations, and (iii) discuss the types and prevalence of selection across a conifer genome.
MATERIALS AND METHODS
Plant material and DNA isolation:
A sample of 24 unrelated trees obtained from six regions located across Washington and Oregon was assembled as a diversity panel (supporting information, Table S1) for the discovery of single-nucleotide polymorphisms (SNPs). These regions are situated along temperature, precipitation, and genecological gradients identified previously for coastal Douglas fir in Oregon and Washington (St. Clair et al. 2005; St. Clair 2006). A single sample of bigcone Douglas fir [P. macrocarpa (Vasey) Mayr] collected from the Angeles National Forest was used as an outgroup. Total genomic DNA was isolated from haploid megagametophyte tissues dissected out from multiple open-pollinated seeds collected from each tree, using a DNeasy 96 plant kit (QIAGEN, Valencia, CA). All DNA extractions and quantifications were performed at the U.S. Department of Agriculture National Forest Genetics Laboratory (Placerville, CA).
Choice of candidate genes:
Candidate genes with a putative role in conferring tolerance to cold temperatures were selected according to three criteria: (i) genes found to collocate with QTL for cold hardiness in Douglas fir (Wheeler et al. 2005), (ii) genes with physiological roles in cold tolerance response, and (iii) genes showing differential expression in microarray studies in A. thaliana L. (Lee et al. 2005). Standard BLAST tools were used to mine the Douglas fir expressed sequence tags (EST) collections housed at Virginia Polytechnic University (http://staff.vbi.vt.edu/estap) for orthologous sequences to selected candidate genes. Those collections represent four EST libraries, three of which are cold-hardiness libraries derived from 1-year-old seedlings subjected to fall, winter, and spring ambient air conditions in Corvallis, Oregon during 2003. A threshold of 1E-10 was used to define homology during BLAST analyses. In total, 553 putative homologs were selected for primer design, of which 378 were able to have primers designed successfully. A total of 121 of those 378 genes were selected on the basis of diversity of function and sequence-based primer validation from a single Douglas fir sample (Table S2 and Table S3).
PCR, DNA sequencing, and sequence analysis:
Sequence data for each candidate gene were obtained using standard PCR and Sanger sequencing protocols (Table S4). The Pine Alignment and SNP Identification Pipeline (PineSAP) was used to assemble, call bases for, align, and identify SNPs from the 5220 sequence reads (Wegrzyn et al. 2009). The final alignments for each candidate gene were generated using a minimum Phred quality score of 20 for base calls associated with SNPs. All alignments were further trimmed, edited, and validated using CodonCode Aligner v. 2.0.4 (CodonCode, Dedham, MA). Gene annotation, data summaries, and laboratory protocols are given at our website (http://dendrome.ucdavis.edu/dfgp).
SNP genotyping and linkage mapping analyses:
A subset of the 933 discovered SNPs (n = 384) was chosen to validate polymorphism identifications made by PineSAP and to investigate the distribution of those polymorphisms across existing linkage maps of Douglas fir through the genotyping of 192 progeny from a three-generation outbred pedigree (cf. Jermstad et al. 1998) using Illumina's GoldenGate SNP genotyping platform. This high-throughput platform works well for the large and complex genomes of conifers (Pavy et al. 2008). Multiplexed genotyping was conducted at the DNA Technologies Core facility located at the University of California, Davis Genome Center. Signal intensities were quantified and matched to specific alleles, using the BeadStudio v 3.1.14 software (Illumina). A minimum GenCall50 (GC50) score of 0.20 was chosen for inclusion of SNP loci in the final data set and genotypic clusters were edited manually as needed (Eckert et al. 2009a).
The existing linkage maps for coastal Douglas fir are based largely on anonymous RFLP markers. Here, we used 189 fully informative RFLP markers, 3 isozyme markers, and the 77 segregating SNPs to infer a sex-averaged genetic linkage map using JoinMap v. 1.4 (Stam 1993). All SNP loci were assessed for conformity to Mendelian segregation ratios prior to linkage mapping, using goodness-of-fit χ2-tests. We removed SNPs from the same candidate gene that mapped to <0.5 cM from each other prior to the production of the final genetic map. Generation of linkage maps within this pedigree is described in detail elsewhere (Jermstad et al. 1998, 2001a,b, 2003; Krutovsky et al. 2004). In brief, we used a LOD threshold of 4.0 to assign markers to linkage groups followed by a threshold of 0.10 to order markers within those inferred linkage groups. Recombination frequencies were converted to map distances in centimorgans, using the Kosambi mapping function. Linkage map results are deposited and freely available at the forest tree CMAP website (http://dendrome.ucdavis.edu/cmap/; accession no. TG05).
Linkage disequilibrium analyses:
LD between pairs of sites within each candidate gene and between pairs of sites in different candidate genes mapped to the same linkage group were evaluated by using DnaSP v. 4.50.3 (Rozas et al. 2003). We used the squared allelic correlation coefficient (r2) between pairs of parsimony informative sites to estimate LD. We used Fisher's exact tests to calculate P-values under the null hypothesis that LD estimates at least as extreme as those observed could have arisen under linkage equilibrium. The decay of LD with distance in base pairs between sites within the same candidate gene was evaluated with nonlinear regression, using the Gauss–Newton algorithm as implemented in the nls function of R (Hill and Weir 1988; Remington et al. 2001).
Population structure and historical demography:
The extent of structure among populations of coastal Douglas fir observed at chloroplast and nuclear microsatellites, as well as allozymes, is low and consistent with weak effects of isolation by distance (Li and Adams 1989; Viard et al. 2001; Krutovsky et al. 2009). We analyzed patterns of differentiation for all 115 polymorphic candidate genes, using pairwise measures of divergence (Dxy) among the six populations, and related this measure to geographical distance using Mantel tests.
To assess the effects of historical demography, especially given the weak patterns of population structure, on the ability to identify candidate genes under selection, we used a likelihood-free Markov chain Monte Carlo (MCMC) approach to estimate parameters under an instantaneous growth model (IGM) (Marjoram et al. 2003; Marjoram and Tavaré 2006). This model is specified by two parameters, the ratio (f) of the current effective population size (Nec) to the effective population size prior to the growth event (Nep) and the time in units of 4Nec generations at which the growth event began (τ). We assigned uniform priors to both parameters, used the average Tajima's D over the18 loci analyzed by Krutovsky and Neale (2005) as the metric (D = −0.248), and specified a rejection threshold (ɛ) of 0.05.
Parameters were estimated with Markov chains that were run for 1.2 × 106 steps with the first 0.2 × 106 steps being discarded as burn-in and a thinning interval of 100 steps. The resulting 10,000 samples for each parameter were taken as an approximation of their unknown posterior distributions. The joint posterior distribution of f and τ was smoothed, using the kernel smoothing functions available in R. Convergence was assessed through three independent MCMC runs by analyzing similarities between the resulting approximate posterior distributions and using effective sample size (ESS) estimates calculated using the CODA ver. 0.13-2 package in R.
To understand the effects of more complicated demographic scenarios on our abilities to detect loci under selection, we also analyzed two bottleneck models, where we set the time of the bottleneck to occur at 10,000 (BIM_10; τ = 0.001) or 100,000 (BIM_100; τ = 0.015) years ago with bottleneck sizes that were 1% of the current population size. The duration of the bottleneck was set to 0.005 coalescent time units, and the estimate of Θ upon which the simulations were conditioned was adjusted so that the average simulated value of θπ per locus was equal to the observed value for each of the 18 loci used to fit the model. Model fit was assessed by simulating 10,000 sets of 18 loci for which we calculated the average and standard deviation of Tajima's D. We used these simulated sets of data to differentiate among models (instantaneous growth or the two bottleneck models), using rejection sampling (ɛ = 0.05 for both D and the standard deviation of D) and the relationship between acceptance rates and Bayes factors (BF = P(S|BIM_i)/P(S|IGM), where P(S|Model) is given by the acceptance rate and represents the probability of the observed statistics given the model). The observed values of average D and the standard deviation of D were −0.248 and 0.837, respectively (Krutovsky and Neale 2005). Following Jeffreys (1998), we considered values >10−1/2 for the BF as “substantial” evidence for a given model (cf. Ross-Ibarra et al. 2009). This scheme conditions all comparisons on the estimates of τ and f from the instantaneous growth model (i.e., these were not estimated again) given added demographic effects (e.g., bottlenecks).
Nucleotide diversity and neutrality tests:
Several estimators of Θ = 4Neu, where Ne is the effective population size and u is the mutation rate, were used to assess the magnitude of nucleotide diversity across candidate genes. A likelihood-ratio test was performed subsequently to test the null hypothesis that Θ is constant across loci (Hudson 1991). The number of haplotypes (h) and haplotypic diversity (Hd) were calculated for comparison with diversity estimators based on the site frequency spectrum. Pairwise divergence (Dxy) for each candidate gene was calculated by comparing the sequences of coastal Douglas fir to a single sequence from bigcone Douglas-fir. We assumed for all calculations that our data conformed to the infinite-sites model. Sites violating these assumptions, or those with missing data, were ignored.
A suite of summary statistics was used to detect departures of the site frequency spectrum from neutral expectations within each candidate gene. The significance of each statistic was determined under three different null models, using 10,000 coalescent simulations conditional on the observed value of θπ. When simulating data under the demographic models, we adjusted the value of Θ so that the median value of the simulated θπ was equal to the observed data. This was done to account for the fact that θπ is only an unbiased estimator of Θ under genetic drift. The positive false discovery rate (FDR) method was used to correct for multiple testing (Storey 2003). We chose to focus on positive selection, because many of the tests we employed are most powerful in differentiating positive from background selection (Zeng et al. 2006, 2007a,b). We employed the DHEW test to search further for departures from neutrality. This test uses the combination of Tajima's D, Fay and Wu's normalized H, and the Ewens–Watterson test of neutrality to detect positive selection. The rejection region for this test was determined through 50,000 coalescent simulations conditional on the observed value of θπ. We used a nominal threshold of P = 0.0005 in the calculation of this region for each locus to account for the fact that we are performing 115 independent tests.
Finally, we used polymorphism–divergence-based tests to search for candidate genes with reduced levels of polymorphism relative to divergence. A maximum-likelihood implementation of the Hudson–Kreitman–Aguade (HKA) test was carried out with MLHKA ver. 2.0 (Wright and Charlesworth 2004). We implemented a likelihood-ratio test by choosing randomly 20 candidate genes exhibiting neutral site frequency spectra and 3 candidate genes exhibiting reduced levels of polymorphism relative to divergence. These 3 genes were chosen because they had reduced levels of polymorphism relative to divergence (θπ/Dxy < 0.15). The likelihood function was evaluated and parameters were estimated under both models, using MCMC with 1.0 × 108 steps. Statistical significance was determined by comparing twice the difference between the log-likelihood scores to a χ2-distribution with 3 d.f., where in model 1 it was assumed that all 23 loci were neutral and in model 2 it was assumed that the 3 focal genes were nonneutral. Three independent MCMC simulations were used to check for convergence of the likelihood score and parameter estimates.
We also computed the ratio of nonsynonymous (Ka) to synonymous (Ks) divergence for each gene using DnaSP and correlated the Ka/Ks ratio to the polymorphism/divergence ratio using simple linear regression in R. This ratio is the average of each pairwise estimate of Ka/Ks of the coastal Douglas fir samples to the single bigcone Douglas fir. We used bootstrapping (n = 10,000 replicates) across samples to construct 95 and 99% confidence intervals for our measure of Ka/Ks.
RESULTS
Patterns of nucleotide diversity and divergence:
A total of 59,173 bp of aligned sequence data, of which 932 bp had missing, ambiguous, or indel data, were generated through the partial resequencing of 121 candidate genes (GenBank accession nos. EU864545–EU867209). The average length of these gene fragments was 489 bp (± 204 bp). These genes are distributed throughout the genome of coastal Douglas fir (Figure S1), with 64 of the 77 segregating SNPs being placed on the existing linkage map. These 64 SNPs represent 42 candidate genes, 36 from this study and 6 from Krutovsky and Neale (2005). Sequence data were obtained for 21 of the 24 samples per gene on average. Of the 121 candidate genes, 115 were polymorphic, yielding a total of 933 SNPs. The number of polymorphisms varied greatly across genes with an average of 8 SNPs per locus (range: 0–29). The majority of SNPs were silent (n = 732), with 478 of those being located in noncoding regions. The remaining 201 SNPs were nonsynonymous, with an average of 2 nonsynonymous SNPs per gene (range: 0–12). The average SNP density ranged from a maximum of 1 SNP per 43 bp for silent sites to a minimum of 1 SNP per 112 bp for nonsynonymous sites.
Average estimates of nucleotide diversity were on the order of 4.0 × 10−3 per site, with silent sites having the greatest (θW = 0.00777, θπ = 0.00756) and nonsynonymous sites the smallest (θW = 0.00214, θπ = 0.00200) average estimates of diversity (Figure 1, Table 1 and Table S5). Diversity at synonymous (θπs) and silent (θπsil) sites was approximately fourfold greater than at nonsynonymous sites (θπa), with most genes exhibiting a θπa/θπs ratio less than one (n = 77). Six genes, however, had θπa/θπs ratios greater than one, with values ranging from 1.13 to 3.09 (Table S2).
Figure 1.—

The distribution of diversity and divergence across different categories of sites. Values are given on a per site scale. Divergence is measured against bigcone Douglas-fir. Vertical lines extend to 1.5 times the interquartile range. Syn, synonymous sites; Sil, silent sites; NSyn, nonsynonymous sites; Total, all sites.
TABLE 1.
Measures of diversity and divergence across 121 candidate genes putatively affecting cold hardiness in Douglas fir
| S | θW | θπ | Dxy | |
|---|---|---|---|---|
| All | 933 (8 ± 7) | 0.00450 (0.00331) | 0.00435 (0.00358) | 0.00852 (0.00619) |
| Silent | 732 (6 ± 6) | 0.00777 (0.00677) | 0.00756 (0.00765) | 0.01296 (0.01059) |
| Synonymous | 254 (2 ± 2) | 0.00791 (0.00821) | 0.00760 (0.00901) | 0.01413 (0.01498) |
| Nonsynonymous | 201 (2 ± 2) | 0.00214 (0.00281) | 0.00200 (0.00297) | 0.00580 (0.00806) |
Values in boldface type represent totals for the number of segregating sites (S) and averages for measures of diversity (θW, θπ) and divergence (Dxy). Numbers in parentheses are averages ± one standard deviation for S and standard deviations for θW, θπ, and Dxy. Estimates of diversity and divergence are scaled to length of the aligned data, ignoring sites with missing data or insertion–deletion polymorphisms (indels).
Nucleotide diversity was also heterogeneous across candidate genes. A likelihood-ratio test indicated that Θ significantly varied across genes when considering all sites (χ2 = 156.59, d.f. = 114, P = 0.005), with the same result occurring for different categories of sites (silent, χ2 = 189.11, d.f. = 111, P < 0.001; nonsynonymous, χ2 = 95.67, d.f. = 71, P = 0.027). In general, θW was larger than θπ, illustrating an excess of rare SNP alleles relative to expectations under neutrality.
All candidate genes exhibited an excess of divergence relative to diversity, with the average divergence estimate approximately twofold greater than the average diversity estimate (Figure 1, Table 1, and Table S6). The magnitudes of divergence also differed by category of sites. Divergence at synonymous (Ks) and silent (Ksil) sites was greater than at nonsynonymous (Ka) sites, with 76 genes exhibiting a Ka/Ks ratio less than one or zero. However, 18 genes had a Ka/Ks ratio that was greater than one, with values ranging from 1.01 to 14.48. Two of these 18 genes had bootstrap confidence intervals excluding one (Table 1). The remaining 27 genes had undefined values for Ka/Ks.
Patterns of intra- and intergenic linkage disequilibrium:
The number of haplotypes at each polymorphic locus varied from 2 (Hd = 0.087) to 20 (Hd = 0.991), with an average of 5 (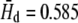 ) per locus. The estimated minimum number of recombination events (RM) was positively correlated with the number of haplotypes and the number of segregating sites (Kendall's τ > 0.409), whereas divergence for all, silent, synonymous, and nonsynonymous sites was uncorrelated with RM (c.f. Hudson and Kaplan 1985). Nucleotide diversity at synonymous and nonsynonymous sites was also uncorrelated with RM.
) per locus. The estimated minimum number of recombination events (RM) was positively correlated with the number of haplotypes and the number of segregating sites (Kendall's τ > 0.409), whereas divergence for all, silent, synonymous, and nonsynonymous sites was uncorrelated with RM (c.f. Hudson and Kaplan 1985). Nucleotide diversity at synonymous and nonsynonymous sites was also uncorrelated with RM.
The relationship of the squared allelic correlation coefficient (r2) with physical distance illustrated that LD decayed ∼50% over 1100 bp, with r2 dropping to <0.25 by 900 bp (Figure 2A). Using Fisher's exact tests, the value of r2 for 866 of the 2837 intragenic site pairs remained significant after a Bonferroni correction. Significant intergenic LD was also apparent for candidate genes that were located proximally on linkage groups. For example, linkage group 17 has two candidate genes whose polymorphisms are almost in complete LD despite being ∼30 cM apart, while genes farther apart show little LD (Figure 2B).
Figure 2.—

Patterns of intra- and interlocus linkage disequilibrium (LD) for coastal Douglas fir. (A) Intragenic patterns of LD using only parsimony informative sites. The fitted lines are the expectation of r2 (plus a 95% confidence interval for C), using the nonlinear least squares estimate of C = 4Ner, where Ne is the effective population size and r is the recombination rate. The thick line is that given for the decay of LD in a previous study of coastal Douglas fir (Krutovsky and Neale 2005). (B) Patterns of intergenic LD along linkage group 17. Solid lines delineate comparisons within and among candidate genes, with letters indicating placement of the candidate genes within the matrix.
Historical demography:
The IGM yielded well-defined approximate posterior probability distributions for the parameters of interest. Replicated runs of the MCMC sampler converged to similar posterior probability distributions, as well as to roughly similar point estimates of the parameters (Figure S2, Table S7). Parameter estimates for this model are consistent with population growth producing a modest increase in effective population size (f = 0.60–0.73), beginning ∼0.039–0.045 coalescent time units ago. These translate into minimum age estimates for the onset of growth of ∼270,000–310,000 years ago if we assume a mutation rate on the order of 1.0 × 10−9 mutations/bp/year (Willyard et al. 2006), a generation time of 50 years, and a genomewide estimate for Θ of 5.92.
We also fit the data to two extreme bottleneck models, where the effective population size during the bottleneck was 5% of the current size (Figure 3). All other parameters were set to the mean values across the MCMC replications (Table S7). When this event was set to occur at 10,000 years ago, the Bayes factor with respect to the instantaneous growth model was 0.15. When the bottleneck, however, was set to be 100,000 years ago, the Bayes factor was 2.5. Following Jeffreys (1998), the IGM fits the data substantially better than the BIM_10 (BF = 6.67, i.e., 1/0.15), but the IGM and BIM_100 models are not differentiated substantially by the data (BF = 2.50). We focus subsequently on the two best models for the purpose of neutrality testing.
Figure 3.—

The distribution of and historical demographic inferences for coastal Douglas fir. (A) The distribution of coastal Douglas fir in Oregon and Washington. Locations for samples are indicated with solid circles and grouped into local populations. (B) Patterns of population structure in coastal Douglas fir. Data and results are from Krutovsky et al. (2009), who used 1283 samples and 31 allozyme and microsatellite loci. Illustrated is the smallest value of K judged as optimal using the ΔK method (Evanno et al. 2005) for categories corresponding to individuals (top) and populations (bottom). (C) Demographic inferences for coastal Douglas fir. The solid line represents the average value of Tajima's D across the 18 loci used to fit the model (cf. Krutovsky and Neale 2005), whereas the dashed line denotes the mean of the simulated distribution under each model (IGM, instantaneous growth model; BIM_10, bottleneck 10,000 years ago; BIM_100, bottleneck model 100,000 years ago). Listed for the BIM models are Bayes factors relative to the IGM.
Neutrality tests:
Six candidate genes exhibited clear patterns of polymorphism and divergence consistent with natural selection. The results from univariate tests are given in Table S8 and Table S9, as well as P-values under null models corresponding to Wright–Fisher mating, the instantaneous growth model, and the extreme bottleneck model set to occur at 100,000 years ago. On average, the summary statistics reflected the excess of low-frequency SNP alleles (D = −0.142) and the slight excess of high-frequency (Fay and Wu's H = −0.106) derived SNP alleles.
A compound test based on D, H, and the Ewens–Watterson haplotype test (DHEW) identified four genes as having patterns of diversity consistent with selective sweeps (Table 2, Figure S3). Products from all four genes function as structural components of the plasma membrane or provide protection against temperature-dependent denaturation of proteins. Inclusion of demographic models (IGM and BIM_100) into the simulations caused one of these loci (CN634517.1) to become nonsignificant.
TABLE 2.
A list of candidate genes putatively affected by positive natural selection
| Locus | Gene product | Resulta |
|---|---|---|
| Compound DHEW test | ||
| Pm_CL908Contig1 | GRAM-containing/ABA-responsive protein | PD = 0.001, PH < 0.001, PEW = 0.080 |
| ES420171.1 | Cold-regulated plasma membrane protein | PD = 0.009, PH = 0.050, PEW = 0.035 |
| ES420250.1 | Dehydrin-like protein | PD = 0.072, PH = 0.083, PEW = 0.042 |
| CN634517.1 | Lumenal binding protein | PD = 0.034, PH = 0.148, PEW = 0.076 |
| Polymorphism-to-divergence | ||
| Pm_CL61Contig1 | Cyclosporin A-binding protein | k = 0.32 |
| Pm_CL908Contig1 | GRAM-containing/ABA-responsive protein | k = 0.58 |
| CN638556.1 | Transcription regulation protein | k = 0.41 |
| Synonymous-to-nonsynonymous divergence | ||
| Pm_CL922Contig1 | Thaumatin-like protein | Ka/Ks = 14.48**, θπ/Dxy = 0.087 |
| CN634677.1 | LRR receptor-like protein kinase | Ka/Ks = 10.78*, θπ/Dxy = 0.066 |
Results for the DHEW test are given as P-values (P) for each of the component tests (D, Tajima's D; H, Fay and Wu's H; EW, Ewens–Watterson test) comprising the joint test. Values for the EW test are one minus the left tailed probabilities (cf. Zeng et al. 2007b). Listed are P-values for drift within a constant size population. Loci significant when demographic models are in included in the simulations are in boldface type. For polymorphism-to-divergence tests, parameter estimates for a maximum-likelihood implementation of the HKA test are listed. Estimates of k are from a nested model where all three putative targets of selection are allowed to have free parameters. The parameter k specifies the level of elevation (k > 1) or reduction (k < 1) in diversity relative to divergence. Asterisks denote significance differences from 1.0 for the Ka/Ks ratio using bootstrapping. **P < 0.01, *P < 0.05 (cf. materials and methods).
Three genes exhibited a significant excess of divergence relative to diversity, using a maximum-likelihood implementation of the HKA test (χ2 = 12.60, d.f. = 3, P = 0.006). These excesses ranged from a two- to threefold reduction in diversity relative to divergence (Table S10, Figure S3), which was also apparent across different categories of sites (Figure S4). All three loci function in generalized stress responses at the levels of protein folding and transcriptional regulation, with locus Pm_CL908Contig1 also being significant for the DHEW test. A weak, yet statistically significant, relationship also existed between Ka/Ks and θπ/Dxy (F1,91 = 4.79, P = 0.031, r2 = 0.05). The driver behind this result was three loci with extreme values of Ka/Ks (i.e., >5, cf. Table 1), all of which had θπ/Dxy < 0.15 (Table 2). Removal of these three loci changed the relationship to be nonsignificant.
Several additional genes exhibited patterns of polymorphism consistent with balancing or diversifying selection, depending upon the null model and summary statistic that was used (Table S8 and Table S9). None of these were significant using the DHEW test. For three of these five genes, a significant and positive relationship existed between geographical distance and pairwise Dxy among populations (Figure S5). This pattern is suggestive of diversifying selection along environmental gradients, especially given that only five candidate genes had a significant value of Mantel's r and three of those five had positive values of D that were nominally significant. Two of these genes remained significant at the FDR threshold of Q = 0.10 for Tajima's test when an extreme bottleneck was included as part of the null model. These loci, CN636901.1 and Pm_CL1400Contig1, encode proteins that are involved with abiotic stress response and cell wall architecture, respectively.
DISCUSSION
Nucleotide diversity and linkage disequilibrium:
Nucleotide diversity in coastal Douglas fir is similar to, but slightly higher than, that observed in other conifer species (Savolainen and Pyhäjärvi 2007). The average nucleotide diversities for all, silent, and nonsynonymous sites were similar to those reported previously (Krutovsky and Neale 2005). Magnitudes of intragenic LD were moderate as compared to those reported for conifers. Our estimate of recombination (ρ/Θ = 0.86) was similar to that reported in loblolly pine (ρ/Θ = 0.29; Brown et al. 2004), but almost an order of magnitude less than that reported in Norway spruce (ρ/Θ = 1.23–4.05; Heuertz et al. 2006) and the central populations of Scots pine (ρ/Θ = 3.80–5.80; Pyhäjärvi et al. 2007).
Interlocus LD was higher than expected on the basis of results from loblolly pine and the previous study in coastal Douglas fir (Brown et al. 2004; Krutovsky and Neale 2005). While genomic variation in recombination rates has been documented across plants (Gaut et al. 2007), which could reflect repetitive regions of the genome where recombination is suppressed, several additional processes such as natural selection, mutation rate variation, population structure, and extreme bottlenecks can also account for these patterns. This is important given that interlocus LD has dramatic effects on the amount and pattern of genetic diversity and population differentiation, as well as on the response to and efficacy of natural selection, across the genome of an organism (Le Corre and Kremer 2003).
Patterns of selection:
Most genes exhibited an excess of nucleotide diversity at synonymous relative to nonsynonymous sites, suggesting the widespread occurrence of purifying selection in coastal Douglas fir (cf. Palmé et al. 2009). Values of Tajima's D across different categories of sites were consistent with this pattern as well (Figure S6). These patterns have been noted extensively in forest trees and have driven the hypothesis that detectable patterns of positive selection in conifers are rare. This hypothesis has been confirmed in a recent examination of genomewide patterns of population differentiation in white spruce (Namroud et al. 2008; but see Eveno et al. 2008). Further examination using phylogenetic approaches, however, has detected patterns consistent with directional selection (Palmé et al. 2008, 2009), suggesting that consideration of relevant timescales is important to the search for selection.
We concur with this hypothesis showing that ∼5% of the genes exhibited patterns consistent with positive selection. Our estimate is likely to be a lower bound due to low statistical power and the conservative assumptions that we made, as well as to the fact that causal variants may exhibit patterns of polymorphism that are indistinguishable from neutral variants due strictly to the quantitative genetic mechanisms underlying the trait (Le Corre and Kremer 2003). Also of consideration are other models of selective sweeps incorporating selection on a standing crop of neutral variants (cf. Innan and Kim 2004). In those models, linked neutral variants are less likely to show the traditional signs of selective sweeps. Contrary to expectations based on patterns of low levels of intragenic LD, the strongest signals came from those genes where patterns were consistent with selective sweeps. The vast majority of candidate genes showed little evidence of elevated levels of intermediate-frequency SNP alleles as expected under diversifying selection and recurrent selective sweep models. Under the bottleneck scenarios, however, two genes had significantly positive values of Tajima's D.
It is likely that these patterns represent relatively recent and single, not recurrent, selective sweep events. A genomewide phenomenon of recurrent selective sweeps would produce average values of D and H that were negative and positive, respectively (Przeworski 2002). Our genes, however, are not random genomic samples, and 42 of the 121 candidate genes exhibit this predicted pattern, producing average values of D = −0.83 and H = 0.55. A conclusion to rule out all occurrences of recurrent sweep models (Gillespie 2001) within conifer genomes, therefore, is premature, especially given our ignorance about large-scale genomic patterns of LD, the spatial distribution of genes across chromosomes, and the historical demography of these lineages.
The simplicity of our demographic models may not account for all of the confounding effects of demography on tests for selection. It also is possible that we have missed evidence supportive of other demographic models due to our limited sampling with respect to the range of coastal Douglas fir. Expansion from bottlenecks could produce patterns consistent with signatures of single and recurrent selective sweeps, depending on the timing and severity of the bottleneck. This is the most common form of demographic model fitted to European forest trees (Heuertz et al. 2006; Pyhäjärvi et al. 2007). The severity of the bottleneck would have to be extreme to produce genomewide patterns mirroring single selective sweeps. Incorporation of an extreme bottleneck (99% reduction of Nec) occurring 100,000 years ago into the null model caused one of the four genes significant previously in the DHEW test to become nonsignificant. It remains significant, however, at a nominal threshold of P = 0.001.
Paleobotanical and geological evidence indicate that Pleistocene glaciation in North America differed dramatically from that in Europe, making an extreme bottleneck model unlikely for coastal Douglas fir. The spatial extent of continental glaciers was smaller and vegetation was not compressed to the same degree, especially on the west coast (Graham 1999). The most parsimonious demographic model for coastal Douglas fir is one of population expansion. Inclusion of a bottleneck following the instantaneous expansion, however, did provide a marginally better fit to the data (Figure 3). The data used to test the demographic models did not have outgroup information, and further work, therefore, should examine rigorously a broad range of demographic models (cf. Ingvarsson 2008).
Putative targets of selection:
The most significant departures from neutrality came from genes that encoded proteins that are structural components of or associated with cellular membranes or those that function as or regulate chaperones. Locus Pm_CL908Contig1 contains a GRAM domain, which characterizes membrane-associated proteins such as glucosyltransferases (Doerks et al. 2000; Caro et al. 2007). The Arabidopsis homolog of locus ES420171.1 is a multispanning G-protein coupled receptor that stabilizes the plasma membrane in response to freezing temperatures (Breton et al. 2003). Both CN634517.1 and Pm_CL61Contig1 have homologs whose primary functions are to act as or in the regulation of chaperones (Romano et al. 2004). Locus Pm_CL922Contig1 encodes a thaumatin-like protein, whose homolog in Arabidopsis has gene expression induced by fungal infection (Hu and Reddy 1997). While the link between cold hardiness and disease resistance is speculative, it is tempting to correlate overall health with the ability to grow and tolerate cold efficiently. Several of these genes are also responsive to abscisic acid (ABA). Controversy has surrounded the role of ABA during cold acclimation in Arabidopsis, however, due to its general effect on plant growth in response to stress (Gilmour and Thomashow 1991).
Limitations and implications:
There are several limitations to our analyses. We used a single sequence from a recently diverged outgroup so that misspecification of ancestral states is possible. This would reduce greatly the power of the HKA test employed here and significantly affect estimation of H (cf. Baudry and Depaulis 2003). This also justifies our avoidance of McDonald–Kreitman-type analyses. Background selection could have produced signatures of selective sweeps, but not with respect to H (Hudson and Kaplan 1995). Finally, we did not incorporate effects of population structure into our hypothesis testing. The effects of undetected structure on inference of selection from summary statistics are tied closely to sampling design and the strength of substructure (Przeworski 2002; Städler et al. 2009), and thus conclusions regarding the conservative or liberal nature of our estimates are premature.
Selective sweeps, while not likely the dominant type of selection across the genome of coastal Douglas fir, are nevertheless important. Here, we have identified several genes whose patterns of diversity and divergence are inconsistent with the standard neutral model, as well as simple demographic scenarios involving instantaneous growth and bottlenecks. Depending upon demographic assumptions, we have identified three to eight such loci. These genes are prime targets for further association genetic (cf. Eckert et al. 2009b) and functional studies, thus beginning to bring molecular population and quantitative genetic approaches full circle with respect to the dissection of complex traits.
Acknowledgments
The authors thank F. Thomas Ledig for contributing bigcone Douglas fir seeds, Valerie Hipkins and her staff at National Forest Genetics Laboratory for performing the DNA extractions, Katie Tsang and Jacqueline Silva for helping to obtain sequence data, and Jeff Ross-Ibarra for helpful discussion about demographic inference. The manuscript was much improved by comments from two anonymous reviewers. Funding for this project was made available through a U.S. Department of Agriculture National Research Initiative Plant Genome grant (04-712-0084).
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.109.103895/DC1.
References
- Aitken, S. N., and W. T. Adams, 1996. Genetics of fall and winter cold hardiness of coastal Douglas-fir in Oregon. Can. J. For. Res. 26: 1828–1837. [Google Scholar]
- Aitken, S. N., and W. T. Adams, 1997. Cold hardiness under strong genetic control in Oregon populations of Pseudotsuga menziesii var. menziesii. Can. J. For. Res. 27: 1773–1780. [Google Scholar]
- Baudry, E., and F. Depaulis, 2003. Effect of misoriented sites on neutrality tests with outgroup. Genetics 165: 1619–1622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Begun, D. J., A. K. Holloway, K. Stevens, L. W. Hillier, Y.-P. Poh et al., 2007. Population genomics: whole-genome analysis of polymorphism and divergence in Drosophila simulans. PLoS Biol. 5: e310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breton, G., J. Danyluk, J. B. F. Charron and F. Sarhan, 2003. Expression profiling and bioinformatic analyses of a novel stress-regulated multispanning transmembrane protein family from cereals and Arabidopsis. Plant Physiol. 132: 64–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, G. R., G. P. Gill, R. J. Kuntz, C. H. Langley and D. B. Neale, 2004. Nucleotide diversity and linkage disequilibrium in loblolly pine. Proc. Natl. Acad. Sci. USA 101: 15255–15260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell, R. K., 1979. Genecology of Douglas-fir in a watershed in the Oregon Cascades. Ecology 60: 1036–1050. [Google Scholar]
- Campbell, R. K., and F. C. Sorensen, 1973. Cold acclimation in seedling Douglas-fir related to phenology and provenance. Ecology 54: 1148–1151. [Google Scholar]
- Caro, E., M. M. Castellano and C. Gutierrez, 2007. A chromatin link that couples cell division to root epidermis patterning in Arabidopsis. Nature 447: 213–217. [DOI] [PubMed] [Google Scholar]
- Clark, R. M., G. Schweikert, C. Toomajian, S. Ossowski, G. Zeller et al., 2007. Common sequence polymorphisms shaping genetic diversity in Arabidopsis thaliana. Science 317: 338–342. [DOI] [PubMed] [Google Scholar]
- Doerks, T., M. Strauss, M. Brendel and P. Bork, 2000. GRAM, a novel domain in glucosyltransferases, myotubularins and other putative membrane-associated proteins. Trends Biochem. Sci. 25: 483–485. [DOI] [PubMed] [Google Scholar]
- Eckert, A. J., B. Pande, E. S. Ersoz, M. H. Wright, V. K. Rashbrook et al., 2009. a High-throughput genotyping and mapping of single nucleotide polymorphisms in loblolly pine (Pinus taeda L.). Tree Genet. Genomes 5: 225–234. [Google Scholar]
- Eckert, A. J., A. D. Bower, J. L. Wegrzyn, B. Pande, K. D. Jermstad et al., 2009. b Association genetics of coastal Douglas-fir (Pseudotsuga menziesii var. menziesii, Pinaceae) I. Cold-hardiness related traits. Genetics 182: 1289–1302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evanno, G., S. Regnaut and J. Goudet, 2005. Detecting the number of clusters of individuals using the software STRUCTURE: a simulation study. Mol. Ecol. 14: 2611–2620. [DOI] [PubMed] [Google Scholar]
- Eveno, E., C. Collada, M. A. Guevara, V. Léger, A. Soto et al., 2008. Contrasting patterns of selection at Pinus pinaster Ait. drought stress candidate genes as revealed by genetic differentiation analyses. Mol. Biol. Evol. 25: 417–437. [DOI] [PubMed] [Google Scholar]
- Gaut, B. S., S. I. Wright, C. Rizzon, J. Dvorak and L. K. Anderson, 2007. Recombination: an underappreciated factor in the evolution of plant genomes. Nat. Rev. Genet. 8: 77–84. [DOI] [PubMed] [Google Scholar]
- Gillespie, J. H., 2001. Is the population size of a species relevant to its evolution? Evolution 55: 2161–2169. [DOI] [PubMed] [Google Scholar]
- Gilmour, S. J., and M. F. Thomashow, 1991. Cold acclimation and cold-regulated gene expression in ABA mutants of Arabidopsis thaliana. Plant Mol. Biol. 17: 1233–1240. [DOI] [PubMed] [Google Scholar]
- González-Martínez, S. C., K. V. Krutovsky and D. B. Neale, 2006. Forest tree population genomics and adaptive evolution. New Phytol. 170: 227–238. [DOI] [PubMed] [Google Scholar]
- Graham, A., 1999. Late Cretaceous and Cenozoic History of North American Vegetation. Oxford University Press, New York.
- Heuertz, M., E. De Paoli, T. Källman, H. Larsson, I. Jurman et al., 2006. Multilocus patterns of nucleotide diversity, linkage disequilibrium and demographic history of Norway spruce [Picea abies (L.) Karst]. Genetics 174: 2095–2105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill, W. G., and B. S. Weir, 1988. Variances and covariances of squared linkage disequilibria in finite populations. Theor. Popul. Biol. 33: 54–78. [DOI] [PubMed] [Google Scholar]
- Holliday, J. A., S. Ralph, R. White, J. Bohlmann and S. N. Aitken, 2008. Global monitoring of gene expression during autumn cold acclimation among rangewide populations of Sitka spruce [Picea sitchensis (Bong.) Carr.]. New Phytol. 178: 103–122. [DOI] [PubMed] [Google Scholar]
- Howe, G. T., S. N. Aitken, D. B. Neale, K. D. Jermstad, N. C. Wheeler et al., 2003. From genotype to phenotype: unraveling the complexities of cold adaptation in forest trees. Can. J. Bot. 81: 1247–1266. [Google Scholar]
- Hu, X., and A. S. Reddy, 1997. Cloning and expression of a PR5-like protein from Arabidopsis: inhibition of fungal growth by bacterially expressed protein. Plant Mol. Biol. 34: 949–959. [DOI] [PubMed] [Google Scholar]
- Hudson, R. R., 1991. Gene genealogies and the coalescent process. Oxf. Surv. Evol. Biol. 7: 1–44. [Google Scholar]
- Hudson, R. R., and N. L. Kaplan, 1985. Statistical properties of the number of recombination events in the history of a sample of DNA sequences. Genetics 111: 147–164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson, R. R., and N. L. Kaplan, 1995. The coalescent process and background selection. Philos. Trans. R. Soc. Lond. B Biol. Sci. 349: 19–23. [DOI] [PubMed] [Google Scholar]
- Ingvarsson, P., 2008. Multilocus patterns of nucleotide polymorphism and the demographic history of Populus tremula. Genetics 180: 329–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Innan, H., and Y. Kim, 2004. Pattern of polymorphism after strong artificial selection in a domestication event. Proc. Natl. Acad. Sci. USA 101: 10667–10672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeffreys, H., 1998. Theory of Probability. Oxford University Press, Oxford.
- Jermstad, K. D., D. L. Bassoni, N. C. Wheeler and D. B. Neale, 1998. A sex-averaged linkage map in coastal Douglas-fir (Pseudotsuga menziesii [Mirb.] Franco) based on RFLP and RAPD markers. Theor. Appl. Genet. 97: 762–770. [Google Scholar]
- Jermstad, K. D., D. L. Bassoni, K. S. Jech, N. C. Wheeler and D. B. Neale, 2001. a Mapping of quantitative trait loci controlling adaptive traits in coastal Douglas-fir. I. Timing of vegetative bud flush. Theor. Appl. Genet. 102: 1142–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jermstad, K. D., D. L. Bassoni, N. C. Wheeler, T. S. Anekonda, S. N. Aitken et al., 2001. b Mapping of quantitative trait loci controlling adaptive traits in coastal Douglas-fir. II. Spring and fall cold-hardiness. Theor. Appl. Genet. 102: 1152–1158. [Google Scholar]
- Jermstad, K. D., D. L. Bassoni, K. S. Jech, G. A. Ritchie, N. C. Wheeler et al., 2003. Mapping of quantitative trait loci controlling adaptive traits in coastal Douglas-fir. III. Quantitative trait loci-by-environment interactions. Genetics 165: 1489–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutovsky, K.V., and D. B. Neale, 2005. Nucleotide diversity and linkage disequilibrium in cold–hardiness and wood-quality related candidate genes in Douglas-fir. Genetics 171: 2029–2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutovsky, K. V., M. Troggio, G. R. Brown, K. D. Jermstad and D. B. Neale, 2004. Comparative mapping in the Pinaceae. Genetics 168: 447–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krutovsky, K. V., J. B. St. Clair, R. Saich, V. D. Hipkins and D. B. Neale, 2009. Estimation of population structure in coastal Douglas-fir [Pseudotsuga menziesii (Mirb.) Franco var. menziesii] using allozyme and microsatellite markers. Tree Genet. Genomes (in press).
- Le Corre, V, and A. Kremer, 2003. Genetic variability at neutral markers, quantitative trait loci and trait in a subdivided population under selection. Genetics 164: 1205–1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee, B.-H., D. A. Henderson and J.-K. Zhu, 2005. The Arabidopsis cold-responsive transcriptome and its regulation by ICE1. Plant Cell 17: 3155–3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, P., and W. T. Adams, 1989. Range-wide patterns of allozyme variation in Douglas-fir (Pseudotsuga menziesii). Can. J. For. Res. 19: 149–161. [Google Scholar]
- Marjoram, P., and S. Tavare, 2006. Modern computational approaches for analyzing molecular genetic variation data. Nat. Rev. Genet. 7: 759–770. [DOI] [PubMed] [Google Scholar]
- Marjoram, P., J. Molitor, V. Plagnol and S. Tavaré, 2003. Markov chain Monte Carlo without likelihoods. Proc. Natl. Acad. Sci. USA 100: 15324–15328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McKay, J. K., and R. G. Latta, 2002. Adaptive population divergence: markers, QTL and traits. Trends Ecol. Evol. 17: 285–291. [Google Scholar]
- Merkle, S. A., and W. T. Adams, 1987. Patterns of allozyme variation within and among Douglas-fir breeding zones in southwest Oregon. Can. J. For. Res. 17: 402–407. [Google Scholar]
- Morgenstern, E. K., 1996. Geographic Variation in Forest Trees. UBC Press, Vancouver, British Columbia, Canada.
- Namroud, M.-C., J. Beaulieu, N. Juge, J. Laroche and J. Bousquet, 2008. Scanning the genome for gene single nucleotide polymorphisms involved in adaptive population differentiation in white spruce. Mol. Ecol. 17: 3599–3613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmé, A. E., M. Wright and O. Savolainen, 2008. Patterns of divergence among conifer ESTs and polymorphism in Pinus sylvestris identify putative selective sweeps. Mol. Biol. Evol. 25: 2567–2577. [DOI] [PubMed] [Google Scholar]
- Palmé, A. E., T. Pyhäjärvi, W. Wachowiak and O. Savolainen, 2009. Selection on nuclear genes in a Pinus phylogeny. Mol. Biol. Evol. 26: 893–905. [DOI] [PubMed] [Google Scholar]
- Pavy, N., B. Pelgas, S. Beauseigle, S. Blais, F. Gagnon et al., 2008. Enhancing genetic mapping of complex genomes through the design of highly-multiplexed SNP arrays: application to the large and unsequenced genomes of white and black spruce. BMC Genomics 9: 21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Przeworski, M., 2002. The signature of positive selection at randomly chosen loci. Genetics 160: 1179–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pyhäjärvi, T., M. R. García-Gil, T. Knürr, M. Mikkonen, W. Wachowiak et al., 2007. Demographic history has influenced nucleotide diversity in European Pinus sylvestris populations. Genetics 177: 1713–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Remington, D. L., J. M. Thornsberry, Y. Matsuoka, L. M. Wilson, S. R. Whitt et al., 2001. Structure of linkage disequilibrium and phenotypic associations in the maize genome. Proc. Natl. Acad. Sci. USA 98: 11479–11484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano, P. G. N., P. Horton and J. E. Gray, 2004. The Arabidopsis cyclophilin gene family. Plant Physiol. 134: 1268–1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross-Ibarra, J., M. Tenaillon and B. S. Gaut, 2009. Historical divergence and gene flow in the genus Zea. Genetics 181: 1399–1413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas, J., J. C. Sánchez-DelBarrio, X. Messegyer and R. Rozas, 2003. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19: 2496–2497. [DOI] [PubMed] [Google Scholar]
- Santiago, E., and A. Caballero, 2005. Variation after a selective sweep in a subdivided population. Genetics 169: 485–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Savolainen, O., and T. Pyhäjärvi, 2007. Genomic diversity in forest trees. Curr. Opin. Plant Biol. 10: 162–167. [DOI] [PubMed] [Google Scholar]
- Slatkin, M., and T. Wiehe, 1998. Genetic hitch-hiking in a subdivided population. Genet. Res. 71: 155–160. [DOI] [PubMed] [Google Scholar]
- St. Clair, J. B., 2006. Genetic variation in fall cold hardiness in coastal Douglas-fir in western Oregon and Washington. Can. J. Bot. 84: 1110–1181. [Google Scholar]
- St. Clair, J. B., N. L. Mandel and K. W. Vance-Borland, 2005. Genecology of Douglas-fir in western Oregon and Washington. Ann. Bot. 96: 1199–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Städler, T., B. Haubold, C. Merino, W. Stephan and P. Pfaffelhuber, 2009. The impact of sampling schemes on the site frequency spectrum in nonequilibrium subdivided populations. Genetics 182: 205–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stam, P., 1993. Construction of integrated genetic linkage maps by means of a new computer package: JoinMap. Plant J. 3: 739–744. [Google Scholar]
- Storey, J. D., 2003. The positive false discovery rate: a Bayesian interpretation and the q-value. Ann. Stat. 31: 2013–2035. [Google Scholar]
- Thomashow, M. F., 1999. Plant cold acclimation: freezing tolerance genes and regulatory mechanisms. Annu. Rev. Plant Physiol. Plant Mol. Biol. 50: 571–599. [DOI] [PubMed] [Google Scholar]
- Viard, F., Y. A. El-Kassaby and K. Ritland, 2001. Diversity and genetic structure in populations of Pseudotsuga menziesii (Pinaceae) at chloroplast microsatellite loci. Genome 44: 336–344. [DOI] [PubMed] [Google Scholar]
- Voight, B. F., S. Kudaravalli, X. Wen and J. K. Pritchard, 2006. A map of recent positive selection in the human genome. PLoS Biol. 4: e72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wegrzyn, J. L., J. M. Lee, J. D. Liechty and D. B. Neale, 2009. PineSAP–Pine alignment and SNP Identification Pipeline. Bioinformatics (in press). [DOI] [PMC free article] [PubMed]
- Wheeler, N. C., K. D. Jermstad, K. V. Krutovsky, S. N. Aitken, G. T. Howe et al., 2005. Mapping of quantitative trait loci controlling adaptive traits in coastal Douglas-fir. IV. Cold-hardiness QTL verification and candidate gene mapping. Mol. Breed. 15: 145–156. [Google Scholar]
- Willyard, A., J. Syring, D. S. Gernandt, A. Liston and R. Cronn, 2006. Fossil calibration of molecular divergence infers a moderate mutation rate and recent radiations for Pinus. Mol. Biol. Evol. 24: 90–101. [DOI] [PubMed] [Google Scholar]
- Wright, S. I., and B. Charlesworth, 2004. The HKA test revisited: a maximum likelihood ratio test of the standard neutral model. Genetics 168: 1071–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yakovlev, I. A., C.-G. Fossdal, Ø. Johnson, O. Junttlia and T. Skrøppa, 2006. Analysis of gene expression during bud burst initiation in Norway spruce via ESTs from subtracted cDNA libraries. Tree Genet. Genomes 2: 39–52. [Google Scholar]
- Zeng, K., Y. X. Fu, S. Shi and C.-I. Wu, 2006. Statistical tests for detecting positive selection by utilizing high-frequency variants. Genetics 174: 1431–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, K., S. Mano, S. Shi and C.-I. Wu, 2007. a Comparisons of site- and haplotype-frequency methods for detecting positive selection. Mol. Biol. Evol. 24: 1562–1574. [DOI] [PubMed] [Google Scholar]
- Zeng, K., S. Suhua and C.-I. Wu, 2007. b Compound tests for the detection of hitchhiking under positive selection. Mol. Biol. Evol. 24: 1898–1908. [DOI] [PubMed] [Google Scholar]