

Summary
Research on PI 3-Kinase (PI3K) is undergoing significant shifts in emphasis. Questions that have been dormant for some time are coming to the forefront, such as the relationship of PTEN to PI3K and the role of AKT in PI3K-driven oncogenesis. Two non-alpha isoforms of Class I PI3K are now established as important determinants in cancer: p110β and p110δ. The oncogenic activities of p110β include a non-catalytic function, a finding that will have immediate consequences for drug development.
Introduction
The PI 3-Kinase (PI3K) field has entered a phase of rapid and dynamic development. Although the basics remain unchanged, there are significant shifts in accents and emphasis. This is particularly true of Class I PI3K which has great significance for cancer and which will be the subject of this review. Among the questions that have recently come under scrutiny is the role of AKT (murine thymoma viral oncoprotein homolog) in the oncogenic signals from PI3K and the relationship between loss of PTEN (phosphatase and tensin homolog) and activation of PI3K. The non-alpha isoforms of Class I p110 are shedding their tentative and subordinate roles in oncogenesis and are emerging as important factors in cancer. In this paper, we will discuss questions that are raised by these recent developments.
PI3K was initially linked to cancer in studies of oncogenic viruses. The middle T antigen of polyoma virus, the Src oncoprotein of Rous sarcoma virus and the Ros oncoprotein of the avian sarcoma virus UR2 are associated with PI3K activity [1–3]. More direct evidence for the oncogenic potential of PI3K comes from avian sarcoma virus 16 which carries a homolog of the PIK3CA gene, coding for p110α, the catalytic subunit of PI3K, as its tumorigenic determinant [4]. In human cancer, deregulation of the PI3K signaling pathway has been recorded with increasing frequency, caused by gain of function in receptor tyrosine kinases, amplification of PIK3CA, activation of the serine-threonine kinase AKT or loss of function of the tumor suppressor phosphatase PTEN that is the catalytic antagonist of PI3K [5–8]. But it was the discovery of cancer-specific mutations in PIK3CA that moved PI3K into the limelight [9]. These mutations confer a gain of function as measured by enzymatic activity, constitutive downstream signaling and oncogenic potential [10–15]. About 80 % of the mutations occur in three hot spots in the gene, each represented by a single nucleotide substitution. The existence of these hot spots strongly suggests that the mutations provide a replicative advantage to the cell which is in accord with the gain of function detected by diverse assays of activity [9]. The mutant p110α proteins would appear as ideal therapeutic targets: they are restricted to cancer cells and, as enzymes, are readily controllable by small-molecule compounds, but mutant-specific inhibitors have not yet been generated [16].
Mutants and mechanisms
The mutant p110α proteins have raised the question of the molecular mechanisms that are responsible for the gain of function. Definitive answers to this question must await specific structural information on the mutants. Genetic experiments suggest the existence of several such mechanisms. Thus, combining kinase domain and helical domain hot spot mutations in the same molecule has a strong synergistic effect on signaling and oncogenicity. Kinase and helical domain mutations also differ in their requirements for interaction with RAS (rat sarcoma virus oncoprotein homolog) and with the PI3K regulatory subunit p85. The helical domain mutations of PIK3CA depend on interaction with RAS for full oncogenic activity but are independent of binding to the regulatory subunit p85. The kinase domain mutation shows the opposite requirements. It is oncogenic in the absence of RAS binding but fails to transform cells if the interaction with p85 is disabled [17]. In addition to the frequent hot spot mutations, almost 100 rare mutations have been identified in PIK3CA (Catalogue of somatic mutations in cancer, Wellcome Trust Sanger Institute; URL: http://www.sanger.ac.uk/genetics/CGP/cosmic/). A study of 15 of these rare mutations revealed varying degrees of increased function and of oncogenicity in all but one [18]. Most cancer-specific mutations in PIK3CA, regardless of their incidence may therefore contribute to the oncogenic phenotype of the cancer cell.
Cancer-specific mutations map over the entire coding sequence of PIK3CA, with the exception of the RAS-binding domain. The exceedingly broad distribution of mutations and the fact that many are located on the surface of the protein and affect electrostatic charge suggest that interactions with other proteins are involved. Changes in the binding to regulatory proteins may be the critical mediators of the gain of function in p110α mutants [19]. A well documented example for an altered interaction are the hot spot mutants located in the helical domain of p110α. They relieve an inhibitory interaction between that domain and the N-terminal SH2 domain of p85 [20]. The absence of mutations in the RAS-binding domain may indicate an importance of that domain not only in RAS binding [21], but also in the structural integrity of the protein.
Mutants of a different kind have been investigated by Zunder and coworkers [22]. Using a battery of PI3K inhibitors and an efficient and rapid yeast-based screen, they generated and analyzed p110α mutations that confer resistance to specific inhibitors. PI3K inhibitors, like protein kinase inhibitors, bind to the ATP affinity pocket. Surprisingly, most single-amino acid substitutions at this site in p110α lead to loss of activity, including the “gate-keeper” mutation that ranks prominently in drug-resistant mutants of protein kinases. In the ATP-binding cavity of p110α, only a single residue (I 800) was capable of inducing resistance when mutated.
PI3K, PTEN and AKT: Questions of correlations and connections
Figure 1 presents basic elements of PI3K signaling from upstream input by activated receptor tyrosine kinases or G-protein-coupled receptors to one of the major downstream targets, the TOR (target of rapamycin) kinase. Although didactically useful, such simplistic renderings could inadvertently lead to incorrect assumptions. For instance, on paper, a loss of function in PTEN is equivalent to a gain of function in PI3K, as both lead to increased levels of PIP3 which are regarded as the determining factor in PI3K signaling. This view is almost certainly incorrect. PI3K and PTEN both affect numerous cellular activities, but their target spectra are only partially overlapping. The effect of varied cellular localization on PI3K and PTEN functions, nonenzymatic scaffolding activities of PTEN and the differential effect of PTEN loss on p110 isoforms are currently active topics of research and discussion [23–27]. A recent analysis of human cancers at various sites strongly suggests that PIK3CA gain-of-function mutations and PTEN loss are not equivalent [8]. Thus, mutations in PIK3CA and loss of PTEN often coexist in human cancers. Therefore, they must be independently selected for, and make distinct, non-redundant contributions to the oncogenic phenotype. In contrast, double mutations in PIK3CA that affect the same enzymatic and signaling activity are very rare. Another problematic point in the canonical PI3K signaling scheme is AKT. It is widely assumed that AKT is an obligatory component of the oncogenic signal from PI3K to downstream targets. However, there are observations that do not fit this assumption, suggesting that the link between PI3K and AKT can be uncoupled. For instance, there are p110α mutations that induce oncogenic transformation in the absence of detectable phosphorylation of AKT, and, vice versa, p110α mutants exist that fail to transform despite robust AKT phosphorylation (Figure 2) [17,18]. Furthermore, mutants of p110α in general differ widely in their ability to induce phosphorylation of AKT at T308 and S473, and these differences are not correlated with oncogenic activity. These unexplained observations make it clear that the role of AKT in PI3K signaling needs to be defined more precisely.
Figure 1.
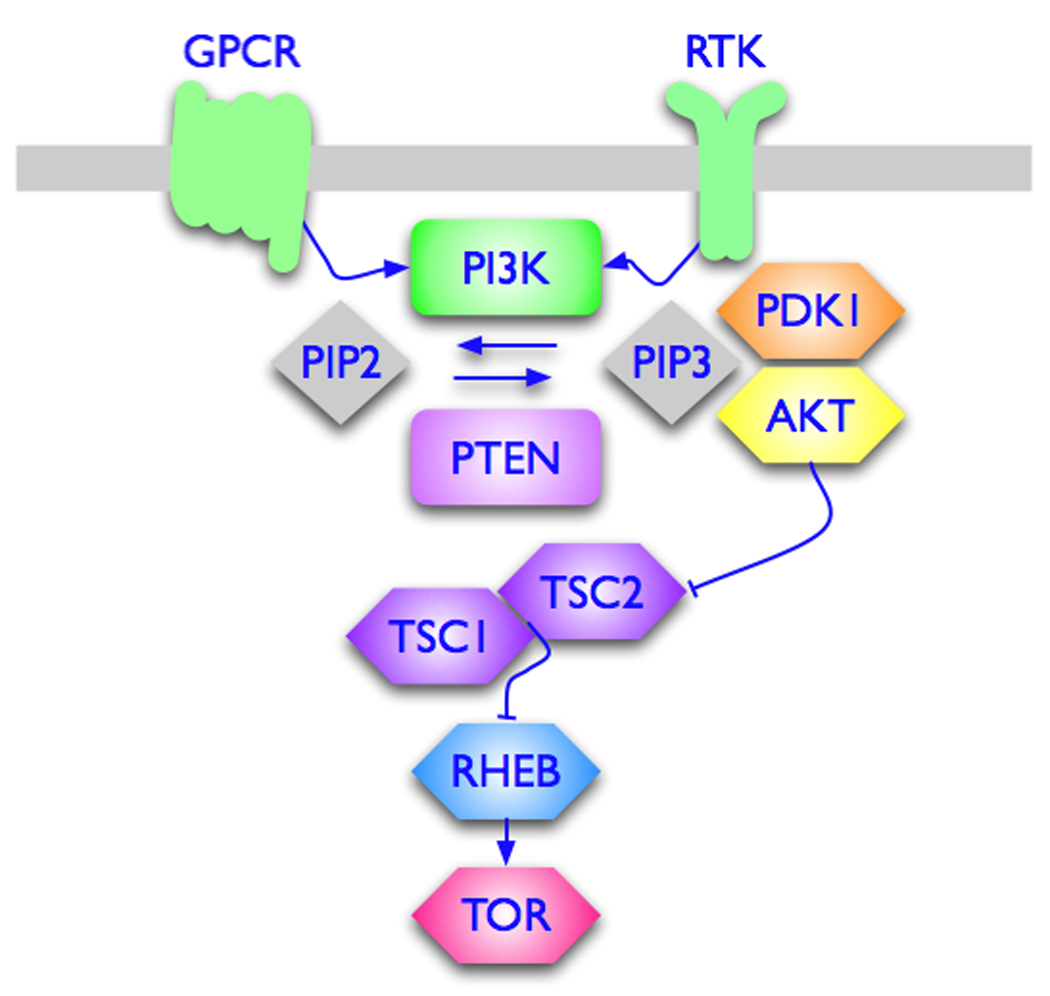
The pathway from PI3K to TOR. Recent publications have focused on the PI3K-PTEN interactions and on the role of AKT in oncogenic, PI3K-driven signaling. Loss of PTEN has differential effects on PI3K isoforms. The lack of correlation between oncogenic activity of PI3K and signaling through AKT suggests new crosstalks and alternative pathways. PIP2, phosphoinositide 4,5 bisphosphate; PIP3, phosphoinositide 3,4,5 trisphosphate; PDK1, phosphoinositide-dependent kinase; TSC1/TSC2, tuberous sclerosis complex; RHEB, RAS homolog enriched in brain.
Figure 2.
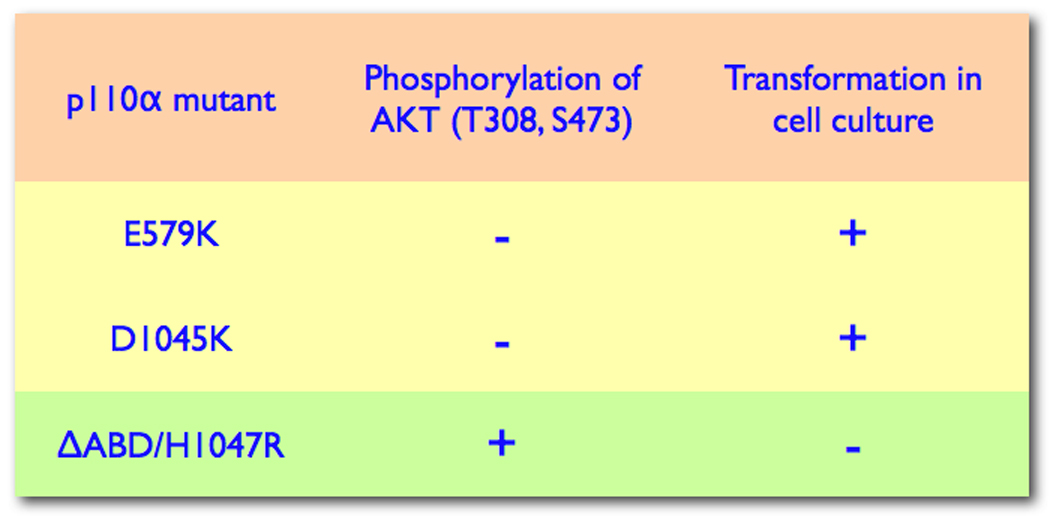
PI3K and AKT uncoupled. The figure shows examples of non-correlation between PI3K activity and phosphorylation of AKT [15,18].
The non-alpha isoforms of Class I p110: emerging roles in cancer
Class I PI3K contains four p110 isoforms, α, β, γ and δ. The association between p110α and cancer is well established and has been greatly strengthened by the occurrence of gain-of-function p110α mutations in human cancer [9]. The relationship of the non-alpha isoforms to cancer and their possible role as oncogenes has been more tenuous, but recent discoveries have changed that situation. A surprising observation was made with the four p110 isoforms overexpressed in avian fibroblasts. Whereas wild-type p110α failed to induce oncogenic transformation, all three non-alpha isoforms proved oncogenic in this cell system [28]. Oncogenic transformation has also been obtained with p110β and p110γ in mouse 10T1/2 cells (Ueno and Vogt, unpublished observation). A more detailed investigation of the p110 isoforms in avian cells uncovered differences in constitutive signaling, requirement for RAS interaction, and sensitivity to inhibitors of the MAP (mitogen-activated protein) kinase pathway. In these cells, only p110δ signals constitutively through AKT; p110δ is also exceptional in that its oncogenic activity is resistant to inhibitors of the MAP kinase pathway and appears not to require binding to RAS. In contrast, p110α, β, and γ lose oncogenic activity when RAS binding is disabled; p110β and p110γ are also highly sensitive to inhibitors of MAP kinase signaling. The loss of function induced in p110β and p110γ by a mutation in the RAS binding domain can be restored by a myristylation signal, suggesting a role of RAS in membrane recruitment [29].
The p110β isoform has now been firmly linked to oncogenesis by several groundbreaking papers published this year (Figure 3). The study of Jia et al. Uses mice with a conditional knockout of the PIK3CB, the gene encoding p110β [30]. The results define a new role of p110β in insulin signaling and show that both kinase-active and kinase-inactive forms of p110β have important functions in cell growth and trafficking. Of particular interest and importance is the observation that cultures of mouse embryo fibroblasts with a PIK3CB knockout cannot be transformed by constitutively active RAS or EGFR. Sensitivity to oncogenic transformation is restored by introducing wild-type p110β into these cells and, amazingly, sensitivity to transformation is also partially re-established by a kinase-inactive mutant of p110β. On the organismic level, Jia et al. found that ablation of p110β blocks prostate tumorigenesis mediated by loss of PTEN. Surprisingly, a prostate-specific knockout of p110α in these mice did not affect tumor formation. With these studies, p110β becomes a therapeutic target in cancer. However, the fact that kinase-inactive p110β can perform an essential role in oncogenesis will require new strategies for drug development. Conventional kinase inhibitors affecting the enzymatic activity of p110β would probably still leave its kinase-independent functions intact. It will be essential to learn more about this scaffolding activity of p110β. The observations of Jia and coworkers on the murine prostate cancer model suggest a connection between loss of PTEN and signaling by p110β. This link is also documented by the investigations of Wee and colleagues [25]. In PTEN-negative human cancers, p110β, but not p110α is essential for signaling and for replication. In another important publication on p110β, Ciraolo and coworkers have investigated mice that carry the homozygous kinase-inactive K805R mutation [31]. Animals expressing high levels of p110β(K805R) go through transient growth retardation but survive to adulthood, suffering from mild defects in insulin signaling. In contrast, low expressor siblings die in utero. Embryonic fibroblasts with low p110β(K805R) expression show inhibition of growth, but cells with high expression of the mutant protein replicate normally, providing additional evidence for a kinase-independent function of p110β in cell growth. In an ERBB2-driven model of breast cancer, the K805R mutation of p110β has a significant protective effect, indicating a role for p110β in the development of these tumors. This connection between p110β and a receptor tyrosine kinase appears in conflict with solid evidence that identifies G-protein-coupled receptors as the exclusive upstream signaling components for p110β [32]. The contradiction could be resolved by postulating crosstalk between ERBB2 and G-protein-coupled receptors. Such crosstalk has indeed been found in certain cell types [33]. A less prominent development on p110 isoforms and cancer concerns p110δ. Unlike p110α and p110β, p110δ is not ubiquitously expressed and is restricted mainly to hematopoietic cells. It has not ranked high as a cancer target, but has been considered for immune disorders [34–37]. However, it has been known for some time that p110δ is overexpressed in acute myeloblastic leukemia and that these leukemia cells are sensitive to isoform-specific inhibitors of p110δ [38,39]. These findings have been confirmed and expanded, and a p110δ-specific inhibitor is currently in phase I clinical trial for non-Hodgkins lymphoma, acute myeloblastic leukemia and chronic lymphoblastic leukemia (ClinicalTrials.gov, identifier NCT0070528).
Figure 3.

Involvement of p110β in oncogenesis [30,31]. A. Cultures of mouse embryo fibroblasts (MEF) lacking p110β cannot be transformed by activated RAS. Re-expression of p110β in these cells makes the cultures permissive for RAS-induced transformation. Surprisingly, MEF cultures expressing the catalytically inactive K805R mutant of p110β can also be transformed by RAS, albeit with lower efficiency. B. In a PTEN¯-driven model of prostate cancer, ablation of p110β, but not of p110α prevents tumor formation. In an ERBB2-driven model of breast cancer, substitution of wildtype p110β with the kinase-inactive K805R mutant has a tumor-protective effect.
Conclusions
We still know far too little about the direct and indirect interactions between PTEN and PI3K and their extensive signaling networks. Recent publications have brought insights and focus to this problem [8,23–25,27,30]. They support the conclusion that loss of PTEN has isoform-specific consequences for PI3K, and these consequences may be cell type-specific. The entry of p110β and p110δ into the realm of cancer marks a milestone for the therapeutic potential of targeting these isoforms. At the same time, the bar for successful drug development is raised by the discovery of catalysis-independent oncogenic functions of p110β.
Catalytically active and inactive p110β in cell replication and oncogenesis.
Mice homozygous for the K805R kinase-inactive mutation of p110β show partial protection from tumorigenesis in an ERBB2-induced model of breast cancer [31]. In a prostate cancer model driven by loss of PTEN, ablation of p110β prevents tumor formation, whereas loss of p110α has no effect on tumorigenesis [30]. Loss of PTEN function generally leads to a dominance of p110β in signaling and regulation of growth. In PTEN¯ human cancer cells, knock-down of p110β, but not of p110α interferes with signaling and cell replication [25]. Mice with the K805R mutation in p110β can survive to adulthood, provided the expression of the catalytically inactive p110β is high. This observation documents non-enzymatic, dosage-dependent functions of p110β in development and growth [31]. In cell culture, ablation of p110β prevents oncogenic transformation by activated RAS and other oncoproteins. Re-expression of p110β in these cultures restores the transforming activity of RAS. The cell cultures also regain partial susceptibility to transformation if instead of the wild-type p110β the K805R mutation is added back onto the p110β¯ background [30].
Acknowledgment
Work of the authors is supported by grants from the National Cancer Institute and by the Stein Fund. This is manuscript number 19791 of The Scripps Research Institute.
References
- 1.Whitman M, Kaplan DR, Schaffhausen B, Cantley L, Roberts TM. Association of phosphatidylinositol kinase activity with polyoma middle-T competent for transformation. Nature. 1985;315:239–242. doi: 10.1038/315239a0. [DOI] [PubMed] [Google Scholar]
- 2.Sugimoto Y, Whitman M, Cantley LC, Erikson RL. Evidence that the Rous sarcoma virus transforming gene product phosphorylates phosphatidylinositol and diacylglycerol. Proc Natl Acad Sci U S A. 1984;81:2117–2121. doi: 10.1073/pnas.81.7.2117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Macara IG, Marinetti GV, Balduzzi PC. Transforming protein of avian sarcoma virus UR2 is associated with phosphatidylinositol kinase activity: possible role in tumorigenesis. Proc Natl Acad Sci U S A. 1984;81:2728–2732. doi: 10.1073/pnas.81.9.2728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Chang HW, Aoki M, Fruman D, Auger KR, Bellacosa A, Tsichlis PN, Cantley LC, Roberts TM, Vogt PK. Transformation of chicken cells by the gene encoding the catalytic subunit of PI 3-kinase. Science. 1997;276:1848–1850. doi: 10.1126/science.276.5320.1848. [DOI] [PubMed] [Google Scholar]
- 5.Vivanco I, Sawyers CL. The phosphatidylinositol 3-Kinase AKT pathway in human cancer. Nat Rev Cancer. 2002;2:489–501. doi: 10.1038/nrc839. [DOI] [PubMed] [Google Scholar]
- 6.Engelman JA, Luo J, Cantley LC. The evolution of phosphatidylinositol 3-kinases as regulators of growth and metabolism. Nat Rev Genet. 2006;7:606–619. doi: 10.1038/nrg1879. [DOI] [PubMed] [Google Scholar]
- 7.Neve RM, Chin K, Fridlyand J, Yeh J, Baehner FL, Fevr T, Clark L, Bayani N, Coppe JP, Tong F, et al. A collection of breast cancer cell lines for the study of functionally distinct cancer subtypes. Cancer Cell. 2006;10:515–527. doi: 10.1016/j.ccr.2006.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Yuan TL, Cantley LC. PI3K pathway alterations in cancer: variations on a theme. Oncogene. 2008;27:5497–5510. doi: 10.1038/onc.2008.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Samuels Y, Wang Z, Bardelli A, Silliman N, Ptak J, Szabo S, Yan H, Gazdar A, Powell SM, Riggins GJ, et al. High frequency of mutations of the PIK3CA gene in human cancers. Science. 2004;304:554. doi: 10.1126/science.1096502. [DOI] [PubMed] [Google Scholar]
- 10.Kang S, Bader AG, Vogt PK. Phosphatidylinositol 3-kinase mutations identified in human cancer are oncogenic. Proc Natl Acad Sci U S A. 2005;102:802–807. doi: 10.1073/pnas.0408864102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ikenoue T, Kanai F, Hikiba Y, Obata T, Tanaka Y, Imamura J, Ohta M, Jazag A, Guleng B, Tateishi K, et al. Functional analysis of PIK3CA gene mutations in human colorectal cancer. Cancer Res. 2005;65:4562–4567. doi: 10.1158/0008-5472.CAN-04-4114. [DOI] [PubMed] [Google Scholar]
- 12. Samuels Y, Diaz LA, Jr, Schmidt-Kittler O, Cummins JM, Delong L, Cheong I, Rago C, Huso DL, Lengauer C, Kinzler KW, et al. Mutant PIK3CA promotes cell growth and invasion of human cancer cells. Cancer Cell. 2005;7:561–573. doi: 10.1016/j.ccr.2005.05.014. This milestone paper reports cancer-specific mutations in PIK3CA occurring at high frequency in common tumors. It intensified the interest in PI3K as a drug target.
- 13.Bader AG, Kang S, Vogt PK. Cancer-specific mutations in PIK3CA are oncogenic in vivo. Proc Natl Acad Sci U S A. 2006;103:1475–1479. doi: 10.1073/pnas.0510857103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Isakoff SJ, Engelman JA, Irie HY, Luo J, Brachmann SM, Pearline RV, Cantley LC, Brugge JS. Breast cancer-associated PIK3CA mutations are oncogenic in mammary epithelial cells. Cancer Res. 2005;65:10992–11000. doi: 10.1158/0008-5472.CAN-05-2612. [DOI] [PubMed] [Google Scholar]
- 15.Zhao JJ, Liu Z, Wang L, Shin E, Loda MF, Roberts TM. The oncogenic properties of mutant p110alpha and p110beta phosphatidylinositol 3-kinases in human mammary epithelial cells. Proc Natl Acad Sci U S A. 2005;102:18443–18448. doi: 10.1073/pnas.0508988102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Marone R, Cmiljanovic V, Giese B, Wymann MP. Targeting phosphoinositide 3-kinase: moving towards therapy. Biochim Biophys Acta. 2008;1784:159–185. doi: 10.1016/j.bbapap.2007.10.003. [DOI] [PubMed] [Google Scholar]
- 17. Zhao L, Vogt PK. Helical domain and kinase domain mutations in p110alpha of phosphatidylinositol 3-kinase induce gain of function by different mechanisms. Proc Natl Acad Sci U S A. 2008;105:2652–2657. doi: 10.1073/pnas.0712169105. Hot spot mutations in PIK3CA map to the helical and to the kinase domain of the enzyme. Combining helical and kinase domain mutations in the same molecule has a strong synergistic effect on oncogenicity and signaling. There is also differential dependence of helical domain and kinase domain mutations on binding to Ras and to the regulatory subunit p85.
- 18.Gymnopoulos M, Elsliger MA, Vogt PK. Rare cancer-specific mutations in PIK3CA show gain of function. Proc Natl Acad Sci U S A. 2007;104:5569–5574. doi: 10.1073/pnas.0701005104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Huang CH, Mandelker D, Gabelli SB, Amzel LM. Insights into the oncogenic effects of PIK3CA mutations from the structure of p110alpha/p85alpha. Cell Cycle. 2008;7:1151–1156. doi: 10.4161/cc.7.9.5817. The recently determined structure of p110α shows unique features that could be utilized for generating isoform-specific and possibly mutant-specific inhibitors.
- 20. Miled N, Yan Y, Hon WC, Perisic O, Zvelebil M, Inbar Y, Schneidman-Duhovny D, Wolfson HJ, Backer JM, Williams RL. Mechanism of two classes of cancer mutations in the phosphoinositide 3-kinase catalytic subunit. Science. 2007;317:239–242. doi: 10.1126/science.1135394. A structure-function analysis of mutations in the helical domain and in the adaptor domain of p110α shows that the former relieve an inhibitory interaction with p85, and the latter probably induce a conformational change affecting p85 binding.
- 21. Wells V, Downward J, Mallucci L. Functional inhibition of PI3K by the betaGBP molecule suppresses RAS-MAPK signalling to block cell proliferation. Oncogene. 2007;26:7709–7714. doi: 10.1038/sj.onc.1210580. This paper documents an interaction between PI3K and RAS that is essential for MAP kinase signaling.
- 22. Zunder ER, Knight ZA, Houseman BT, Apsel B, Shokat KM. Discovery of drug-resistant and drug-sensitizing mutations in the oncogenic PI3K isoform p110 alpha. Cancer Cell. 2008;14:180–192. doi: 10.1016/j.ccr.2008.06.014. Most single amino acid substitutions including the “gate-keeper” mutation in the ATP affinity pocket of p110α lead to loss of function. This intolerance of the lipid kinase ATP pocket to mutations is in contrast to observations on protein kinases where single amino acid substitutions are more often compatible with function.
- 23. Salmena L, Carracedo A, Pandolfi PP. Tenets of PTEN tumor suppression. Cell. 2008;133:403–414. doi: 10.1016/j.cell.2008.04.013. The authors present a critical and up-to-date review of PTEN as a tumor suppressor and of the various catalytic and non-catalytic interactions with other signaling pathways, notably that of PI3K.
- 24.Carracedo A, Salmena L, Pandolfi PP. SnapShot: PTEN signaling pathways. Cell. 2008;133 doi: 10.1016/j.cell.2008.04.023. 550 e551. [DOI] [PubMed] [Google Scholar]
- 25. Wee S, Wiederschain D, Maira SM, Loo A, Miller C, deBeaumont R, Stegmeier F, Yao YM, Lengauer C. PTEN-deficient cancers depend on PIK3CB. Proc Natl Acad Sci U S A. 2008;105:13057–13062. doi: 10.1073/pnas.0802655105. A surprising set of data that show PTEN-negative cancers depend on p110β, but not p110α for signaling and for growth.
- 26.Keniry M, Parsons R. The role of PTEN signaling perturbations in cancer and in targeted therapy. Oncogene. 2008;27:5477–5485. doi: 10.1038/onc.2008.248. [DOI] [PubMed] [Google Scholar]
- 27.Carracedo A, Pandolfi PP. The PTEN-PI3K pathway: of feedbacks and cross-talks. Oncogene. 2008;27:5527–5541. doi: 10.1038/onc.2008.247. [DOI] [PubMed] [Google Scholar]
- 28. Kang S, Denley A, Vanhaesebroeck B, Vogt PK. Oncogenic transformation induced by the p110beta, -gamma, and -delta isoforms of class I phosphoinositide 3-kinase. Proc Natl Acad Sci U S A. 2006;103:1289–1294. doi: 10.1073/pnas.0510772103. This study reports oncogenic transformation of avian cells by wild-type p110β, p110γ and p110δ, revealing an unexpected oncogenic potential of these non-alpha isoforms.
- 29.Denley A, Kang S, Karst U, Vogt PK. Oncogenic signaling of class I PI3K isoforms. Oncogene. 2008;27:2561–2574. doi: 10.1038/sj.onc.1210918. [DOI] [PubMed] [Google Scholar]
- 30. Jia S, Liu Z, Zhang S, Liu P, Zhang L, Lee SH, Zhang J, Signoretti S, Loda M, Roberts TM, et al. Essential roles of PI(3)K-p110beta in cell growth, metabolism and tumorigenesis. Nature. 2008;454:776–779. doi: 10.1038/nature07091. In this seminal paper, the authors establish a firm link between p110β and oncogenesis. They show that loss of p110β induces resistance to tumor formation in a PTEN¯-driven model of prostate cancer and that p110β- negative mouse embryo fibroblasts cannot be transformed by RAS, but sensitivity to transformation can be partially restored by catalytically inactive p110β.
- 31. Ciraolo E, Iezzi M, Marone R, Marengo S, Curcio C, Costa C, Azzolino O, Gonella C, Rubinetto C, Wu H, et al. Phosphoinositide 3-kinase p110beta activity: key role in metabolism and mammary gland cancer but not development. Sci Signal. 2008;1:ra3. doi: 10.1126/scisignal.1161577. This landmark paper re-enforces and complements the conclusions of [30]. It shows that the catalytically inactive p110β can be sufficient to support organismic development to adulthood and that loss of p110β catalytic activity is tumor-protective in an ERBB2-driven model of breast cancer.
- 32. Guillermet-Guibert J, Bjorklof K, Salpekar A, Gonella C, Ramadani F, Bilancio A, Meek S, Smith AJ, Okkenhaug K, Vanhaesebroeck B. The p110beta isoform of phosphoinositide 3-kinase signals downstream of G protein-coupled receptors and is functionally redundant with p110gamma. Proc Natl Acad Sci U S A. 2008;105:8292–8297. doi: 10.1073/pnas.0707761105. In this fundamental study, the authors show that p110β receives upstream signaling from G-protein-coupled receptors and is unresponsive to signaling from receptor tyrosine kinases.
- 33.Daub H, Weiss FU, Wallasch C, Ullrich A. Role of transactivation of the EGF receptor in signalling by G-protein-coupled receptors. Nature. 1996;379:557–560. doi: 10.1038/379557a0. [DOI] [PubMed] [Google Scholar]
- 34.Jou ST, Carpino N, Takahashi Y, Piekorz R, Chao JR, Wang D, Ihle JN. Essential, nonredundant role for the phosphoinositide 3-kinase p110delta in signaling by the B-cell receptor complex. Mol Cell Biol. 2002;22:8580–8591. doi: 10.1128/MCB.22.24.8580-8591.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Patton DT, Garden OA, Pearce WP, Clough LE, Monk CR, Leung E, Rowan WC, Sancho S, Walker LS, Vanhaesebroeck B, et al. Cutting edge: the phosphoinositide 3-kinase p110 delta is critical for the function of CD4+CD25+Foxp3+ regulatory T cells. J Immunol. 2006;177:6598–6602. doi: 10.4049/jimmunol.177.10.6598. [DOI] [PubMed] [Google Scholar]
- 36. Rommel C, Camps M, Ji H. PI3K delta and PI3K gamma: partners in crime in inflammation in rheumatoid arthritis and beyond? Nat Rev Immunol. 2007;7:191–201. doi: 10.1038/nri2036. This review makes an eloquent and well documented case for p110δ and p110γ as targets in inflammation and immune-mediated disorders.
- 37.Ali K, Camps M, Pearce WP, Ji H, Ruckle T, Kuehn N, Pasquali C, Chabert C, Rommel C, Vanhaesebroeck B. Isoform-specific functions of phosphoinositide 3-kinases: p110 delta but not p110 gamma promotes optimal allergic responses in vivo. J Immunol. 2008;180:2538–2544. doi: 10.4049/jimmunol.180.4.2538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Sujobert P, Bardet V, Cornillet-Lefebvre P, Hayflick JS, Prie N, Verdier F, Vanhaesebroeck B, Muller O, Pesce F, Ifrah N, et al. Essential role for the p110delta isoform in phosphoinositide 3-kinase activation and cell proliferation in acute myeloid leukemia. Blood. 2005;106:1063–1066. doi: 10.1182/blood-2004-08-3225. Very little has been published on the involvement of p110δ in cancers of the hematopoietic system. The data presented in this paper suggest a possible role for p110δ-specific inhibitors in leukemia and lymphoma. Ref. [39] adds support to this suggestion.
- 39.Billottet C, Grandage VL, Gale RE, Quattropani A, Rommel C, Vanhaesebroeck B, Khwaja A. A selective inhibitor of the p110delta isoform of PI 3-kinase inhibits AML cell proliferation and survival and increases the cytotoxic effects of VP16. Oncogene. 2006;25:6648–6659. doi: 10.1038/sj.onc.1209670. [DOI] [PubMed] [Google Scholar]