

Abstract
The long-term fates of duplicate genes are well studied both empirically and theoretically, but how the short-term evolution of duplicate genes contributes to phenotypic variation is less well known. Here, we have studied the genetic basis of flowering time variation in the disomic tetraploid Capsella bursa-pastoris. We sequenced four duplicate candidate genes for flowering time and 10 background loci in samples from western Eurasia and China. Using a mixed-model approach that accounts for population structure, we found that polymorphisms at one homeolog of two candidate genes, FLOWERING LOCUS C (FLC) and CRYPTOCHROME1 (CRY1), were associated with natural flowering time variation. No potentially causative polymorphisms were found in the coding region of CRY1; however, at FLC two splice site polymorphisms were associated with early flowering. Accessions harboring nonconsensus splice sites expressed an alternatively spliced transcript or did not express this FLC homeolog. Our results are consistent with the function of FLC as a major repressor of flowering in Arabidopsis thaliana and imply that nonfunctionalization of duplicate genes could provide an important source of phenotypic variation.
GENE duplication has had a major impact on most eukaryote genomes, many of which contain duplicate gene pairs that have been retained for long evolutionary periods of time (Nadeau and Sankoff 1997; Lynch and Conery 2000; Wendel 2000). In the long term, retention of duplicate genes is expected if (1) there is selection for maintenance of dosage balance (Papp et al. 2003) or for increased gene dosage (Conant and Wolfe 2008), (2) each copy accumulates mutations that result in the degeneration of independent subfunctions (subfunctionalization) (Force et al. 1999), or (3) one copy acquires mutations resulting in a new function and the other copy remains under purifying selection (neofunctionalization) (Ohno 1970). The likelihood of each of these outcomes depends on the effective population size, and in most eukaryotes the vast majority of duplicate genes are expected to acquire loss-of-function mutations within a short period of time after they arise (Lynch and Conery 2000). When loss-of-function mutations have been fixed in the population, leaving only one functional copy, the process of nonfunctionalization is complete.
Before fixation occurs, segregating mutations leading to partial or complete loss of function of a duplicate gene could constitute an important source of phenotypic variation. This would be expected especially for genes whose effect on the phenotype is dependent on gene dosage (Osborn et al. 2003). Consistent with this hypothesis, null mutations at duplicated genes have been shown to contribute to variation in photoperiod sensitivity, vernalization requirement, and timing of seed maturity in polyploid wheat (Dubcovsky and Dvorak 2007). Genomic data suggest that newly arisen gene duplicates do indeed undergo a brief period of partially relaxed selection (Lynch and Conery 2000) and a large-scale assessment of intraspecific sequence diversity in Arabidopsis thaliana found that recent duplicates exhibited an excess of changes with a major effect on gene integrity (Clark et al. 2007). Gene duplication has also been shown to alter patterns of gene expression (e.g., Liu and Adams 2007; Ha et al. 2009). For example, interspecific comparison between A. thaliana and its close relative A. arenosa and examination of gene expression patterns in resynthesized and natural allotetraploids of these two species indicate that duplicate genes generally have higher levels of expression divergence than single-copy genes and thereby could have contributed to hybrid vigor (Ha et al. 2009). However, data connecting the early stages of duplicate gene evolution to phenotypic variation are still scarce. Here we have conducted a candidate gene association study of flowering time in the tetraploid Capsella bursa-pastoris with the aim to assess the role of polymorphisms at duplicate genes for phenotypic variation in a wild tetraploid species.
C. bursa-pastoris is a predominantly selfing, disomic tetraploid with a nearly worldwide distribution. Although it has low levels of variation at cpDNA and nuclear genes (Ceplitis et al. 2005; Slotte et al. 2008), it is very variable for many morphological and life-history traits, including flowering time (Hurka and Neuffer 1997). Flowering is induced by long days, and natural accessions differ in their requirement of vernalization (a prolonged cold treatment) for flowering (Slotte et al. 2007). The genetic basis of flowering time variation in C. bursa-pastoris has begun to be elucidated through quantitative trait locus (QTL) mapping and expression studies (Linde et al. 2001; Slotte et al. 2007).
In this study, we chose to focus on four candidate genes in A. thaliana. In C. bursa-pastoris, these genes are duplicated as a result of polyploidy. For two of the candidate genes, CRYPTOCHROME 1 (CRY1) and LUMINIDEPENDENS (LD), one homeolog in C. bursa-pastoris is situated in a region encompassed by a major flowering time QTL (A. Ceplitis, T. Slotte, B. Neuffer, M. Linde, T. Kraft and M. Lascoux, unpublished data). CRY1 encodes a blue-light photoreceptor involved in the photoperiodic flowering time pathway, whereas LD is part of the autonomous flowering time pathway (Lee et al. 1994). We also included homologs of two major determinants of flowering time variation in the model species A. thaliana, FRIGIDA (FRI), and FLOWERING LOCUS C (FLC) (Michaels et al. 2003). In A. thaliana, natural variation at these two genes confers variation in vernalization requirement (Johanson et al. 2000; Lempe et al. 2005; Werner et al. 2005; Shindo et al. 2006). FLC encodes a major repressor of the transition to flowering, which acts in a dosage-dependent manner and is downregulated by vernalization (Michaels and Amasino 1999; Sheldon et al. 1999). FRI in turn acts to upregulate FLC and therefore confers a vernalization requirement for flowering.
We tested for an association between flowering time and sequence polymorphism at these four pairs of duplicate genes in C. bursa-pastoris. Because population structure can lead to spurious associations, we also sequenced 10 background loci to assess population stratification and conducted association analyses in a mixed-model setting, a method that successfully reduced the rate of false positives in A. thaliana (Zhao et al. 2007). Detailed characterization of significantly associated candidate loci was carried out to assess whether associations were likely due to polymorphisms at the candidate loci. Our results suggest that ongoing nonfunctionalization of duplicate genes contributes to potentially adaptive flowering time variation in C. bursa-pastoris.
MATERIALS AND METHODS
Association populations:
Because samples from different parts of the species range differ with regard to the history of introgression from C. rubella (Slotte et al. 2008), we searched for associations in two separate association populations. The association populations consisted of 60 C. bursa-pastoris accessions from Europe, the Middle East, and North Africa (here called western Eurasia for brevity) and 42 C. bursa-pastoris accessions from China (Figure 1). The ploidy level of all accessions was checked by cell flow cytometry as in Slotte et al. (2006). Genomic DNA was extracted from fresh or frozen leaf material, using the QIAGEN (Hilden, Germany) Dneasy Plant Kit.
Figure 1.—

Sampling locations of C. bursa-pastoris accessions from western Eurasia (A) and China (B). An accession from Addis Ababa, Ethiopia, is not shown. (A) The two clusters inferred by STRUCTURE as indicated by open circles (northern group, Q1) and solid circles (southern group, Q2). The number of accessions sampled in the same geographical area, although not from the same populations, is indicated next to circles where it differs from one. (B) In China, two accessions were sampled in each population.
Flowering time:
The flowering time of all accessions from western Eurasia was determined previously (Ceplitis et al. 2005). Briefly, seeds were germinated in petri dishes and seedlings were transferred to individual pots and grown in the greenhouse in fully randomized blocks with a 16-hr photoperiod. Seeds from the Chinese accessions were stratified for 4 days at 4° on moist filter paper and subsequently transferred to soil in pots, which were placed in randomized positions in a growth chamber (16 hr cool white light/8 hr dark, 23°). Flowering time was measured as the number of days from germination to the opening of the first flower.
PCR and sequencing:
To provide information on population structure in C. bursa-pastoris, we sequenced parts of five background genes. We chose background genes that are involved in different physiological processes and unlinked, i.e., situated on different ancestral chromosomes (AK) (Schranz et al. 2006) in C. rubella (Table 1). For each gene, both homeologs, i.e., the two loci duplicated by polyploidy, were sequenced. As candidate loci for flowering time, we obtained partial sequences of both homeologs of CRY1, LD, FRI, and FLC. Sequences for both homeologs of Alcohol dehydrogenase (Adh), CRY1, FLC, FRI, LD, and PISTILLATA (PI) for 50 of the western Eurasian accessions and all Chinese accessions were obtained in a previous study (Slotte et al. 2008). Here, we sequenced these loci in 10 additional western Eurasian accessions and both homeologs of three additional genes (AT1G03560, AT2G20470, and AT2G36530; LOS2; Table 1) in all 60 western Eurasian accessions and all 42 Chinese accessions. For each gene, 0.3–0.9 kb of each homeolog was sequenced, resulting in 18 loci (10 background loci and 8 candidate loci) and a total of ∼9.3 kb per accession.
TABLE 1.
A. thaliana locus tags, location in the ancestral karyotype, gene products, and gene names of the background loci used in this study
| Locus tag | Location (AK) | Gene product | Gene name |
|---|---|---|---|
| AT1G03560 | AK1 | Pentatricopeptide-containing protein | |
| ADH1 | AK2 | Alcohol dehydrogenase | Adh |
| AT2G20470 | AK3 | Protein kinase | |
| AT2G36530 | AK4 | Enolase | LOS2 |
| AT5G20240 | AK6 | Floral homeotic protein PISTILLATA | PISTILLATA (PI) |
AK, ancestral chromosome (Schranz et al. 2006)
All sequences analyzed in this study were amplified using homeolog-specific primers, to avoid PCR-mediated sequence artifacts (Cronn et al. 2002). For PISTILLATA (PI), Alcohol dehydrogenase (Adh), and LD, we used homeolog-specific primers and PCR protocols developed in Slotte et al. (2006) and for CRY1, FLC, and FRI, we used primers and protocols from Slotte et al. (2008). For AT1G03560, AT2G20470, and LOS2, new homeolog-specific primer pairs were developed according to the same strategy. A detailed outline of amplification protocols, primers, and sequencing details is given in supporting information, File S1. Briefly, we initially used primers located in conserved regions (Table S1) to amplify both homeologs of each gene in four accessions of C. bursa-pastoris. The resulting heterogeneous PCR products were cloned. On the basis of clone sequences, homeolog-specific primers (Table S2) were then designed so that the 3′ end of one primer in each primer pair was located in a region where the two homeologs were divergent. Homeologs were designated as A or B on the basis of sequence similarity to C. rubella as in Slotte et al. (2006) or were arbitrarily designated L1 and L2 if no C. rubella sequences were available. Sequences have been deposited in GenBank under accession nos. GQ251527–GQ252249.
Population genetic analyses:
We estimated the population mutation parameter per nucleotide site 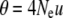 (with Ne the effective population size and u the mutation rate), using Tajima's estimator π (Tajima 1989) and Watterson's estimator θw (Watterson 1975). In addition we obtained an estimate of haplotype diversity (Hd). All the above estimates were obtained in DnaSP V. 4.10.9 (Rozas et al. 2003). Sites containing indels were excluded from all diversity analyses.
(with Ne the effective population size and u the mutation rate), using Tajima's estimator π (Tajima 1989) and Watterson's estimator θw (Watterson 1975). In addition we obtained an estimate of haplotype diversity (Hd). All the above estimates were obtained in DnaSP V. 4.10.9 (Rozas et al. 2003). Sites containing indels were excluded from all diversity analyses.
To assess population stratification, we used the admixture model implemented in STRUCTURE V. 2.2 (Pritchard et al. 2000).This model attempts to infer the proportion of ancestry (q) of each individual in the sample in a number (K) of ancestral populations where unlinked markers are at Hardy–Weinberg and linkage equilibrium. For these analyses, sequences were recoded as haplotypes on the basis of all polymorphic sites, including indels. Only background loci were included, and because C. bursa-pastoris is a selfing species, the Hardy–Weinberg equilibrium assumption was relaxed by treating the data as haploid (see, e.g., Falush et al. 2003). Homeologs were assumed to be independently inherited, as expected in a tetraploid with disomic inheritance. The number of clusters with the highest posterior probability was selected for further analyses. Each run had 1,000,000 iterations, had a burn-in of 200,000, and was repeated three times for K 1–10. Allele frequencies were assumed to be correlated (Falush et al. 2003). For the optimal K, we extracted the Q matrix, i.e., the q values for each individual in each of the inferred clusters, and used it to account for population structure in the mixed-model association analysis (see below).
Mixed-model analysis:
For association analysis, we employed a mixed-model approach (Yu et al. 2006; Zhao et al. 2007). As in Zhao et al. (2007), we modeled the vector of phenotypes, y, as
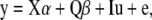 |
where X contains candidate locus genotype information, α is a vector of allelic effects that we wish to estimate, Q contains the proportion of ancestry in each of the K ancestral populations inferred by STRUCTURE, β is a vector of subpopulation effects, I is an identity matrix, u are random deviates due to overall genomewide relatedness, and e is residual error. Flowering time data were analyzed as the number of days from germination to flowering, transformed by the natural logarithm. The proportion of shared haplotypes between each pair of individuals, K*, calculated for all variable background loci, was used to account for kinship in mixed-model analyses. Mixed-model analysis was done in SAS 9.1 (SAS Institute, Cary, NC) using PROC MIXED, and significance of fixed effects was determined by type 3 F-tests with Satterthwaite denominator degrees of freedom.
FLC splicing and expression assays:
The impact of a FLC A polymorphism on splicing variation was assayed in four Chinese accessions harboring a nonconsensus splice donor site 3′ of FLC A exon 5 (FLC A SNP495), four western Eurasian accessions with a nonconsensus splice acceptor site 5′ of FLC A exon 5 (FLC A SNP452), and eight accessions with consensus splice sites 5′ and 3′ of FLC A exon 5. For each accession, total RNA was extracted from a single 2-week-old nonvernalized seedling grown under the same long-day conditions described in the Flowering time section above, using the RNeasy Plant Mini Kit (QIAGEN), with DNaseI digestion. First-strand cDNA was synthesized from 0.5 μg total RNA, with SuperScript III reverse transcriptase (Invitrogen, Carlsbad, CA) and an oligo(dT)20 primer, according to the enzyme manufacturer's instructions. The cDNA was diluted 1:100, and a region of FLC ranging from exon 1 to 7 was amplified using primers FLC3_1F (CTGTTCTCTGTGACGCATCC) and FLC3_7R (GAGTCACCGGAAGATTGTCG), and the product was purified and directly sequenced. We compared transcribed SNPs in cDNA sequences to genomic sequences to determine which homeolog was expressed. A shorter region of FLC ranging from exon 4 to 7 was amplified using primers FLC2_4F (GAGGAACACCTTGAGACTGC) and FLC2_7R (TTTGTCCAGCAGGTGACATC) and size scored on a 1.5% agarose gel. The results were compared to GENSCAN (Burge and Karlin 1997) coding sequence (cds) predictions based on genomic DNA sequences.
We quantified the total level of FLC expression by qRT–PCR, as in Slotte et al. (2007). Primers FLC_5 (CATGAGCTACTTGAACTTGTGGAAA) and FLC_6 (TTCGGCACTCACATTATTGACAT) were used to amplify both homeologs of FLC and primers TUB_1 (ACCACTCCTAGCTTTGGTGATCTG) and TUB_2 (AGGTTCACTGCGAGCTTCCTCA) were used to amplify the reference gene TUB.
RESULTS
Diversity estimates:
Mean diversity levels were higher in western Eurasia than in China (Table 2; Table S3). Nucleotide diversity estimates were somewhat higher for background loci than for candidate loci in both regions, but haplotype diversity estimates were similar (Table 2). The number of haplotypes at each locus ranged from one to four in both regions. In China, one background locus, the B homeolog of AT1G03560, and both homeologs of the candidate locus CRY1 were monomorphic. In western Eurasia, three background loci, Adh B and the L1 homeologs of AT2G20470 and LOS2 were monomorphic, as was FLC B. At each candidate and background gene, we amplified and sequenced two distinct homeologs in each individual, and there was no haplotype sharing among homeologs. Thus, in agreement with mapping data that show disomic inheritance of duplicate genes in C. bursa-pastoris (A. Ceplitis, T. Slotte, B. Neuffer, M. Linde, T. Kraft and M. Lascoux, unpublished data), our data exhibit a pattern of “fixed heterozygosity” as expected in a selfing tetraploid with disomic inheritance, justifying the treatment of homeologs as independently inherited.
TABLE 2.
Mean diversity estimates and haplotype diversity
| Region | Locus type | θw | π | Hd |
|---|---|---|---|---|
| Western Eurasia | Background | 0.0017 | 0.0022 | 0.27 |
| Candidate | 0.00079 | 0.00096 | 0.26 | |
| All | 0.0013 | 0.0017 | 0.27 | |
| China | Background | 0.00092 | 0.00039 | 0.16 |
| Candidate | 0.00054 | 0.00029 | 0.17 | |
| All | 0.00075 | 0.00034 | 0.16 |
θw and π are Watterson's and Tajima's estimators of the population mutation rate and Hd is haplotype diversity.
Population stratification:
We ran STRUCTURE on all variable background loci, i.e., on seven and nine background loci for the western Eurasian and Chinese accessions, respectively. In western Eurasia, the posterior probability of K was highest for two clusters (Figure 2). These clusters consisted of a northern group and a southern group, and each accession was assigned to either cluster with high probability (q > 0.9 for one of the two clusters, Figure S1). There was no detectable population stratification among the Chinese accessions (Figure 2).
Figure 2.—

Log posterior probability of 1–10 clusters, K, in the western Eurasian (left) and Chinese (right) association populations.
Association analysis:
In western Eurasia, there were a total of 20 nonsingleton single-nucleotide polymorphisms (SNPs) in the seven polymorphic candidate loci. We tested for an association between every candidate gene SNP and flowering time, using a mixed model that takes both population structure (Q) and kinship (K*) into account. There was a significant association between 4 SNPs and flowering time, at a 5% nominal significance level. One significant candidate gene SNP was located at position 452 in FLC A and corresponded to a polymorphism resulting in a nonconsensus splice acceptor site (AT instead of AG) 5′ of exon 5. Accessions that had the nonconsensus splice acceptor site flowered earlier than accessions with the consensus splice site (Figure 3, Table 3). The remaining 3 significant SNPs were located in the A homeolog of CRY1 and were in complete linkage disequilibrium. The haplotype defined by these synonymous polymorphisms is shared with the closely related diploid species C. rubella (Slotte et al. 2008). CRY1 A alleles shared with C. rubella were segregating in the northern cluster of C. bursa-pastoris and accessions harboring these alleles flowered later than accessions with C. bursa-pastoris alleles (Table 3). There were no replacement substitutions in the entire CRY1 A coding sequence between two assayed accessions with different CRY1 A genotypes.
Figure 3.—

Estimated mean number of days to flowering (DTF), ±SE for accessions with different candidate SNP genotypes at FLC A. In both western Eurasia (A) and China (B), nonconsensus splice site alleles (in boldface type) are associated with earlier flowering. In western Eurasia, FLC A splice site variation is found only in the northern cluster.
TABLE 3.
Association tests of candidate SNPs
| Region | Locus | SNP position | Allele 1 | Allele 2 | Estimated effect of SNP allele 1 | SE | d.f. | F1,d.f. | P-value | Minor allele frequency |
|---|---|---|---|---|---|---|---|---|---|---|
| Western Eurasia | CRY1 A | 2 | G | A | 0.226 | 0.105 | 48 | 4.623 | 0.036 | 0.28 |
| CRY1 A | 8 | C | T | 0.226 | 0.105 | 48 | 4.623 | 0.036 | 0.28 | |
| CRY1 A | 242 | A | G | 0.226 | 0.104 | 49 | 4.709 | 0.035 | 0.28 | |
| FLC A | 452 | T | G | −0.413 | 0.190 | 48.8 | 4.752 | 0.034 | 0.10 | |
| China | FLC A | 495 | A | G | −0.603 | 0.179 | 38 | 11.290 | 0.002 | 0.10 |
The standard error (SE) of the estimated allelic effect (in log days to flowering), the denominator degrees of freedom (d.f.), the F-value, and the corresponding P-value are given for SNPs that had a significant association with flowering time at the nominal 5% level. Accessions with allele 1 at FLC A position 452 in western Eurasia, or allele 1 at position 495 in China, which result in a nonconsensus splice site, flower earlier than accessions with consensus splice sites.
Because there was no discernible population structure in the Chinese association population, we tested for association in this population using a simpler model without Q and K*. There were a total of four nonsingleton SNPs in three of the candidate genes, and one of these SNPs, at position 495 in the sequenced region of FLC A, exhibited an association with flowering time at a nominal 5% significance level (Table 3). The minor allele at this SNP yielded a nonconsensus splice donor site (AT instead of GT) 3′ of FLC A exon 5. Again, accessions harboring the nonconsensus splice site at FLC A flowered earlier than accessions with the consensus splice site (Figure 3). Haplotype-based tests of each candidate locus yielded similar results in both geographical regions (not shown): CRY1 A and FLC A were significantly associated with flowering time in western Eurasia (two of seven tests significant at a nominal 5% level) and FLC A was significantly associated with flowering time in China (one of three tests significant at a nominal 5% level).
FLC splicing and expression assays:
We performed additional assays to assess the impact of splice site polymorphisms on the FLC A transcript and global FLC expression level.
Chinese accessions with a nonconsensus splice site at FLC A SNP495 either expressed both FLC A and FLC B or expressed FLC A only. Their FLC A transcripts lacked a 42-bp region corresponding to exon 5, in accordance with GENSCAN cds predictions (Figure 4). One of the four assayed western Eurasian accessions with a nonconsensus splice site at FLC A SNP452 also expressed an FLC A transcript that lacked exon 5. We could not detect FLC A but only FLC B in the three other western Eurasian splice mutants (Figure 4). In the eight assayed accessions with consensus FLC splice sites, both homeologs were expressed in four and in two accessions each we could detect only FLC A or FLC B, respectively. There was a significant correlation between flowering time and total FLC expression level in western Eurasian accessions (Spearman's RS = 0.88, P = 0.03); however, the difference in the total level of FLC transcript between western Eurasian splice site mutants and accessions with consensus splice sites was not significant (Figure S2).
Figure 4.—

Effect of different FLC A SNP495 alleles on splicing. (A) The GENSCAN predicted cds in the sequenced fragment of FLC A lacks exon 5 in the Chinese accessions that have a nonconsensus splice donor site at position 495, here represented by GY31. Accessions with the consensus splice donor site (BJB236) have a predicted cds containing exon 5. (B) Size scoring of both homeologous FLC transcripts (results are shown for 10 representative accessions of the 16 assayed). Chinese and European accessions with a nonconsensus splice site at FLC A are indicated with a solid or shaded bar above the accession designation. Chinese accessions with the nonconsensus splice site at position 495 (GY31, GY36, KMB206, and KMB210) produce an alternatively spliced FLC A transcript. European accessions with a nonconsensus splice site at position 452 produce only transcripts of normal length (SE37) or alternatively spliced transcripts (SE12). Accessions with consensus splice sites produce only FLC transcripts of the expected size. (C) FLC transcript sequences for four accessions. Positions where FLC A and FLC B differ are indicated with plus signs below the alignment. The FLC transcript sequence for a Chinese splice site mutant expressing alternatively spliced FLC A (GY31) lacks exon 5, whereas a European splice site mutant expresses FLC B only (SE37) and two accessions with consensus splice sites express both FLC A and FLC B (BJB236 and SE45).
DISCUSSION
In this study, we have obtained DNA sequence data for 10 background loci and four duplicate candidate genes for flowering time in two geographically well-separated samples of C. bursa-pastoris, to assess the role of polymorphisms at duplicate (homeologous) genes for flowering time variation. Specifically, we were interested in testing whether polymorphisms of major effect on gene integrity were segregating and affecting phenotypic variation.
In agreement with studies of variation at chloroplast microsatellites (Ceplitis et al. 2005) and nuclear genes (Slotte et al. 2008), we find overall low levels of nucleotide diversity in C. bursa-pastoris. Reduced diversity levels are expected in a polyploid if the origin of the species was recent and constituted a severe bottleneck. Indeed, nucleotide diversity in C. bursa-pastoris (mean π = 0.001) was lower than in both its diploid congeners [the selfer C. rubella, mean π = 0.002 over all sites; and the outcrosser C. grandiflora, mean π = 0.011 over all sites (Foxe et al. 2009)] and was also lower than in A. thaliana [mean π = 0.0071 over all sites (Schmid et al. 2005)]. Despite the generally low levels of variation, we found robust evidence for population stratification in western Eurasia, where C. bursa-pastoris accessions from northern and southern Europe formed two distinct clusters. Taking the inferred population structure into account, SNPs at two of the candidate genes, the A homeologs of CRY1 and FLC, were significantly associated with flowering time variation in western Eurasia. In China, where STRUCTURE did not detect any population stratification, another SNP at the A homeolog of FLC was significantly associated with flowering time variation.
Although it has been shown that genomewide association studies in highly structured species suffer from elevated false positive rates (Aranzana et al. 2005; Zhao et al. 2007), several lines of evidence suggest that the associations discovered in the present study are not examples of such false positives. For instance, both FLC A SNPs that were associated with flowering time have major effects on gene integrity because they affect splice sites. Chinese accessions with a nonconsensus splice donor site 3′ of exon 5 had an FLC A transcript that lacked exon 5 and flowered earlier than accessions with a consensus splice donor site. Lack of this exon results in a deletion in the K domain, a conserved domain that is involved in protein–protein interaction (Yang et al. 2003). If a deletion in the K domain affects the function of FLC, early flowering would be the expected outcome given that FLC is a potent repressor of the transition to flowering. In A. thaliana, splice site mutations in FLC have been shown to disrupt FLC function (Michaels and Amasino 1999) and naturally occurring splice forms of FLC have been identified, many of which have effects on flowering time variation (Caicedo et al. 2004; Lempe et al. 2005; Werner et al. 2005). The function of FLC as a repressor of the transition to flowering also seems to be conserved at least within the Brassicaceae, although the details of FLC regulation may differ (e.g., Tadege et al. 2001; Wang et al. 2009). Indeed, an induced mutation at a splice site in FLC was recently shown to affect the transition to flowering and polycarpic habit of Arabis alpina (Wang et al. 2009) and naturally occurring splice forms of FLC1, a Brassica ortholog of FLC, are associated with flowering time variation in Brassica rapa (Yuan et al. 2009). Thus, although none of the previously identified splice forms are identical to the ones identified here, it seems reasonable to expect splicing variation at FLC to have effects on flowering time in Capsella.
Early flowering in European accessions with mutations at a different FLC A splice site seems to be partly through a different mechanism. Although one very early-flowering accession (SE12) expressed an alternatively spliced transcript identical to that found in Chinese splice site mutants, the other three assayed accessions did not express FLC A at detectable levels. The fact that accessions harboring FLC splice site mutations were found at relatively low frequencies (10%) and that several additional polymorphisms with effects on FLC transcript integrity are segregating among other accessions from China (H.-R. Huang and X.-J. Ge, unpublished data) suggests relaxed purifying selection on FLC in C. bursa-pastoris. However, the parallel occurrence of identical splicing variants in two separate geographical regions raises the possibility that positive selection for early flowering could have favored accessions harboring major-effect polymorphisms at FLC.
In a previous study we found no differences in the level of FLC expression between two pairs of early- and late-flowering accessions (Slotte et al. 2007); however, this study did not include any accessions with splice site mutations. Because FRI acts to increase the expression of FLC in the absence of vernalization, we hypothesized that coding differences or null mutations in FRI such as the common deletions conferring natural variation in A. thaliana were not important for flowering time differences between the C. bursa-pastoris accessions studied. In agreement with this, we found no association between FRI polymorphisms and flowering time in this study, this time in two large samples of accessions from separate parts of the species range. However, even though FRI may not be important for flowering time variation in C. bursa-pastoris, variation at other genes in the vernalization pathway still appears to play a role, as indicated by the FLC A splice site polymorphisms uncovered in this study.
An inherent limitation of association mapping is that depending on the extent of linkage disequilibrium, associated polymorphisms may be due to polymorphisms at linked genes and not at the candidate gene under study. This seems to be the case for CRY1 A, because there were no differences in amino acid sequence between accessions at CRY1 A, and there was no differential expression of CRY1 between the early- and late-flowering accessions included in a previous study (Slotte et al. 2007), although we cannot exclude the possibility of differential expression of CRY1 among the larger set of accessions in this study. The SNPs at CRY1 A that are associated with late flowering in western Eurasia define a haplotype that is shared with C. rubella. If the CRY1 A haplotype shared with C. rubella is indeed recently introgressed, as we concluded in a previous study (Slotte et al. 2008), linkage disequilibrium could extend a considerable distance. Given the possible involvement of altered circadian rhythms to hybrid vigor in allopolyploids (Ni et al. 2009), differential expression of core circadian clock genes between early- and late-flowering C. bursa-pastoris (Slotte et al. 2007), and the importance of the circadian clock in the control of flowering time, circadian clock genes in this chromosomal region could be good candidates for further study.
In conclusion, this study suggests that major-effect polymorphisms at one homeolog of FLC have evolved repeatedly in C. bursa-pastoris and have an effect on flowering time. Although further work is needed to fully characterize the genetic polymorphisms involved, this study suggests that ongoing nonfunctionalization of duplicate genes may contribute to flowering time variation in this species.
Acknowledgments
We thank Stephen I. Wright for comments on and discussion of this manuscript and Koen Verhoeven and Anna Palmé for comments on an earlier version of this manuscript. Financial support was provided by grants from the Swedish Research Council for Environmental, Agricultural Sciences and Spatial Planning (FORMAS) to M.L., from the Nilsson-Ehle foundation to T.S., from the Carl Trygger Foundation to A.C., from the Swedish Research Council and FORMAS to U.L., and from the National Natural Science Foundation of China (grant no. 30870366) and Swedish Institute to H.H.
Supporting information is available online at http://www.genetics.org/cgi/content/full/genetics.109.103705/DC1.
References
- Aranzana, M. J., S. G. Kim, K. Y. Zhao, E. Bakker, M. Horton et al., 2005. Genome-wide association mapping in Arabidopsis identifies previously known flowering time and pathogen resistance genes. PLoS Genet. 1: 531–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burge, C., and S. Karlin, 1997. Prediction of complete gene structures in human genomic DNA. J. Mol. Biol. 268: 78–94. [DOI] [PubMed] [Google Scholar]
- Caicedo, A. L., J. R. Stinchcombe, K. M. Olsen, J. Schmitt and M. D. Purugganan, 2004. Epistatic interaction between Arabidopsis FRI and FLC flowering time genes generates a latitudinal cline in a life history trait. Proc. Natl. Acad. Sci. USA 101: 15670–15675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ceplitis, A., Y. Su and M. Lascoux, 2005. Bayesian inference of evolutionary history from chloroplast microsatellites in the cosmopolitan weed Capsella bursa-pastoris (Brassicaceae). Mol. Ecol. 14: 4221–4233. [DOI] [PubMed] [Google Scholar]
- Clark, R. M., G. Schweikert, C. Toomajian, S. Ossowski, G. Zeller et al., 2007. Common sequence polymorphisms shaping genetic diversity in Arabidopsis thaliana. Science 317: 338–342. [DOI] [PubMed] [Google Scholar]
- Conant, G. C., and K. H. Wolfe, 2008. Turning a hobby into a job: how duplicated genes find new functions. Nat. Rev. Genet. 9: 938–950. [DOI] [PubMed] [Google Scholar]
- Cronn, R., M. Cedroni, T. Haselkorn, C. Grover and J. F. Wendel, 2002. PCR-mediated recombination in amplification products derived from polyploid cotton. Theor. Appl. Genet. 104: 482–489. [DOI] [PubMed] [Google Scholar]
- Dubcovsky, J., and J. Dvorak, 2007. Genome plasticity a key factor in the success of polyploid wheat under domestication. Science 316: 1862–1866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Falush, D., M. Stephens and J. K. Pritchard, 2003. Inference of population structure using multilocus genotype data: linked loci and correlated allele frequencies. Genetics 164: 1567–1587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Force, A., M. Lynch, F. B. Pickett, A. Amores, Y. L. Yan et al., 1999. Preservation of duplicate genes by complementary, degenerative mutations. Genetics 151: 1531–1545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foxe, J. P., T. Slotte, E. Stahl, B. Neuffer, H. Hurka et al., 2009. Recent speciation associated with the evolution of selfing in Capsella. Proc. Natl. Acad. Sci. USA 31: 5241–5245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ha, M., E. D. Kim and Z. J. Chen, 2009. Duplicate genes increase expression diversity in closely related species and allopolyploids. Proc. Natl. Acad. Sci. USA 106: 2295–2300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurka, H., and B. Neuffer, 1997. Evolutionary processes in the genus Capsella (Brassicaceae). Plant Syst. Evol. 206: 295–316. [Google Scholar]
- Johanson, U., J. West, C. Lister, S. Michaels, R. Amasino et al., 2000. Molecular analysis of FRIGIDA, a major determinant of natural variation in Arabidopsis flowering time. Science 290: 344–347. [DOI] [PubMed] [Google Scholar]
- Lee, I., M. J. Aukerman, S. L. Gore, K. N. Lohman, S. D. Michaels et al., 1994. Isolation of LUMINIDEPENDENS: a gene involved in the control of flowering time in Arabidopsis. Plant Cell 6: 75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lempe, J., S. Balasubramanian, S. Sureshkumar, A. Singh, M. Schmid et al., 2005. Diversity of flowering responses in wild Arabidopsis thaliana strains. PLoS Genet. 1: 109–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Linde, M., S. Diel and B. Neuffer, 2001. Flowering ecotypes of Capsella bursa-pastoris (L.) Medik. (Brassicaceae) analysed by a cosegregation of phenotypic characters (QTL) and molecular markers. Ann. Bot. 87: 91–99. [Google Scholar]
- Liu, Z., and K. L. Adams, 2007. Expression partitioning between genes duplicated by polyploidy under abiotic stress and during organ development. Curr. Biol. 17: 1669–1674. [DOI] [PubMed] [Google Scholar]
- Lynch, M., and J. S. Conery, 2000. The evolutionary fate and consequences of duplicate genes. Science 290: 1151–1155. [DOI] [PubMed] [Google Scholar]
- Michaels, S. D., and R. M. Amasino, 1999. FLOWERING LOCUS C encodes a novel MADS domain protein that acts as a repressor of flowering. Plant Cell 11: 949–956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michaels, S. D., Y. He, K. C. Scortecci and R. M. Amasino, 2003. Attenuation of FLOWERING LOCUS C activity as a mechanism for the evolution of summer-annual flowering behavior in Arabidopsis. Proc. Natl. Acad. Sci. USA 100: 10102–10107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nadeau, J. H., and D. Sankoff, 1997. Comparable rates of gene loss and functional divergence after genome duplications early in vertebrate evolution. Genetics 147: 1259–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni, Z. F., E. D. Kim, M. S. Ha, E. Lackey, J. X. Liu et al., 2009. Altered circadian rhythms regulate growth vigour in hybrids and allopolyploids. Nature 457: 327–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno, S., 1970. Evolution by Gene Duplication. Springer-Verlag, Heidelberg, Germany.
- Osborn, T. C., J. C. Pires, J. A. Birchler, D. L. Auger, Z. J. Chen et al., 2003. Understanding mechanisms of novel gene expression in polyploids. Trends Genet. 19: 141–147. [DOI] [PubMed] [Google Scholar]
- Papp, B., C. Pal and L. D. Hurst, 2003. Dosage sensitivity and the evolution of gene families in yeast. Nature 424: 194–197. [DOI] [PubMed] [Google Scholar]
- Pritchard, J. K., M. Stephens and P. Donnelly, 2000. Inference of population structure using multilocus genotype data. Genetics 155: 945–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozas, J., J. C. Sanchez-DelBarrio, X. Messeguer and R. Rozas, 2003. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19: 2496–2497. [DOI] [PubMed] [Google Scholar]
- Schmid, K. J., S. Ramos-Onsins, H. Ringys-Beckstein, B. Weisshaar and T. Mitchell-Olds, 2005. A multilocus sequence survey in Arabidopsis thaliana reveals a genome-wide departure from a neutral model of DNA sequence polymorphism. Genetics 169: 1601–1615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schranz, M. E., M. A. Lysak and T. Mitchell-Olds, 2006. The ABC's of comparative genomics in the Brassicaceae: building blocks of crucifer genomes. Trends Plant Sci. 11: 535–542. [DOI] [PubMed] [Google Scholar]
- Sheldon, C. C., J. E. Burn, P. P. Perez, J. Metzger, J. A. Edwards et al., 1999. The FLF MADS box gene: a repressor of flowering in Arabidopsis regulated by vernalization and methylation. Plant Cell 11: 445–458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shindo, C., C. Lister, P. Crevillen, M. Nordborg and C. Dean, 2006. Variation in the epigenetic silencing of FLC contributes to natural variation in Arabidopsis vernalization response. Genes Dev. 20: 3079–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotte, T., A. Ceplitis, B. Neuffer, H. Hurka and M. Lascoux, 2006. Intrageneric phylogeny of Capsella (Brassicaceae) and the origin of the tetraploid C. bursa-pastoris. Am. J. Bot. 93: 1714–1724. [DOI] [PubMed] [Google Scholar]
- Slotte, T., K. Holm, L. M. McIntyre, U. Lagercrantz and M. Lascoux, 2007. Differential expression of genes important for adaptation in Capsella bursa-pastoris (Brassicaceae). Plant Physiol. 145: 160–173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Slotte, T., H. Huang, M. Lascoux and A. Ceplitis, 2008. Polyploid speciation did not confer instant reproductive isolation in Capsella (Brassicaceae). Mol. Biol. Evol. 25: 1472–1481. [DOI] [PubMed] [Google Scholar]
- Tadege, M., C. C. Sheldon, C. A. Helliwell, P. Stoutjesdijk, E. S. Dennis et al., 2001. Control of flowering time by FLC orthologues in Brassica napus. Plant J. 28: 545–553. [DOI] [PubMed] [Google Scholar]
- Tajima, F., 1989. The effect of change in population size on DNA polymorphism. Genetics 123: 597–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, R., S. Farrona, C. Vincent, A. Joecker, H. Schoof et al., 2009. PEP1 regulates perennial flowering in Arabis alpina. Nature 459: 423–427. [DOI] [PubMed] [Google Scholar]
- Watterson, G. A., 1975. On the number of segregating sites in genetical models without recombination. Theor. Popul. Biol. 7: 256–276. [DOI] [PubMed] [Google Scholar]
- Wendel, J. F., 2000. Genome evolution in polyploids. Plant Mol. Biol. 42: 225–249. [PubMed] [Google Scholar]
- Werner, J. D., J. O. Borevitz, N. H. Uhlenhaut, J. R. Ecker, J. Chory et al., 2005. FRIGIDA-independent variation in flowering time of natural Arabidopsis thaliana accessions. Genetics 170: 1197–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang, Y., L. Fanning and T. Jack, 2003. The K domain mediates heterodimerization of the Arabidopsis floral organ identity proteins, APETALA3 and PISTILLATA. Plant J. 33: 47–59. [DOI] [PubMed] [Google Scholar]
- Yu, J., G. Pressoir, W. H. Briggs, I. Vroh Bi, M. Yamasaki et al., 2006. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness. Nat. Genet. 38: 203–208. [DOI] [PubMed] [Google Scholar]
- Yuan, Y. X., J. Wu, R. F. Sun, X. W. Zhang, D. H. Xu et al., 2009. A naturally occurring splicing site mutation in the Brassica rapa FLC1 gene is associated with variation in flowering time. J. Exp. Bot. 60: 1299–1308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao, K., M. J. Aranzana, S. Kim, C. Lister, C. Shindo et al., 2007. An Arabidopsis example of association mapping in structured samples. PLoS Genet. 3: e4.. [DOI] [PMC free article] [PubMed] [Google Scholar]