

Abstract
Energy balance to prevent the development of obesity is dependent on energy expenditure. Although physical activity is the dominant mechanism for dissipating excess energy, a system of thermogenesis that evolved to protect the body from hypothermia is based upon the uncoupling of oxidative phosphorylation in brown adipocytes by the mitochondrial uncoupling protein (UCP1). It has been shown that upregulation of UCP1 by genetic manipulations or pharmacological agents can reduce obesity and improve insulin sensitivity. Recent evidence has shown the existence of two sources for brown adipocytes, one appearing as discrete brown fat depots during fetal development and the other appears during post-natal development as diffuse populations in traditional white fat depots. The latter can be induced by adrenergic stimulation depending on the genetic background of the animals and the nutritional environment. Understanding the biological and environmental factors controlling the expression of these two brown adipocyte populations promises to provide new strategies by which enhanced thermogenesis can be used to reduce obesity.
Keywords: thermogenesis, UCP1 transgenic mice, brown adipocytes, transcription synergy, white adipose tissue
Introduction
Mechanisms that prevent excessive storage of lipids in tissues besides adipose tissue are necessary for maintaining a physiological state that achieves optimal insulin sensitivity, thereby avoiding the development of chronic diseases, collectively referred to as the metabolic syndrome. The broad underlying mechanisms to maintain a healthy body composition are described by the energy balance equation whereby energy intake equals energy expenditure. Studies of experimental animals with genetic and surgically induced lesions in centers in the brain have identified molecular mechanisms in major centers in the central nervous system that are involved in the regulation of food intake. It follows that the other side of the equation, the energy expenditure arm, should also have a centrally regulated control system that can be induced when an animal goes into a positive energy balance. This energy expenditure system has been envisioned to include a thermogenic mechanism that can burn off excess fat to re-establish energy balance and reverse insulin resistance.1 At the present time, the nature of a thermogenic mechanism that could be the determinant of the energy expenditure arm of energy balance in the human is completely unknown. Some progress has been made in identifying thermogenic mechanisms in mice, and these will be described in this paper.
During the 1970s when biochemical and physiological studies on brown fat thermogenesis were very active, the idea emerged that an ideal thermogenic mechanism to regulate body weight as well as temperature was the mitochondrial uncoupling protein, UCP1.2,3 Many properties of brown fat nonshivering thermogenesis recommend a function in regulating body weight. UCP1 was specifically expressed in the inner membrane of mitochondria from brown adipocytes to generate heat by uncoupling oxidative phosphorylation, and the induction of thermogenesis was controlled adrenergically by the cold–warm centers in the central nervous system. Linking the feeding centers in the hypothalamus to this thermogenic system for the control of body temperature to body weight regulation could be a very effective dual function for brown fat thermogenesis. The initial evidence for this linkage came from subphenotypes of the leptin-deficient ob/ob mouse; mainly the cold sensitivity of ob/ob mice was interpreted as indicating that the ob gene (leptin deficiency) suppressed Ucp1 expression and precluded activation of brown fat thermogenesis.4 The loss of this thermogenic mechanism, together with an inability to suppress food intake, could account for the obese phenotype of leptin-deficient mice.5 The belief in such a mechanism for body weight regulation persists to the present day; however, studies showing that mice deficient for UCP1 are actually resistant to diet-induced obesity (DIO) indicate that we need to reconsider the basic mechanisms of thermogenesis and how they can be used in the regulation in body weight.6 In addition, new findings showing that the regulation of brown adipocyte differentiation in discrete brown fat depots, such as interscapular brown fat, differs from that of brown adipocytes diffusely located in white fat depots indicate that new strategies to utilize the thermogenic potential of UCP1 will be possible.7,8 Finally, I will discuss UCP1-independent thermogenic mechanisms that promise to completely reverse the direction of strategies for the regulation of body weight by thermogenesis.
Discussion
Regulating body temperature and weight in the absence of UCP1
To understand phenotypes that are caused by manipulating the expression of Ucp1 expression, one must emphasize the characteristics of UCP1 that endow the brown adipocyte with its unparalleled capacity to produce heat with high efficiency and then to distribute the heat to vital organs of the body.
The brown adipocyte has a higher density of mitochondria than just about any other cell in the body to maximize its electron transport capacity for generating the proton motive force to drive thermogenesis by the uncoupling mechanism.
Uncoupling protein-1 constitutes about 10% of the mitochondrial membrane protein, in contrast to UCP2 and UCP3, which, in the cells where UCP1 is expressed, constitute about 0.01–0.1% of the membrane protein.
The ATP synthase content of the brown fat mitochondria is suppressed to increase the proportion of the membrane potential that is directed towards uncoupling as opposed to ATP synthesis.
The brown adipocyte has a very high capacity for fatty acid oxidation and TCA activity, as well as the highest capacity for generating glycerol phosphate from glucose and its subsequent oxidation in the mitochondria. All of this capacity for substrate oxidation is directed towards maximizing respiration when signals from the brain are received.
Uncoupling protein-1 activity in uncoupling mitochondria is exquisitely regulated by the inhibitory action of the purine nucleotide level of ATP that is released by fatty acids when they are generated by the adrenergic stimulated breakdown of triglycerides in the lipid vesicles of the brown adipocyte.
Brown fat is highly innervated and vascularized, both of which are controlled by the sympathetic nervous system, to provide for the rapid activation of thermogenesis followed by the efficient distribution of heat released from brown fat for delivery to vital organs.
Emphasizing these assets and strengths of the brown adipocyte for thermogenesis is necessary to help us to understand how the mouse responds when Ucp1 is inactivated by gene targeting. As the ability to maintain a body temperature of 37 °C is essential to life, in the absence of UCP1, alternative thermogenic mechanisms must be utilized to maintain normal body temperature. Given that the level of brown fat in humans is low, possibly approaching that of genetically deficient mice, the identification of UCP1-independent thermogenic mechanisms in mice may be applicable to the problem of thermogenesis and body weight regulation in humans.
Body temperature in Ucp1−/− mice
As predicted, Ucp1−/− mice are cold-sensitive, thereby establishing a fundamental function of UCP1 in the regulation of body temperature, but the cold-sensitive phenotype of Ucp1−/− mice is highly variable. The genetic background of UCP1-deficient mice strongly affects the cold-sensitivity phenotype. Mice of inbred backgrounds cannot tolerate the cold if the mouse is immediately exposed to an ambient temperature of 4 °C; however, these mice can adapt during a gradual reduction in ambient temperature.9 One of the most striking phenotypes is the tolerance to cold of Ucp1−/− mice on a hybrid genetic background without the need for a period of adaptation.10 At the other extreme is the intolerance to cold exposure of double mutants of Ucp1−/− and Lep−/−. Even though both single mutants can adapt to the cold, the double mutant cannot survive at temperatures below 12 °C, even after slow adaptation, unless the mice are administered leptin or T3.11 These experiments indicate that an animal deficient in a major thermogenic mechanism for maintaining body temperature can induce alternative mechanisms; however, the studies with the double mutant suggests that there are limits.
Body weight in Ucp1−/− mice
Based upon our understanding of the relationship between Ucp1 overexpression and resistance to obesity at the time the Ucp1−/− mouse was generated, it was expected that ablation of Ucp1 expression, the major thermogenic mechanism in the mouse, would result in reduced capacity for burning fat, which would make Ucp1−/− mice obese, and they would have an increased susceptibility to diet-induced and genetic obesity.12 In contrast, our studies on DIO in Ucp1−/− mice showed that in the absence of UCP1, mice were resistant to DIO.6 Furthermore, as it was proposed that the increased metabolic efficiency of ob/ob mice was due in part from the inability of these mice to stimulate UCP1 thermogenesis, obesity in ob/ob mice would be enhanced by UCP1 deficiency. However, UCP1 deficiency neither increased nor decreased the obesity of Lep−/− mice.11 The latter phenotype suggested that the thermogenesis of UCP1 under normal ambient temperatures, that is, 23–28 °C, was not linked to the energy expenditure status of ob/ob mice. However, if the ambient temperature is reduced to the degree that it threatens the capacity of the mouse to maintain body temperature, it is clear that, under cold stress, the absence of leptin seriously compromises the ability of the mouse to generate heat. Curiously, the mice are not able to utilize their fat stores as fuel.
We have interpreted the resistance to obesity in Ucp1−/− as a logical outcome of the characteristics of brown fat thermogenesis described above. In the absence of UCP1, the mouse is forced to use alternative thermogenic metabolic systems. Although the exothermic properties of these systems are able to contribute to the thermal requirements of the animal; nevertheless, they have not been designed for this purpose with the consequence that more fuel is required to be burned to maintain a normal body temperature. They are not under stringent, rapid adrenergic regulation of the fatty acid oxidation and genetic induction of expression, nor do they have the dense vascularization to rapidly distribute generated heat. Therefore, UCP1-deficient mice are resistant to obesity, but only under conditions in which a reduced ambient temperature increases the demand for heat production. As the ambient temperature increases towards thermoneutrality, body weight regulation in the UCP1-deficient mice is virtually identical to the wild-type mouse. A caveat to the phenotype observed in young adult mice up to 6 months of age is the evidence that during aging, Ucp1−/− mice show a modest tendency to have increased adiposity compared with the wild-type mice.13
To develop a more sensitive model system for identifying alternative thermogenic mechanisms, we combined Ucp1−/− with the inactivated gene for the mitochondrial glycerol-3-phosphate dehydrogenase, Gdm1−/−.14 The latter is part of a thermogenic mechanism, based on the glycerol-phosphate shuttle, for transporting cytoplasmic NADH into the mitochondria.15 The glycerol phosphate enters the mitochondria where it is oxidized by the mitochondrial flavin-linked enzyme. The generation of only two ATPs per NADH by this pathway rather than three ATPs introduces metabolic inefficiency. The double mutant model further reduces thermogenic capacity and causes the mouse to utilize even less robust thermogenic mechanisms with the net effect being an even greater resistance to DIO than observed in the Ucp1−/− mouse. The site for the alternative thermogenesis appears to be determined by enhanced mitochondrial expression and capacity for β-oxidation in the inguinal fat depot (R Anunciado-Koza and LP Kozak, unpublished).
An interpretation of the body temperature and weight phenotypes of Ucp1−/− mice leads to the conclusion that UCP1 thermogenesis does not play a major direct role in body weight regulation, rather that the effects of changes in UCP1 on body weight are a consequence of the status of mechanisms to maintain body temperature. This conclusion may be applicable to mice and humans. Maintenance of body temperature has always been indispensable for survival; however, the need to burn off energy to maintain a body composition free of insulin resistance only became important in modern times. The physical activity necessary to gather food generated more than enough thermogenesis to burn off fat and maintain a body free of insulin resistance. Accordingly, there was simply no need to evolve a specialized thermogenic system to control body weight. However, this parsimonious explanation for UCP1 function and body weight regulation is challenged by the function that may be associated with brown adipocytes that are inducible in white fat depots.
Ectopic constitutive overexpression of UCP1 reduces DIO and insulin resistance
Despite the evidence suggesting that UCP1 is not normally involved in body weight regulation, many transgenic animals have been developed that are highly resistant to both DIO and genetic obesity. These genetically modified mice fall into one of two groups, those in which the Ucp1 cDNA has been ectopically expressed in white fat and muscle from a constitutively expressed promoter such as the aP2 promoter or the mysosin light-chain promoter.16 Although the reduction of adiposity is certainly significant, the mechanism may reflect the effects of a specific proton leak that is regulatable by purine nucleotides and fatty acids, as described for the normal function of UCP1 in the brown adipocyte, or it may be a nonspecific proton leak that is mechanistically similar to uncoupling by dinitrophenol and other chemical uncouplers. The fact that homozyogous aP2-Ucp1-transgenic mice have such a high level of expression of UCP1 in brown adipocytes that it becomes cytotoxic to at least 90% of the brown adipocytes, creating a mouse with a phenotype virtually indistinguishable from the Ucp1−/− mouse, strongly argues for nonspecific proton leaks in these transgenic mice and the muscle overexpression of Ucp3.17,18
Although the ectopic expression of UCPs is effective in reducing obesity, the observation that a simple doubling of expression can lead to cell death does not recommend the strategy as applicable to human obesity.
Brown adipocyte induction in white fat depots
The other kind of transgenic mouse overexpression of UCP1 in brown adipocytes is almost always caused by overexpression of signaling molecules and transcription factors that lead to the induction of brown adipocytes in the traditional white fat depots. The overexpression of the β1-adrenergic receptor increases the signaling of the PKA pathway. The transgenic overexpression of FoxC2 in the PKA RIIβ knockout mouse also augments PKA signaling by upregulation of the RIα-regulatory subunit, which increases the differentiation pathway for brown adipocytes in white fat depots, by virtue of its lower affinity constant for cAMP.19–21
Unlike the transgenic mice described above, the upregulation of Ucp1 through induction of brown adipocytes maintains control of normal mechanisms for controlling expression and function. Given the potential for brown fat thermogenesis to reduce obesity and diabetes, it is important to identify mechanisms that can activate brown adipocyte differentiation in response to drug treatment. Transcriptional regulation of the Ucp1 gene is controlled by regulatory elements that have been shown to be critical for both white and brown fat adipogenesis. This includes members of the peroxisome proliferator-activated receptor (PPAR) and CCAAT/enhancer-binding protein (C/EBP) families and cAMP response-binding protein (CREB).22 Pivotal to this regulation is a peroxisome proliferator response element site in the distal Ucp1 enhancer that forms a complex with PPARγ2-RXR and the co-activator PGC-1α,23–25 as well as half-site cAMP response elements (CRE) in both the proximal promoter and the distal enhancer. These CRE sites interact with CREB and activating transcription factor-2, and mutations within them abolish Ucp1 expression in transient expression assays.26,27 The role of PPARγ in adipogenesis has been extensively documented in both tissue culture and in vivo models of adipogenesis and brown adipocyte expression.24,28,29 Although C/EBPα appears not to be required for brown fat expression,30 C/EBPβ and δ expression have critical roles in brown fat differentiation.31 Also located in the Ucp1 enhancer region is a site for regulation by the thyroid hormone receptor,32 which accordingly links Ucp1 regulation to the potential influence of thyroid hormone on thermogenesis.33
The adrenergic signaling mechanism for regulation of Ucp1 expression and brown adipocyte differentiation is a G-protein receptor mechanism coupling cAMP production to protein kinase A (PKA)-dependent phosphorylation of CREB and p38 MAP kinase. The latter has been implicated in regulating activating transcription factor-2 by phosphorylation, with subsequent effects on both Pgc-1α and Ucp1 transcription.27 Interactions between retinoblastoma protein and FOXC2, which has been postulated to act in the signaling cascade by inducing the level of the PKA-RIα-regulatory protein, have also been implicated in the regulation of Ucp1.21,34 More recently, a previously unidentified transcription factor, PRMD16, which is expressed in brown adipocytes but not white adipocytes, has been proposed to be essential for the induction of the brown adipocyte differentiation program through its effects on PGC1α.35
Accordingly, the number of potential sites for regulation of Ucp1 and brown adipocyte induction is large, and many of the signaling molecules and transcription factors could be involved in other aspects of adipocyte biology, as well as muscle and liver structure and function. A fundamental question is how can one modulate these pathways pharmacologically to selectively induce brown fat differentiation in white fat depots without adversely affecting regulation in other organ systems? To address this question, we have pursued a genetic analysis based on the variation in brown adipocyte differentiation observed between A/J and C57BL/6J (B6) mice.7,36–38 The large differences in Ucp1 mRNA and protein levels (in some experiments up to 80-fold) between these strains suggest that the quantitative genetic analysis of signaling and transcription pathways would reveal how natural genetic variation is able to achieve selective induction of brown adipocytes in white fat depots by adrenergic signaling. In addition, this analysis summarized in Figure 1 shows how small variations in individual transcription and signaling molecules interact synergistically to amplify the final expression of Ucp1. An additional conclusion of this study is that it illustrates that the most effective strategy for induction of brown adipocytes will probably come from activation of more that one regulatory site. Selecting sites that are specific for adipose tissue and do not affect other vital functions, such as the liver and cardiovascular system, will be essential.
Figure 1.

Sites of genetic variation in the signaling and transcription factors associated with induction of Ucp1 in retroperitoneal fat depots. The ratio in expression between expression in A/J versus C57BL/6 J mice is shown in blue. The analysis suggests that the synergy among several regulatory sites with modest differences in expression leads to the 80-fold ratio in the level of Ucp1 expression.
Most investigators will agree that if brown fat thermogenesis is to be used as an antiobesity agent, it will be achieved through the induction of brown adipocytes into white fat depots. However, if the capacity for induction of brown adipocytes in human white fat depots resembles that of mice by showing high genetic variability among strains, the effective use of this strategy will probably require the use of a personalized genomic medicine approach. Whether a mouse is able to induce brown adipocytes in white fat depends on the type of fat depot, diet, environmental temperature, and probably nutritional programming, but the most important variable is the genotype of the individual. Mice of some strains, such as C57BL/6J, are highly resistant to the induction of brown adipocytes by adrenergic stimulation, whereas others, such as A/J and 129, are very sensitive with levels of Ucp1 in retroperitoneal fat approaching that of interscapular brown fat. In fact, genotype can have an impact on both nutritionally related, that is, high-fat diets, and pharmacological aspects of brown adipocyte induction.38
The capacity for induction of brown adipocytes in white adipose tissue of the adult mouse reflects processes that occur in post-natal development between 15 and 30 days of age as shown in Figure 2. The developmental time course for visceral and subcutaneous white fat depots differs, with inguinal subcutaneous fat having fully differentiated adipocytes within a few days of birth, whereas visceral depot adipocytes do not begin to differentiate until about 9 days of age. However, a transient appearance of brown adipocytes can be observed in both visceral and subcutaneous fat between 15 and 30 days of age.8 The function of these brown adipocytes is not known, but we have recently found that UCP1 expression in the brown adipocytes of inguinal fat is very sensitive to the nutritional environment, as undernutrition, caused by food restriction to the mother with suckling pups, severely suppresses the expression of Ucp1 in the fat depots of the pups (LP Kozak and R Anunciado-Koza, unpublished observations). This finding on the effects of the nutritional environment during early development suggesting that nutritional factors are involved in Ucp1 regulation in white fat, together with additional data on the strong effects of diets high or low in fat on the QTL analysis of Ucp1, indicate that the induction of brown adipocytes in white fat diet may have a function in the regulation of energy balance.
Figure 2.
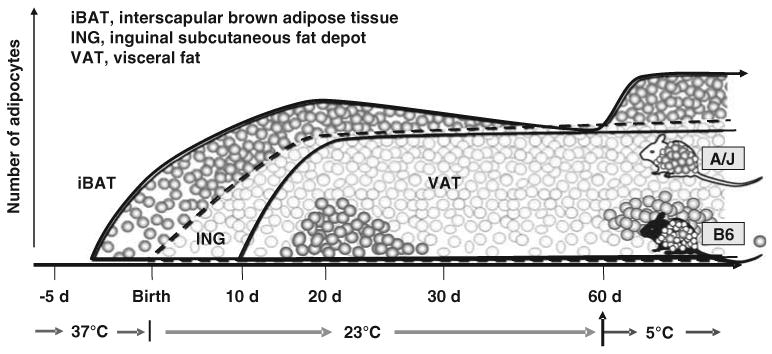
Developmental profiles illustrating the appearance of brown adipocytes in interscapular brown adipose tissue (iBAT) initially at about 4 days before birth. Although the inguinal fat depot (ING) first appears at birth, but the visceral fat (VAT) not until ∼ 10 days of age, brown adipocytes appear in both depots between 15 and 30 days of age. The induction of brown adipocyte at 20 days of age is higher in A/J than C57Bl/6J (B6) mice and is induced in adult animals by adrenergic stimulation in A/J mice, but not B6 mice. This genetic difference in the induction of brown adipocytes among white fat depots is not found in iBAT. This figure has been modified from Xue et al.,39 courtesy of Nina Laidlaw.
We have recently provided genetic and developmental evidence that the brown adipocytes of interscapular fat and those of white fat depots are fundamentally different.8 We hypothesize that the discrete brown adipocyte depots, epitomized by interscapular fat, which develop precociously during prenatal development function, principally and perhaps almost exclusively in the maintenance of body temperature, whereas those in white fat depots, which are not observed until 15–20 days of age in the mouse, function predominantly in the regulation of energy balance. Determining the origins of these genetic and developmentally distinct brown adipocytes promises to be an important research goal during the next decade of obesity research. Recent studies on the developmental origins of interscapular brown adipocytes indicate an association with a mesenchymal cell related to cells of the muscle lineage.40 It also appears from studies described above that the brown adipocytes in the white fat depots will have a different developmental origin.
Conclusion
The maintenance of a healthy body composition is dependent on a balance between energy intake and expenditure. Although mechanisms controlling energy intake are rapidly advancing, in both humans and experimental animals, those contributing to stimulation of energy expenditure have lagged behind. The problem is serious, and its resolution is important. We need to know whether or not there are well-defined thermogenic mechanisms that respond to a positive energy balance to bring it into balance. It may be that the physical and nutrient environment during human evolution did not evolve such a thermogenic mechanism because physical activity was more than sufficient to maintain energy balance. In fact, the more serious problem of human existence was the avoidance of a negative energy balance, which is consistent with an evolutionary strategy that led to a metabolism to maximize food intake and deposition of fat mass. In this presentation, we describe thermogenic mechanisms for the regulation of body fat in the mouse that have emerged from genetic manipulation of the mouse. These studies suggest that it may be possible to induce thermogenic mechanisms that could lead to energy balance. The most promising mechanisms for their application to human physiology lie in the potential for induction of brown adipocytes in human white fat depots. If this induction could be achieved, thermogenesis could be stimulated with selective β3-adrenergic agonists to reduce obesity.
Acknowledgments
Starting with Anders Jacobsson, a graduate student from the Wenner-Gren Institute in Stockholm, who came to my laboratory in 1983 with an antibody against UCP1 to work on cloning of Ucp1 cDNA, I wish to thank the large number of post-doctoral associates and collaborators who have worked with me defining the structure and function of the Ucp1 gene and its role in thermogenesis and body weight regulation. I also acknowledge the financial support through R01 HD08431 from the NIH throughout this period.
Footnotes
Conflict of interest: The authors have declared no financial interests.
References
- 1.Schwartz MW, Woods SC, Seeley RJ, Barsh GS, Baskin DG, Leibel RL. Is the energy homeostasis system inherently biased toward weight gain? Diabetes. 2003;52:232–238. doi: 10.2337/diabetes.52.2.232. [DOI] [PubMed] [Google Scholar]
- 2.Rothwell NJ, Stock MJ. A role for brown adipose tissue in diet-induced thermogenesis. Nature. 1979;281:31–35. doi: 10.1038/281031a0. [DOI] [PubMed] [Google Scholar]
- 3.Cannon B, Nedergaard J. Brown adipose tissue: function and physiological significance. Physiol Rev. 2004;84:277–359. doi: 10.1152/physrev.00015.2003. [DOI] [PubMed] [Google Scholar]
- 4.Trayhurn P, Thurlby PL, James WPT. Thermogenic defect in pre-obese ob/ob mice. Nature. 1977;266:60–62. doi: 10.1038/266060a0. [DOI] [PubMed] [Google Scholar]
- 5.Himms-Hagen J, Desautels M. A mitochondrial defect in brown adipose tissue of the obese (ob/ob) mouse: reduced binding of purine nucleotides and a failure to respond to cold by an increase in binding. Biochem Biophys Res Commun. 1978;83:628–634. doi: 10.1016/0006-291x(78)91036-7. [DOI] [PubMed] [Google Scholar]
- 6.Liu X, Rossmeisl M, McClaine J, Riachi M, Harper ME, Kozak LP. Paradoxical resistance to diet-induced obesity in UCP1-deficient mice. J Clin Invest. 2003;111:399–407. doi: 10.1172/JCI15737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coulter AA, Bearden CM, Liu X, Koza RA, Kozak LP. Dietary fat interacts with QTLs controlling induction of Pgc-1 alpha and Ucp1 during conversion of white to brown fat. Physiol Genomics. 2003;14:139–147. doi: 10.1152/physiolgenomics.00057.2003. [DOI] [PubMed] [Google Scholar]
- 8.Xue B, Rim JS, Hogan JC, Coulter AA, Koza RA, Kozak LP. Genetic variability affects the development of brown adipocytes in white fat but not in interscapular brown fat. J Lipid Res. 2007;48:41–51. doi: 10.1194/jlr.M600287-JLR200. [DOI] [PubMed] [Google Scholar]
- 9.Ukropec J, Anunciado RP, Ravussin Y, Hulver MW, Kozak LP. UCP1-independent thermogenesis in white adipose tissue of cold-acclimated Ucp1−/− mice. J Biol Chem. 2006;281:31894–31908. doi: 10.1074/jbc.M606114200. [DOI] [PubMed] [Google Scholar]
- 10.Hofmann WE, Liu X, Bearden CM, Harper ME, Kozak LP. Effects of genetic background on thermoregulation and fatty acid-induced uncoupling of mitochondria in UCP1-deficient mice. J Biol Chem. 2001;276:12460–12465. doi: 10.1074/jbc.M100466200. [DOI] [PubMed] [Google Scholar]
- 11.Ukropec J, Anunciado RV, Ravussin Y, Kozak LP. Leptin is required for uncoupling protein-1-independent thermogenesis during cold stress. Endocrinology. 2006;147:2468–2480. doi: 10.1210/en.2005-1216. [DOI] [PubMed] [Google Scholar]
- 12.Hirsch J. Obesity. Some heat but not enough light. Nature. 1997;387:27–28. doi: 10.1038/387027a0. [DOI] [PubMed] [Google Scholar]
- 13.Kontani Y, Wang Y, Kimura K, Inokuma KI, Saito M, Suzuki-Miura T, et al. UCP1 deficiency increases susceptibility to diet-induced obesity with age. Aging Cell. 2005;4:147–155. doi: 10.1111/j.1474-9726.2005.00157.x. [DOI] [PubMed] [Google Scholar]
- 14.Brown LJ, Koza RA, Everett C, Reitman ML, Marshall L, Fahien LA, et al. Normal thyroid thermogenesis but reduced viability and adiposity in mice lacking the mitochondrial glycerol phosphate dehydrogenase. J Biol Chem. 2002;277:32892–32898. doi: 10.1074/jbc.M202408200. [DOI] [PubMed] [Google Scholar]
- 15.Lardy H, Su CY, Kneer N, Wielgus S. Dehydroepiandosterone induces enzymes that permit thermogenesis and decrease metabolic efficiency. In: Lardy H, Stratman F, editors. Hormones, thermogenesis, and obesity. Elsevier; New York: 1989. pp. 415–426. [Google Scholar]
- 16.Kopecky J, Clarke G, Enerback S, Spiegelman B, Kozak LP. Expression of the mitochondrial uncoupling protein gene from the aP2 gene promoter prevents genetic obesity. J Clin Invest. 1995;96:2914–2923. doi: 10.1172/JCI118363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kopecky J, Hodny Z, Rossmeisl M, Syrovy I, Kozak LP. Reduction of dietary obesity in aP2-Ucp transgenic mice: physiology and adipose tissue distribution. Am J Physiol. 1996;270:E768–E775. doi: 10.1152/ajpendo.1996.270.5.E768. [DOI] [PubMed] [Google Scholar]
- 18.Clapham JC, Arch JR, Chapman H, Haynes A, Lister C, Moore GB, et al. Mice overexpressing human uncoupling protein-3 in skeletal muscle are hyperphagic and lean. Nature. 2000;406:415–418. doi: 10.1038/35019082. [DOI] [PubMed] [Google Scholar]
- 19.Soloveva V, Graves RA, Rasenick MM, Spiegelman BM, Ross SR. Transgenic mice overexpressing the beta 1-adrenergic receptor in adipose tissue are resistant to obesity. Mol Endocrinol. 1997;11:27–38. doi: 10.1210/mend.11.1.9870. [DOI] [PubMed] [Google Scholar]
- 20.Cummings DE, Brandon EP, Planas JV, Motamed K, Idzerda RL, McKnight GS. Genetically lean mice result from targeted disruption of the RII beta subunit of protein kinase A. Nature. 1996;382:622–626. doi: 10.1038/382622a0. [DOI] [PubMed] [Google Scholar]
- 21.Cederberg A, Gronning LM, Ahren B, Tasken P, Carlsson P, Enerback S. FOXC2 is a winged helix gene that counteracts obesity, hypertriglceridemia, and diet-induced insulin resistance. Cell. 2001;106:563–573. doi: 10.1016/s0092-8674(01)00474-3. [DOI] [PubMed] [Google Scholar]
- 22.Rosen ED, Walkey CJ, Puigserver P, Spiegelman BM. Transcriptional regulation of adipogenesis. Genes Dev. 2000;14:1293–1307. [PubMed] [Google Scholar]
- 23.Tontonoz P, Hu E, Spiegelman BM. Stimulation of adipogenesis in fibroblasts by PPARg2, a lipid-activated transcription factor. Cell. 1994;79:1147–1156. doi: 10.1016/0092-8674(94)90006-x. [DOI] [PubMed] [Google Scholar]
- 24.Sears IB, MacGinnitie MA, Kovacs LG, Graves RA. Differentiation-dependent expression of the brown adipocyte uncoupling protein gene: regulation by peroxisome proliferator-activated receptor gamma. Mol Cell Biol. 1996;16:3410–3419. doi: 10.1128/mcb.16.7.3410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Puigserver P, Wu Z, Park CW, Graves R, Wright M, Spiegelman BM. A cold-inducible coactivator of nuclear receptors linked to adaptive thermogenesis. Cell. 1998;92:829–839. doi: 10.1016/s0092-8674(00)81410-5. [DOI] [PubMed] [Google Scholar]
- 26.Kozak UC, Kopecky J, Teisinger J, Enerback S, Boyer B, Kozak LP. An upstream enhancer regulating brown-fat-specific expression of the mitochondrial uncoupling protein gene. Mol Cell Biol. 1994;14:59–67. doi: 10.1128/mcb.14.1.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cao W, Daniel KW, Robidoux J, Puigserver P, Medvedev AV, Bai X, et al. p38 mitogen-activated protein kinase is the central regulator of cyclic AMP-dependent transcription of the brown fat uncoupling protein 1 gene. Mol Cell Biol. 2004;24:3057–3067. doi: 10.1128/MCB.24.7.3057-3067.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Graves RA, Tontonoz P, Spiegelman BM. Analysis of a tissue-specific enhancer: ARF6 regulates adipogenic gene expression. Mol Cell Biol. 1992;12:1202–1208. doi: 10.1128/mcb.12.3.1202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Barak Y, Nelson MC, Ong ES, Jones YZ, Ruiz-Lozano P, Chien KR, et al. PPAR gamma is required for placental, cardiac, and adipose tissue development. Mol Cell. 1999;4:585–595. doi: 10.1016/s1097-2765(00)80209-9. [DOI] [PubMed] [Google Scholar]
- 30.Linhart HG, Ishimura-Oka K, DeMayo F, Kibe T, Repka D, Poindexter B, et al. C/EBPalpha is required for differentiation of white, but not brown, adipose tissue. Proc Natl Acad Sci USA. 2001;98:12532–12537. doi: 10.1073/pnas.211416898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Tanaka T, Yoshida N, Kishimoto T, Akira S. Defective adipocyte differentiation in mice lacking the C/EBPb and C/EPBd gene. EMBO J. 1997;16:7432–7443. doi: 10.1093/emboj/16.24.7432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cassard-Doulcier AM, Larose M, Matamala JC, Champigny O, Bouillaud F, Ricquier D. In vitro interactions between nuclear proteins and uncoupling protein gene promoter reveal several putative transactivating factors including Ets1, retinoid X receptor, thyroid hormone receptor, and a CACCC box-binding protein. J Biol Chem. 1994;269:24335–24342. [PubMed] [Google Scholar]
- 33.Silva JE. The multiple contributions of thyroid hormone to heat production. J Clin Invest. 2001;108:35–37. doi: 10.1172/JCI13397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hansen JB, Jorgensen C, Petersen RK, Hallenborg P, De Matteis R, Boye HA, et al. Retinoblastoma protein functions as a molecular switch determining white versus brown adipocyte differentiation. Proc Natl Acad Sci USA. 2004;101:4112–4117. doi: 10.1073/pnas.0301964101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Seale P, Kajimura S, Yang W, Chin S, Rohas LM, Uldry M, et al. Transcriptional control of brown fat determination by PRDM16. Cell Metab. 2007;6:38–54. doi: 10.1016/j.cmet.2007.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Guerra C, Koza RA, Yamashita H, Walsh K, Kozak LP. Emergence of brown adipocytes in white fat in mice is under genetic control. Effects on body weight and adiposity. J Clin Invest. 1998;102:412–420. doi: 10.1172/JCI3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Koza RA, Hohmann SM, Guerra C, Rossmeisl M, Kozak LP. Synergistic gene interactions control the induction of the mitochondrial uncoupling protein (Ucp1) gene in white fat tissue. J Biol Chem. 2000;275:34486–34492. doi: 10.1074/jbc.M002136200. [DOI] [PubMed] [Google Scholar]
- 38.Xue B, Coulter A, Rim JS, Koza RA, Kozak LP. Transcriptional synergy and the regulation of Ucp1 during brown adipocyte induction in white fat depots. Mol Cell Biol. 2005;25:8311–8322. doi: 10.1128/MCB.25.18.8311-8322.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xue B, Rim JS, Hogan JC, Coulter AA, Koza RA, Kozak LP. Genetic variability affects the development of brown adipocytes in white fat but not in interscapular brown fat. J Lipid Res. 2007;48(1):41–51. doi: 10.1194/jlr.M600287-JLR200. [DOI] [PubMed] [Google Scholar]
- 40.Timmons JA, Wennmalm K, Larsson O, Walden TB, Lassmann T, Petrovic N, et al. Myogenic gene expression signature establishes that brown and white adipocytes originate from distinct cell lineages. Proc Natl Acad Sci USA. 2007;104:4401–4406. doi: 10.1073/pnas.0610615104. [DOI] [PMC free article] [PubMed] [Google Scholar]