

Abstract
Ventricular trabeculation and compaction are two of the many essential steps for generating a functionally competent ventricular wall. A significant reduction in trabeculation is usually associated with ventricular compact zone deficiencies (hypoplastic wall), which commonly leads to embryonic heart failure and early embryonic lethality. In contrast, hypertrabeculation and lack of ventricular wall compaction (noncompaction) are closely related defects in cardiac embryogenesis associated with left ventricular noncompaction (LVNC), a genetically heterogenous disorder. Here we summarize our recent findings through the analyses of several genetically engineered mouse models that have defects in cardiac trabeculation and compaction. Our data indicate that cellular growth and differentiation signaling pathways are keys in these ventricular morphogenetic events.
During mammalian embryonic heart development, the ventricles undergo a series of morphogenesis [1-3]. Ventricular trabeculation and compaction are two of the many essential steps for generating a functionally competent ventricular wall [4]. Simplistically, development of the ventricular wall has 4 distinct stages; Stage 1, formation of single cell layered myocardium at an early developmental stage: Following induction via adjacent endoderm, lateral mesoderm gives rise to an early tubular heart. The heart at this stage is composed of one cell layer of myocardium and one cell layer of endocardium lining the lumen [1, 5]; Stage 2, formation of a trabeculated and compact myocardium at early midgestation stage: As the myocardium thickens, cardiomyocytes along the inner wall form sheet-like protrusions into the lumen to give rise to trabecular myocardium, while the outside layer of myocardium becomes organized into compact myocardium. Ventricular trabeculation has been suggested to facilitate oxygen and nutrient exchange and to enhance force generation to match the increasing blood flow in the developing embryos [1, 4]; Stage 3, myocardial compaction at late midgestation stage: As development proceeds, the trabecular myocardium becomes compacted towards the myocardial wall and contributes to forming a thicker, compact ventricular wall. The majority of trabeculae have become compacted after E14.5 in mouse embryos [4, 6]; Stage 4, Formation of a mature and multilayered spiral myocardium during late fetal and neonatal stage [2, 7]. Following the formation of primitive trabecular ridges (about E9.5 in mouse embryo, start of stage 2), the myocardium undergoes extensive expansion by either recruiting cardiomyocytes from myocardial wall into the trabecular ridges or via cellular proliferation within the trabecular cardiomyocytes. In support of the cellular recruitment mechanism, proliferative activity is consistently higher within the compact myocardium as there is a gradient of decreasing proliferation and increased differentiation from the outside of the heart towards the lumen and trabeculae side [8-11]. This balance of proliferation and differentiation is critical to the formation of a functionally competent ventricular wall. Fate mapping experiments using retroviral tagging experiments using β-galactosidase in developing chick hearts provided important insights into the interplay between cellular proliferation and ventricular wall formation [12, 13]. Upon myogenic differentiation, single labeled cardiomyocytes give rise to transmural and cone-shaped growth units. This pattern of growth is closely associated with the formation of trabecular myocardium [14, 15].
A significant reduction in trabeculation (stage 2 above) is usually associated with ventricular compact zone deficiencies (hypoplastic wall), which commonly leads to embryonic heart failure and early embryonic lethality. In contrast, hypertrabeculation and lack of ventricular wall compaction (noncompaction) are closely related defects in cardiac embryogenesis associated with left ventricular noncompaction (LVNC). Although LVNC has been considered rare, recent surveys indicate that it is much more common than previously thought and a considerable number of patients survive into the adult ages [16]. Indeed, LVNC can even be considered a third form of cardiomyopathy in addition to dilated and hypertrophic cardiomyopathies [17, 18]. LVNC is clinically defined as a cardiomyopathy characterized by excessively thickened ventricular trabeculae and deep intertrabecular recesses based on echocardiogram diagnosis. It appears that LVNC is a genetically heterogenous disorder [19, 20]. However, the etiology and pathogenesis of LVNC are largely unknown and reflect the lack of understanding of the underlying molecular and cellular mechanism(s) responsible for regulating ventricular trabeculation and compaction. Recently, several genetically engineered mouse models exhibiting various aspects of LVNC have been generated [21-23]. The FKBP12-deficient mouse line is one of them [24].
FKBP12 is a ubiquitously expressed 12kDa cytoplasmic protein and belongs to the immunophilin family [25, 26]. FKBP12 has been shown to be associated with multiple intracellular protein complexes, including BMP/activin/TGFβ type-I receptors [27] and Ca2+-release channels, such as inositol trisphosphate receptor (IP3R) and ryanodine receptor (RyR) [28-31]. FKBP12-deficient mice die between embryonic day 14.5 (E14.5) and birth due to severe cardiac defects including a characteristic increase in the number and thickness of ventricular trabeculae, deep inter-trabecular recesses, lack of compaction, thin ventricular wall, and a prominent ventricular septal defect (VSD) (Figure 1) [24]. These cardiac defects are typical for LVNC. Therefore, the FKBP12-deficient mouse is a valuable model for studying the underlying mechanisms regulating ventricular trabeculation, compaction, and LVNC.
Figure 1.

Systemic FKBP12-null mice develop hypertrabeculated and noncompacted hearts, similar to NLVM in patients. (A-D) Cardiac histology of FKBP12-null hearts at E14.5 (B) and E18.5 (D) as compared to wildtype littermates at E14.5 (A) and E18.5 (C). Arrows point the characteristic excessive and thickened null ventricular trabeculae, which is accompanied by VSDs. (E & F) Echocardiographic images acquired from the 4-chamber view of a 3-month old patient with NLVM (F) and an age-matched unaffected patient (E). This image demonstrates excessive trabeculation in the posterior-lateral left ventricle wall (red arrows), which is very similar to that seen in FKBP12-deficient hearts. LV: left ventricle; RV: right ventricle; LA: left atrium; RA: right atrium; vs: ventricular septum.
Previous studies on the morphogenetic process of ventricular trabeculation and compaction have so far relied heavily upon histological descriptions, based on either light or scanning electron microscopic analyses [32-35]. These conventional approaches are less reliable in analyzing and documenting cardiac defects in trabeculation and/or compaction in mutant mice because of difficulties in quantification. Inconsistencies using these tools have led to confusion in describing hypertrabeculation and noncompaction phenotypes. In order to systematically quantify and compare the dynamic processes of trabecular myocardial formation and compaction, we developed a dual florescence imaging technique that is capable of quantifying the thickness of trabecular ridges and sheets, the overall thickness of the compact wall, and the ratio of trabecular myocardium to compact wall thickness. This methodology is based on confocal imaging of hearts immunostained with florescence labeled anti-CD31 (PCAM, labeled with Alexa Fluor 488) and MF-20 (myosin heavy chain, labeled with Alexa Fluor 647) antibodies. Anti-CD31 immune reactivity highlights endothelial cells, including the endocardium lining the trabecular myocardium. MF-20 immune reactivity labels the myocardium. These immunostained hearts are serially cryosectioned (25 μm) in a four-chamber orientation. Sections with a similar orientation plane are used to generate a series of consecutive laser scanned images (Z series) can be obtained by imaging serial confocal planes. This imaging system allows a precise visualization of the architecture of the developing ventricular wall and the overall distribution and orientation of myocardial and endocardial cells, and distinguishes the endothelial cells from the myocardial cells for measurement of the thickness of trabecular ridges and sheets, and the compact wall. In addition, these digitized serial images can be further processed for 3 dimensional reconstruction using Voxx software (Volume Rendering Program for 3D Microscopy)[36]. Most significantly, this quantitative imaging analysis methodology is capable of unbiased statistical testing and is able to unambiguously identify even subtle defects.
Figure 2 illustrates the use of this approach to analyze abnormal ventricular trabeculation and compaction in FKBP12-deficient hearts at E10.5 to E14.5. These images show that each trabecular myocardial sheet is wrapped by a thin endothelial cell layer. Remarkably, in addition to the dynamic process of trabecular growth, these images demonstrate for the first time the dramatic “layer-by-layer” process of normal ventricular compaction (arrows in figure 2A-d: arrows in red indicate the layers having significant amount of newly formed capillaries, highlighted by the CD31 positive endocardium endothelium; arrows in blue indicate the layers having much less endocardium endothelium). In the FKBP12 mutant heart, this compaction process is defective. By tracing the trabeculae and compact walls, we are able to calculate the average thickness and overall areas for trabecular myocardium and compact myocardium (Figure 2B). Significant differences in cardiac trabeculation and compaction between wild type and FKBP12-deficient heart can be seen as early as E11.5. Interestingly, as development proceeds, the ratio of trabecular myocardium thickness to compact wall thickness is reversed in FKBP12-deficient hearts when compare to that in wild-type hearts. This analysis allows us to confirm that FKBP12-deficient mice are abnormal in both the processes of trabeculation and compaction.
Figure 2.
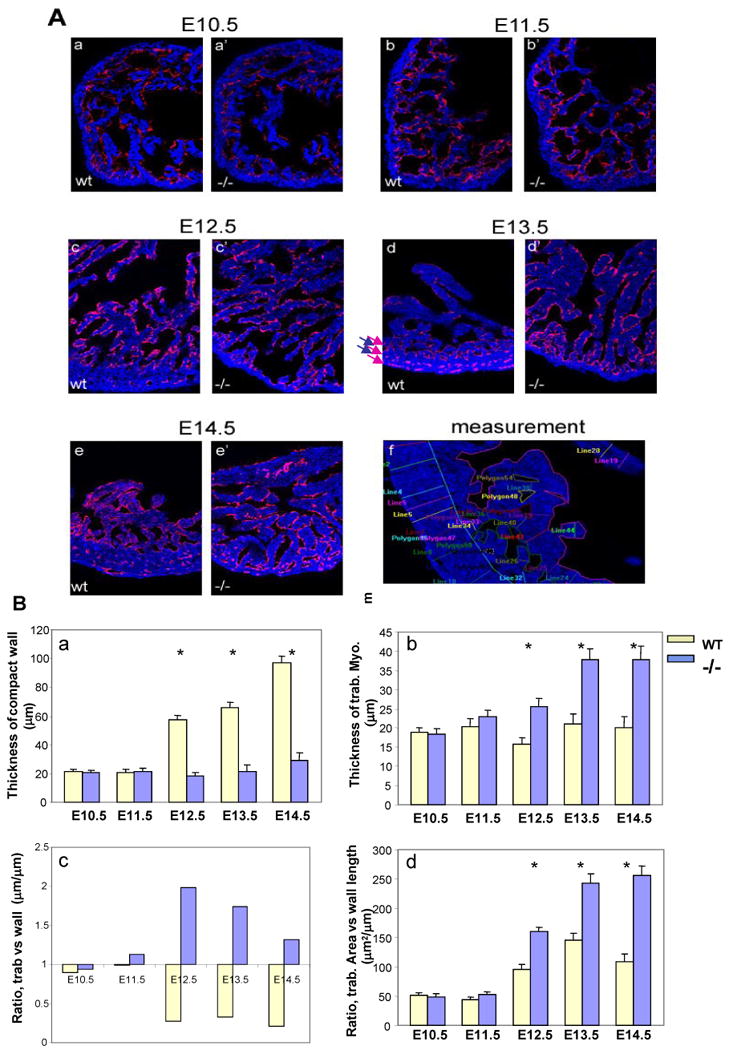
Confocal image analysis of trabecular and compact myocardium. (A) Dual florescence merged images acquired from a confocal microscope. All images (200X) are from single laser layer scanning (un-stacked). CD31 positive (in red) cells are representing endocardium, while MF20 (in blue) cells are myocardium. Cardiac trabeculation and compact can be visually and quantitatively compared via this imaging analysis. (f) Represents an example of measurement for the quantitative comparison. In addition, these images demonstrate that the endocardial endothelial cells form a thin endothelial layer extending up and around each trabecular sheet in both the wild type and FKBP12 null heart. This suggests that the physical contact between endocardium and myocardium is normal in the FKBP12 null heart, whereas the intercellular molecular signaling is likely disrupted. (B) Comparison of thickness of wall (a) and trabecular sheet (b), ratio of the thickness between trabecular sheet and compact wall (c), and ratio of overall trabecular area versus the length of compact wall (d). *P<0.01.
Importantly, Bmp10 was dramatically up-regulated in FKBP12-null hearts, which was initially discovered during our gene profiling analysis, and was later further confirmed via Northern blot, RT-PCR, and in situ hybridization. Bmp10 is a peptide growth factor that belongs to the TGF-β superfamily [37, 38]. The spatial and temporal expression pattern of Bmp10 in mouse embryogenesis was assessed using whole mount and section in situ hybridization (Figure 3). During cardiac development, Bmp10 is expressed transiently in the ventricular trabecular myocardium from E9.0 to E13.5, a critical time span when cardiac development shifts from patterning to growth and chamber maturation. By E16.5-E18.5, Bmp10 was only detectable in atria, and then only in right atria in postnatal hearts. Interestingly, the up-regulation of Bmp10 expression in myocardium is associated with another hypertrabeculation mouse model, Nkx2.5-myocardial specific knockout mice [39], suggesting that Bmp10 is a key morphogenetic growth factor involved in the regulation of cardiac trabeculation and/or compaction.
Figure 3.
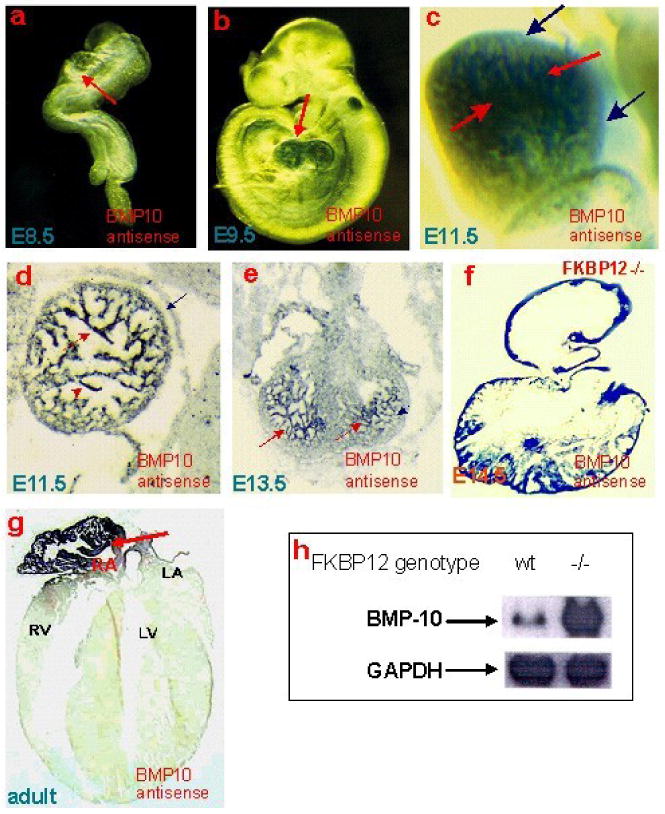
Bmp10 expression is present in only the trabecular myocardium in mouse embryonic heart, whereas it is restricted to right atrium in adult hearts. (a-c) Whole mount and section (d-g) in situ analysis. Northern analysis revealed that Bmp10 expression is significantly over-expressed in FKBP12-null hearts when compared to wild-type littermates (h).
To determine whether up-regulated Bmp10 expression would directly impact trabeculation and/or compaction in the developing myocardium, we used the human atrial natriuretic factor (hANF) promoter to drive exogenous Bmp10 expression in mouse embryonic hearts. No viable hANF-Bmp10 mice were found. In situ hybridization demonstrated a significant up-regulation of Bmp10 in hANF-Bmp10 hearts. Histological examination showed that the eight highest expressing hANF-Bmp10 positive hearts all exhibited hypertrabeculation and noncompaction (Figure 4) [39]. These data demonstrate that overexpression of Bmp10 alone is sufficient to cause cardiac hypertrabeculation and noncompaction as seen in the FKBP12-deficient hearts.
Figure 4.
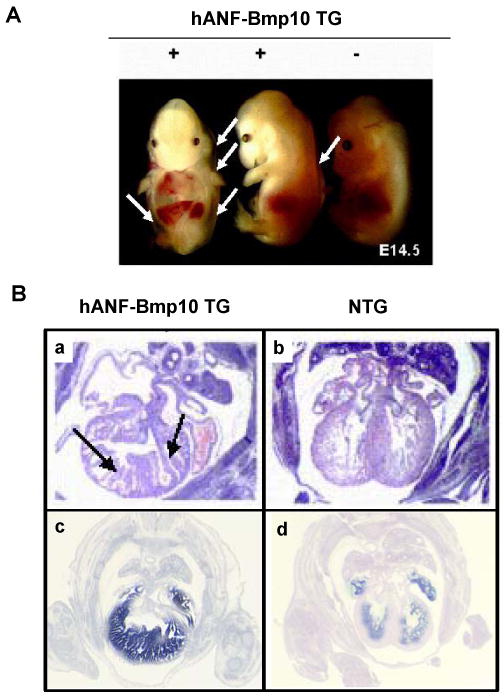
Transgenic over-expression of Bmp10 in developing heart leads to excessive trabeculation and severe heart failure. A) Gross morphology of hANF-BMP10 embryos. White arrows indicate body wall edema. B) Cardiac histology (a & b) and BMP10 in situ hybridization confirm BMP10 expression in transgenic heart (c & d). Black arrows point the abnormal trabeculae.
Bmp10-deficient mice were generated to analyze its role in ventricular development [37]. Bmp10-deficient embryos appear normal at E8.5-E9.0, suggesting that Bmp10 is not required for initial cardiac patterning and cardiac looping. At E9.0-E9.5, cardiogenesis appears arrested and null embryos die in utero at E10.5 due to poorly developed hearts. These mutant embryos display cardiac dysgenesis with profound hypoplastic ventricular walls and an absence of ventricular trabeculae. Using immunohistochemical staining and confocal microscopy, we analyzed formation of the primitive trabecular ridge and the endocardial structure in Bmp10-deficient hearts. As shown in Figure 5, using Flk-1 (endothelial receptor for VEGF) as an endocardial marker (green) and MF20 as a myocardial marker (blue), we have observed that the developing endocardium is in normal proximity to the ventricular wall and the primitive trabecular ridges are actually formed in mutant hearts. This finding indicates that Bmp10 is not critical to the initiation of cardiac trabeculation, but is essential for subsequent growth. Consistent with this observation, there was a marked reduction in proliferation (using 3H-thymidine to monitor DNA synthesis) in Bmp10-deficient cardiomyocytes [37].
Figure 5.
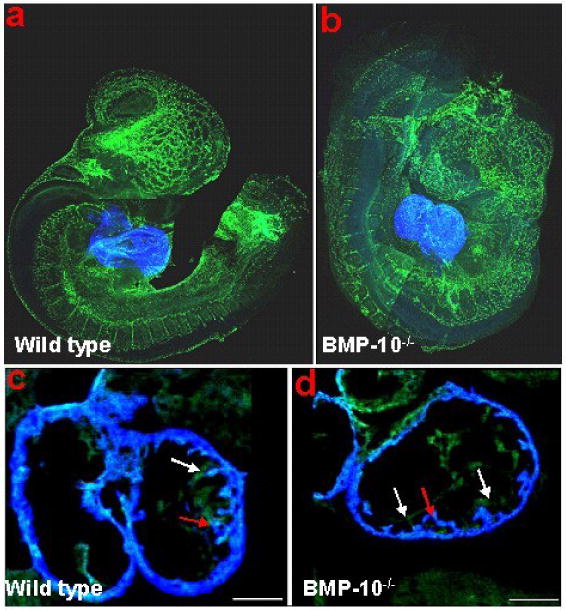
Whole-mount immunostaining and confocal analysis of control (A & C) and Bmp10-deficient embryos (B & D) at E9.25. The Bmp10-deficient heart displays a much thinner wall. However, the primitive trabecular ridges are formed and the endocardium is in normal proximity to the myocardium. Red arrows: trabecular ridge; while arrows: endocardium.
p57kip2 is a critical negative cell cycle regulator [40]. Interestingly, p57kip2 expression is first detectable in the developing mouse heart at E10.5 and is restricted to the ventricular trabecular myocardium [41]. Therefore, p57kip2 is considered a key negative regulator involved in cardiac cell cycle exit within the developing ventricular trabeculae during chamber maturation. Immunohistochemistry staining reveals that p57kip2 is up-regulated and ectopically expressed throughout the ventricular wall in Bmp10-null hearts at E9.5 compared to wild type littermate controls (Figure 6). Significantly, FKBP12-deficient hearts (E13.5), which have elevated Bmp10 expression and an over-production of trabeculae, exhibit significantly lower p57kip2 levels in trabecular myocardium when compared to control littermate (Figure 6). Using the 3H-thymidine incorporation assay, there is a significant enhancement of proliferative activity in FKBP12-deficient trabecular myocardium, while there is no obvious difference in compact myocardial wall as compared to normal controls (Figure 7). Consistent with the increase in proliferative activity of trabecular myocardium, the process of cellular differentiation and maturation of cardiomyocytes in trabecular myocardium is also defective in the FKBP12-deficient hearts. In normal myocardium at E9.5, the trabecular cells have a more advanced state of cellular differentiation, as reflected by a more mature and organized sarcomere with a higher density of Z-lines/bodies, while cardiomyocytes in the compact wall have significantly less Z-lines/bodies [42, 43]. By E13.5, both the trabecular and compact myocardium will have normally matured and have organized sarcomeres. In FKBP12-deficient embryonic hearts (E13.5), the organization of sarcomeres in trabecular cardiomyocytes are not yet established when compared to normal littermate control (Figure 8). This suggests that the normal balance of proliferation and differentiation is altered in FKBP12-deficient myocardium (i.e. higher proliferative activity and a lower differentiation state).
Figure 6.

Distribution and expression of p57kip2 in BMP-10-deficient and FKBP12-deficient hearts using immunohistochemistry staining. At E9.5, p57kip2 expression was undetectable in wild type heart (a), but was ectopically present in the BMP-10-deficient heart (b & c) under identical staining conditions. The expression of p57kip2 was abundant in E13.5 ventricular trabecular myocardium of wild type heart (d), but was significantly down-regulated in the E13.5 FKBP12-deficient trabecular myocardium (e). Arrows indicate areas of positive staining.
Figure 7.

Comparison of 3H-thymidine labeling index of FKBP12-deficient heart versus littermate control heart (E14.5). Boxed areas in panels A to F highlight the ventricular trabeculae. FKBP12-deficient heart has a significantly higher labeling index, suggesting a higher proliferation activity in the FKBP12-deficient heart. (A & B) 3H-thymidine labeled heart; (C & D) Phase contrast of the identical ventricular region shown in A and B; (E & F) Hoechst staining to show the nucleus of the identical region of A and B; (G) Statistic comparison of the cardiac 3H-thymidine labeling index in the trabecular myocardium. There is no significant difference in compact wall.
Figure 8.

Comparison of myofibrillogenesis in FKBP12-deficient and wild type ventricular trabecular myocardium (E13.5). Frozen cardiac sections were stained with anti-α-actinin monoclonal antibody for Z-line (FITC labeled) and were counter-stained with anti-F-actin (rhodamine labeled). Confocal images were acquired using a Zeiss LSM 510 laser microscope. (A) Wild type trabecular myocardium contains well organized sarcomeres (white arrows). (B) There are no mature sarcomeres in FKBP12-deficient trabecular myocardium. In addition, instead of an elongated cellular morphology in wild type, most of FKBP12-deficient cardiomyocytes appear to be rounded, an indication of under-differentiation.
Taken together, these data indicate that hypertrabeculation is likely the result of altered regulation in cell proliferation, differentiation, and maturation during ventricular wall formation. In addition, Bmp10 is an important regulator for this spatial-temporally controlled cell cycle activity in cardiac trabeculation via its either direct or indirect regulation of p57kip2. However, it is not entirely clear whether noncompaction is a result of hypertrabeculation or an independent pathological event. Our recent study analyzing the Smad7-deficient mouse heart indicates that noncompaction of myocardium can occur without an overall increase in cell proliferative activity in trabecular myocardium [44], suggesting that the failure of myocardial compaction is likely an independent pathological event. Interestingly, Smad7 is an important negative regulator for the BMP/TGFβ mediated signaling pathway [45, 46]. Ablation of Smad7 leads to elevated levels of activated Smad1/5/8, but not Smad2/3, in the developing endocardial endothelial cells [44], suggesting that the cardiac endothelium plays an important role in ventricular myocardial compaction, likely via BMP-mediated signaling. Future studies will address the molecular mechanism by which FKBP12 regulates the expression of Bmp10. It is also important to determine the spatial and temporal regulation of FKBP12 activity and BMP10 expression in LVNC patients. The knowledge gaining from these studies will help to understand the etiology of LVNC in patients.
Acknowledgments
The study was support in part by the National Institutes of Health HL81092 (WS), HL70259 (WS), and HL85098 (WS, MP).
References
- 1.Bartman T, Hove J. Mechanics and function in heart morphogenesis. Developmental Dynamics. 2005;233(2):373–81. doi: 10.1002/dvdy.20367. [DOI] [PubMed] [Google Scholar]
- 2.Taber LA. Mechanical aspects of cardiac development. Progress in Biophysics & Molecular Biology. 1998;69(23):237–55. doi: 10.1016/s0079-6107(98)00010-8. [DOI] [PubMed] [Google Scholar]
- 3.Moorman AF, et al. The heart-forming fields: one or multiple? Philosophical Transactions of the Royal Society of London - Series B: Biological Sciences. 2007;362(1484):1257–65. doi: 10.1098/rstb.2007.2113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sedmera D, et al. Developmental patterning of the myocardium. Anatomical Record. 2000;258(4):319–37. doi: 10.1002/(SICI)1097-0185(20000401)258:4<319::AID-AR1>3.0.CO;2-O. [DOI] [PubMed] [Google Scholar]
- 5.Brutsaert DL, Andries LJ. The endocardial endothelium. American Journal of Physiology. 1992;263(4 Pt 2):H985–1002. doi: 10.1152/ajpheart.1992.263.4.H985. [DOI] [PubMed] [Google Scholar]
- 6.Risebro CA, Riley PR. Formation of the ventricles. The scientific world journal. 2006;6:1862–80. doi: 10.1100/tsw.2006.316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mikawa T, et al. Induction and patterning of the Purkinje fibre network. Novartis Foundation Symposium. 250:142–53. discussion 153-6. [PubMed] [Google Scholar]
- 8.Icardo JM. Heart anatomy and developmental biology. Experientia. 1988;44(1112):910–9. doi: 10.1007/BF01939884. [DOI] [PubMed] [Google Scholar]
- 9.Icardo JM, Fernandez-Teran A. Morphologic study of ventricular trabeculation in the embryonic chick heart. Acta Anat (Basel) 1987;130(3):264–74. doi: 10.1159/000146455. [DOI] [PubMed] [Google Scholar]
- 10.Rumyantsev PP, Krylova MI. Ultrastructure of myofibers and cells synthesizing DNA in the developing and regenerating lymph-heart muscles. Int Rev Cytol. 1990;120:1–52. doi: 10.1016/s0074-7696(08)61598-3. [DOI] [PubMed] [Google Scholar]
- 11.Pasumarthi KB, Field LJ. Cardiomyocyte cell cycle regulation. Circulation Research. 2002;90(10):1044–54. doi: 10.1161/01.res.0000020201.44772.67. [DOI] [PubMed] [Google Scholar]
- 12.Mikawa T, et al. Clonal analysis of cardiac morphogenesis in the chicken embryo using a replication-defective retrovirus: I. Formation of the ventricular myocardium. Developmental Dynamics. 1992;193(1):11–23. doi: 10.1002/aja.1001930104. [DOI] [PubMed] [Google Scholar]
- 13.Mikawa T, Cohen-Gould L, Fischman DA. Clonal analysis of cardiac morphogenesis in the chicken embryo using a replication-defective retrovirus. III: Polyclonal origin of adjacent ventricular myocytes. Developmental Dynamics. 1992;195(2):133–41. doi: 10.1002/aja.1001950208. [DOI] [PubMed] [Google Scholar]
- 14.Gourdie RG, Kubalak S, Mikawa T. Conducting the embryonic heart: orchestrating development of specialized cardiac tissues. Trends in Cardiovascular Medicine. 1999;9(12):18–26. doi: 10.1016/s1050-1738(98)00035-8. [DOI] [PubMed] [Google Scholar]
- 15.Meilhac SM, et al. A retrospective clonal analysis of the myocardium reveals two phases of clonal growth in the developing mouse heart. Development. 2003;130(16):3877–89. doi: 10.1242/dev.00580. [DOI] [PubMed] [Google Scholar]
- 16.Weiford BC, Subbarao VD, Mulhern KM. Noncompaction of the ventricular myocardium. Circulation. 2004;109(24):2965–71. doi: 10.1161/01.CIR.0000132478.60674.D0. [DOI] [PubMed] [Google Scholar]
- 17.Pignatelli RH, et al. Clinical characterization of left ventricular noncompaction in children: a relatively common form of cardiomyopathy. Circulation. 2003;108(21):2672–8. doi: 10.1161/01.CIR.0000100664.10777.B8. [DOI] [PubMed] [Google Scholar]
- 18.Sandhu R, et al. Prevalence and characteristics of left ventricular noncompaction in a community hospital cohort of patients with systolic dysfunction. Echocardiography. 2008;25(1):8–12. doi: 10.1111/j.1540-8175.2007.00560.x. [DOI] [PubMed] [Google Scholar]
- 19.Xing Y, et al. Genetic analysis in patients with left ventricular noncompaction and evidence for genetic heterogeneity. Mol Genet Metab. 2006;88(1):71–7. doi: 10.1016/j.ymgme.2005.11.009. [DOI] [PubMed] [Google Scholar]
- 20.Klaassen S, et al. Mutations in sarcomere protein genes in left ventricular noncompaction. Circulation. 2008;117(22):2893–901. doi: 10.1161/CIRCULATIONAHA.107.746164. [DOI] [PubMed] [Google Scholar]
- 21.Shi W, et al. TACE is required for fetal murine cardiac development and modeling. Developmental Biology. 2003;261(2):371–80. doi: 10.1016/s0012-1606(03)00315-4. [DOI] [PubMed] [Google Scholar]
- 22.Lee Y, et al. Jumonji, a nuclear protein that is necessary for normal heart development. Circulation Research. 2000;86(9):932–8. doi: 10.1161/01.res.86.9.932. [DOI] [PubMed] [Google Scholar]
- 23.King T, et al. Expression of Peg1 (Mest) in the developing mouse heart: involvement in trabeculation. Developmental Dynamics. 2002;225(2):212–5. doi: 10.1002/dvdy.10142. [DOI] [PubMed] [Google Scholar]
- 24.Shou W, et al. Cardiac defects and altered ryanodine receptor function in mice lacking FKBP12. Nature. 1998;391(6666):489–92. doi: 10.1038/35146. [DOI] [PubMed] [Google Scholar]
- 25.Bierer BE, et al. Two distinct signal transmission pathways in T lymphocytes are inhibited by complexes formed between an immunophilin and either FK506 or rapamycin. Proceedings of the National Academy of Sciences of the United States of America. 1990;87(23):9231–5. doi: 10.1073/pnas.87.23.9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Schreiber SL, Crabtree GR. Immunophilins, ligands, and the control of signal transduction. Harvey Lectures. 1995;91:99–114. [PubMed] [Google Scholar]
- 27.Wang T, et al. The immunophilin FKBP12 functions as a common inhibitor of the TGF beta family type I receptors. Cell. 1996;86(3):435–44. doi: 10.1016/s0092-8674(00)80116-6. [DOI] [PubMed] [Google Scholar]
- 28.Timerman AP, et al. The calcium release channel of sarcoplasmic reticulum is modulated by FK-506-binding protein. Dissociation and reconstitution of FKBP-12 to the calcium release channel of skeletal muscle sarcoplasmic reticulum. Journal of Biological Chemistry. 1993;268(31):22992–9. [PubMed] [Google Scholar]
- 29.Jayaraman T, et al. FK506 binding protein associated with the calcium release channel (ryanodine receptor) Journal of Biological Chemistry. 1992;267(14):9474–7. [PubMed] [Google Scholar]
- 30.Cameron AM, et al. Calcineurin associated with the inositol 1,4,5-trisphosphate receptor-FKBP12 complex modulates Ca2+ flux. Cell. 1995;83(3):463–72. doi: 10.1016/0092-8674(95)90124-8. [DOI] [PubMed] [Google Scholar]
- 31.Cameron AM, et al. FKBP12 binds the inositol 1,4,5-trisphosphate receptor at leucine-proline (1400-1401) and anchors calcineurin to this FK506-like domain. Journal of Biological Chemistry. 1997;272(44):27582–8. doi: 10.1074/jbc.272.44.27582. [DOI] [PubMed] [Google Scholar]
- 32.Moorman A, et al. Development of the heart: (1) formation of the cardiac chambers and arterial trunks. Heart. 2003;89(7):806–14. doi: 10.1136/heart.89.7.806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Anderson RH, et al. Development of the heart: (2) Septation of the atriums and ventricles. Heart. 2003;89(8):949–58. doi: 10.1136/heart.89.8.949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anderson RH, et al. Development of the heart: (3) formation of the ventricular outflow tracts, arterial valves, and intrapericardial arterial trunks. Heart. 2003;89(9):1110–8. doi: 10.1136/heart.89.9.1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Cook AC, Yates RW, Anderson RH. Normal and abnormal fetal cardiac anatomy. Prenat Diagn. 2004;24(13):1032–48. doi: 10.1002/pd.1061. [DOI] [PubMed] [Google Scholar]
- 36.Clendenon JL, et al. Voxx: a PC-based, near real-time volume rendering system for biological microscopy. Am J Physiol Cell Physiol. 2002;282(1):C213–8. doi: 10.1152/ajpcell.2002.282.1.C213. [DOI] [PubMed] [Google Scholar]
- 37.Chen H, et al. BMP10 is essential for maintaining cardiac growth during murine cardiogenesis. Development. 2004;131(9):2219–31. doi: 10.1242/dev.01094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Neuhaus H, Rosen V, Thies RS. Heart specific expression of mouse BMP-10 a novel member of the TGF-beta superfamily. Mechanisms of Development. 1999;80(2):181–4. doi: 10.1016/s0925-4773(98)00221-4. [DOI] [PubMed] [Google Scholar]
- 39.Pashmforoush M, et al. Nkx2-5 pathways and congenital heart disease; loss of ventricular myocyte lineage specification leads to progressive cardiomyopathy and complete heart block. Cell. 2004;117(3):373–86. doi: 10.1016/s0092-8674(04)00405-2. [DOI] [PubMed] [Google Scholar]
- 40.Besson A, Dowdy SF, Roberts JM. CDK inhibitors: cell cycle regulators and beyond. Developmental Cell. 2008;14(2):159–69. doi: 10.1016/j.devcel.2008.01.013. [DOI] [PubMed] [Google Scholar]
- 41.Kochilas LK, et al. p57Kip2 expression is enhanced during mid-cardiac murine development and is restricted to trabecular myocardium. Pediatric Research. 1999;45(5 Pt 1):635–42. doi: 10.1203/00006450-199905010-00004. [DOI] [PubMed] [Google Scholar]
- 42.Hagopian M, Spiro D. Derivation of the Z line in the embryonic chick heart. Journal of Cell Biology. 1970;44(3):683–7. doi: 10.1083/jcb.44.3.683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ronna KC. Myogenesis and contraction in the early embryonic heart of the rainbow trout. An electron microscopic study. Cell & Tissue Research. 1977;180(1):123–32. [PubMed] [Google Scholar]
- 44.Chen Q, et al. Smad7 is required for the development and function of heart. Journal of Biological Chemistry. 2008 doi: 10.1074/jbc.M807233200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shi Y, Massague J. Mechanisms of TGF-beta signaling from cell membrane to the nucleus. Cell. 2003;113(6):685–700. doi: 10.1016/s0092-8674(03)00432-x. [DOI] [PubMed] [Google Scholar]
- 46.Heldin CH, Miyazono K, ten Dijke P. TGF-beta signalling from cell membrane to nucleus through SMAD proteins. Nature. 1997;390(6659):465–71. doi: 10.1038/37284. [DOI] [PubMed] [Google Scholar]