

Abstract
The endoplasmic reticulum (ER) consists of tubules that are shaped by the reticulons and DP1/Yop1p, but how the tubules form an interconnected network is unknown. Here, we show that mammalian atlastins, which are dynamin-like, integral membrane GTPases, interact with the tubule-shaping proteins. The atlastins localize to the tubular ER and are required for proper network formation in vivo and in vitro. Depletion of the atlastins or overexpression of dominant-negative forms inhibits tubule interconnections. The Sey1p GTPase in S. cerevisiae is likely a functional ortholog of the atlastins; it shares the same signature motifs and membrane topology and interacts genetically and physically with the tubule-shaping proteins. Cells simultaneously lacking Sey1p and a tubule-shaping protein have ER morphology defects. These results indicate that formation of the tubular ER network depends on conserved dynamin-like GTPases. Since atlastin-1 mutations cause a common form of hereditary spastic paraplegia, we suggest ER shaping defects as a novel neuropathogenic mechanism.
Introduction
The atlastins comprise a family of highly-related, integral membrane GTPases (Zhao et al., 2001; Rismanchi et al., 2008; Zhu et al., 2003). They belong to the dynamin family of GTPases that associate with different intracellular membranes (for review, see Praefcke and McMahon, 2004). The prototypical member of this family, dynamin-1, is involved in vesicle budding from the plasma membrane during clathrin-mediated endocytosis; dynamin-related proteins are also required for the fusion and fission of mitochondria (Hoppins et al., 2007; Praefcke and McMahon, 2004). The functions of the atlastins are largely unknown. Mutations in atlastin-1 (SPG3A) are the second most frequent cause of pure hereditary spastic paraplegia (HSP), and the most common cause of early-onset HSP (for review, see Salinas et al., 2008). The cardinal feature of these disorders is progressive spasticity and weakness of the lower limbs due to a length-dependent, retrograde axonopathy of the corticospinal motor neurons. Pure forms of HSP are characterized by spastic paraplegia alone, while additional neurological features are present in complicated forms. The two other genes most frequently associated with pure HSP are spastin, a microtubule-severing ATPase, and REEP1 (Beetz et al., 2008), a member of the DP1/Yop1p protein family involved in ER tubule formation (Voeltz et al., 2006). Because atlastin-1 and REEP1 are both associated with pure HSPs, this raises the possibility that the disease is caused by ER morphology defects.
The ER is a continuous membrane system that is comprised of the inner and outer nuclear membranes as well as peripheral ER sheets and a network of interconnected tubules (for review, see Shibata et al., 2006). The ER network is very dynamic, with tubules undergoing continuous fusion and fission to generate and eliminate three-way junctions (Lee and Chen, 1988; for review, see Du et al., 2004). Some insight has been gained into how the tubular ER is generated and maintained. ER tubules are frequently pulled out from a membrane reservoir by molecular motors as they move along microtubules in mammalian cells, or along actin filaments in plant and yeast cells, or else tubules can be pulled out by their association with the tips of microtubules or actin filaments as they grow by polymerization (Prinz et al., 2000; Waterman-Storer and Salmon, 1998). However, the tubules are ultimately stabilized by cytoskeleton-independent mechanisms, since the alignment of membrane tubules with the cytoskeleton is not perfect (Terasaki et al., 1986), and actin depolymerization in yeast does not result in the disruption of the tubular network (Prinz et al., 2000). In addition, ER tubules can be generated in vitro through the fusion of small vesicles without involvement of the cytoskeleton (Dreier and Rapoport, 2000).
Two families of integral membrane proteins were recently identified which appear responsible for shaping the tubular ER (Voeltz et al., 2006). The first is the reticulons, comprising four reticulon genes in mammals (RTN1-4) and two in yeast (RTN1 and RTN2). The other family consists of the DP1/Yop1p proteins, which includes six mammalian DP1/REEP genes and the yeast ortholog YOP1. Members of both families are ubiquitously expressed in all eukaryotes and localize predominantly to the tubular ER (Voeltz et al., 2006). Their overexpression renders the mammalian ER network resistant to the rearrangement that follows microtubule depolymerization (Shibata et al., 2008), and overexpression of certain reticulon isoforms leads to long and unbranched tubules (Voeltz et al., 2006). Conversely, the lack of reticulons and Yop1p in yeast results in the loss of tubular ER (Voeltz et al., 2006). In addition, purified yeast Rtn1p and Yop1p deform reconstituted proteoliposomes into tubules (Hu et al., 2008). Together, these results indicate that the reticulons and DP1/Yop1p are both necessary and sufficient for ER tubule formation.
The reticulons and DP1/Yop1p do not share primary sequence homology, but both have a conserved domain of ∼200 amino acid residues, comprising two long hydrophobic segments that sit in the membrane as hairpins (Voeltz et al., 2006). This domain is also responsible for oligomerization of these proteins (Shibata et al., 2008). The reticulons and DP1/Yop1p probably deform the lipid bilayer into tubules through “wedging” and “scaffolding” mechanisms (Hu et al., 2008; Shibata et al., 2008). The double-hairpin transmembrane segments may form a wedge that occupies more space in the outer leaflet of the bilayer than in the inner leaflet, thus generating high curvature that is characteristic of cross-sections of tubules. Oligomerization of the reticulons and DP1/Yop1p may generate arc-like structures that could serve as scaffolds along the tubules. Although the reticulons and DP1/Yop1p appear to be the minimal components required for ER tubule formation, in vivo there are likely additional factors that determine the shape of the ER network. These components might be involved in forming branched interconnections or in modulating ER morphology during the cell cycle or in response to external signals.
Here, we demonstrate that the atlastin GTPases interact with the ER tubule-shaping proteins, the reticulons and DP1, and provide evidence supporting a role for atlastins in the formation of an interconnected tubular network. We propose the GTPase Sey1p as a functional ortholog of the atlastins in S. cerevisiae, since it has similar signature motifs and membrane topology, interacts genetically and physically with the tubule-shaping proteins, and is involved in ER network formation. These results indicate that the morphology of the tubular ER network depends on a ubiquitous class of dynamin-like, membrane-bound GTPases and suggest that pure HSP may be caused by defects in ER morphology.
Results
Atlastins interact with the reticulons and DP1
An early indication that atlastins play a role in ER network formation came from experiments seeking to identify interaction partners of the atlastins. We used detergent extracts from rat brain, where the atlastin-1 (ATL1) isoform is abundantly expressed (Zhu et al., 2003). The extracts were incubated with anti-peptide antibodies against ATL1, and immunoprecipitated proteins were analyzed by SDS-PAGE and Coomassie staining (Figure 1A, lane 2). A small number of co-precipitated proteins were absent from samples in which the antibodies were preincubated with the antigenic peptide (Figure 1A, lane 1). Mass spectrometry identified reticulon 4a (Rtn4a) as an ATL1-interacting protein. This protein was also found in similar pull-down experiments using extracts from rat spinal cord (Figure 1A, lane 4).
Figure 1. Interaction of ATL1 with Rtn4a in neuronal cells.
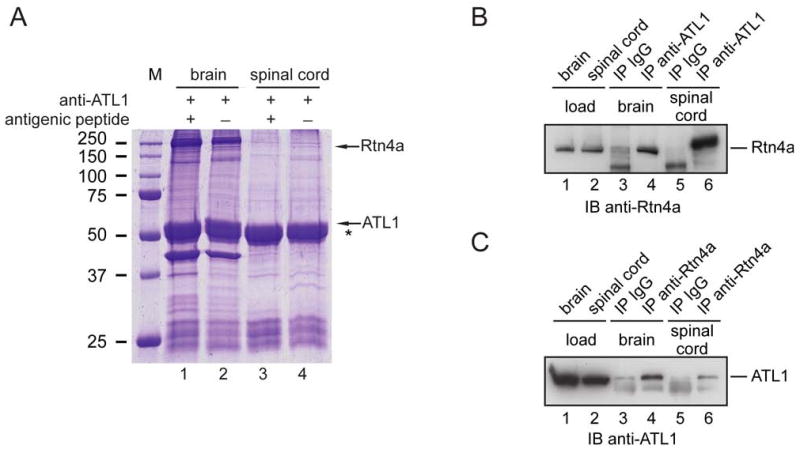
(A) Detergent extracts from mouse brain or spinal cord were incubated with peptide-specific antibodies to ATL1. Where indicated, antibodies were preincubated with the antigenic peptide. Immunoprecipitated proteins were analyzed by SDS-PAGE and Coomassie staining. Rtn4a was identified by mass spectrometry (5 distinct peptides covering 74 of the 1163 amino acids). An asterisk (*) indicates the position of the IgG heavy chain. M, molecular mass standards (in kDa).
(B) Proteins immunoprecipitated (IP) with ATL1 antibodies or control IgG were analyzed by immunoblotting (IB) with Rtn4a antibodies. Lanes 1 and 2 (loads) show 10% of the starting material used for immunoprecipitation.
(C) Proteins immunoprecipitated with Rtn4a antibodies or control IgG were immunoblotted with ATL1 antibodies. Lanes 1 and 2 (loads) contain 10% of the starting material used for the immunoprecipitations.
To confirm the interaction of ATL1 and Rtn4a, brain or spinal cord extracts were incubated with ATL1 antibodies, and precipitated proteins were resolved by SDS-PAGE. Immunoblot analysis demonstrated that Rtn4a was co-precipitated (Figure 1B, lanes 4 and 6; non-immune IgG controls are shown in lanes 3 and 5), in contrast to two abundant control membrane proteins (Figure S1). Conversely, antibodies to Rtn4a co-precipitated ATL1 (Figure 1C, lanes 4 and 6; controls in lanes 3 and 5). Together, these experiments show that ATL1 interacts specifically with Rtn4a, an ER tubule-shaping protein.
To test whether ATL1 also interacts with other tubule-shaping proteins, we performed co-transfection experiments. Myc-tagged ATL1 (Myc-ATL1) and hemagglutinin (HA)-tagged reticulon 3c (HA-Rtn3c) were expressed either individually or together in COS-7 cells, and cell extracts were subjected to immunoprecipitation. In cells expressing both proteins, HA-antibodies precipitated a fraction of Myc-ATL1 (Figure 2A, lane 19) and, conversely, Myc-antibodies precipitated a fraction of HA-Rtn3c (Figure 2A, lane 21). No co-precipitation was seen with control anti-FLAG antibodies (Figure 2A, lane 17) or with extracts from cells expressing only one of the two proteins (Figure 2A, lanes 1-14). Two unrelated, endogenous ER membrane proteins (TRAPalpha and calnexin) were not co-precipitated with either Myc- or HA-antibodies (Figures 2A, bottom panel, and S2A).
Figure 2. ATL1 interacts with the reticulons and DP1.
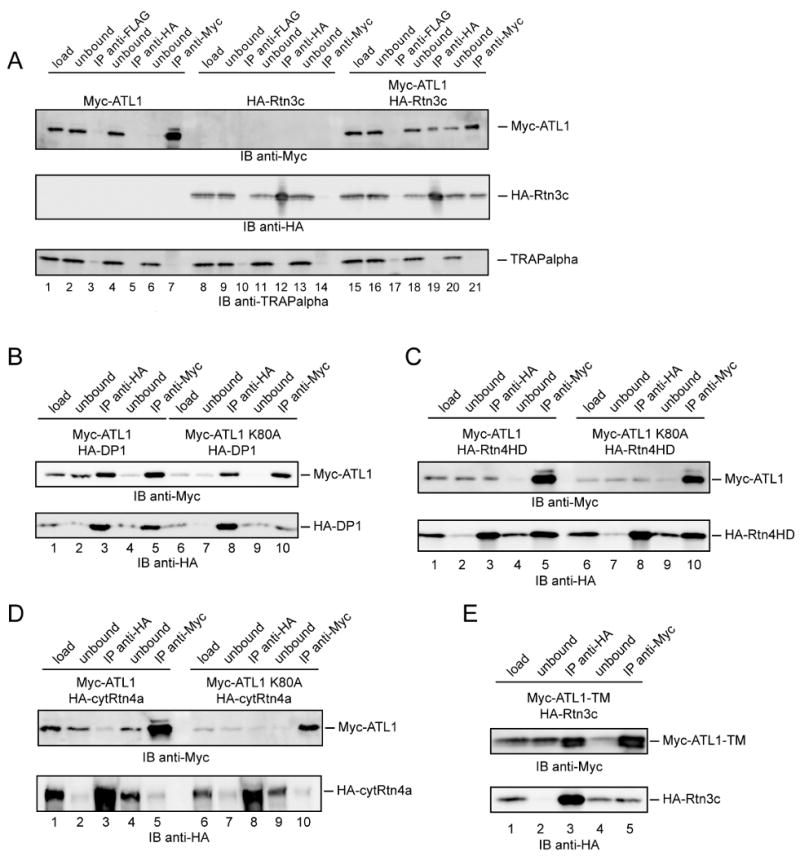
(A) Myc-ATL1 and HA-Rtn3c were expressed either individually or together in COS-7 cells. Digitonin-extracts were used for immunoprecipitation (IP) with anti-HA, anti-Myc, or anti-FLAG antibodies. Ten percent of the starting material (load) and of the material not bound to the antibodies (unbound), as well as the precipitates were analyzed by SDS-PAGE and immunoblotting (IB) with anti-HA or anti-Myc antibodies. As a control, the blot was also probed with antibodies to the integral ER membrane protein TRAPalpha.
(B) Myc-ATL1 or Myc-ATL1 K80A was expressed together with HA-tagged DP1 (HA-DP1) in COS-7 cells. Immunoprecipitation was performed as in (A).
(C) As in (B), but with COS-7 cells expressing Myc-ATL1 and the HA-tagged reticulon homology domain of Rtn4a (HA-Rtn4HD).
(D) As in (B), but with cells expressing Myc-ATL1 and the HA-tagged N-terminal, cytoplasmic segment of Rtn4a (HA-cytRtn4a).
(E) As in (B), but with cells expressing the transmembrane domain of ATL1 (Myc-ATL1TM) and HA-Rtn3c.
Moreover, when Myc-ATL1 or HA-Rtn3c was co-expressed with tagged control ER membrane proteins, no interaction was detected (Figures S2B-2D). These experiments indicate that ATL1 interacts specifically with Rtn3c, another isoform of the reticulon family. Similar experiments demonstrated that ATL1 also interacts with DP1, a member of the other tubule-shaping protein family (Figure 2B, lanes 3 and 5). Furthermore, a Myc-tagged version of another atlastin isoform, ATL2, also interacted with HA-Rtn3c and HA-DP1 (Figures S3A and S3B). Together, these experiments suggest that the atlastins interact generally with ER tubule-shaping proteins. Although these proteins could interact indirectly through another protein, it seems more likely that they bind directly to one another because the interactions can be observed when both partners are highly expressed.
Next, we tested whether the atlastins interact with the reticulons through the conserved reticulon homology domain. This was suggested by the fact that Rtn3c contains a relatively small cytoplasmic N-terminus preceding this domain. Indeed, when the cytoplasmic N-terminus was deleted from Rtn4a, leaving only the reticulon homology domain (Rtn4HD), the interaction with ATL1 was maintained (Figure 2C, lanes 3 and 5). Conversely, the cytoplasmic domain of Rtn4a (cytRtn4a) did not interact with ATL1 (Figure 2D, lanes 3 and 5). Because the reticulon homology domain harbors the two hairpin membrane anchors, these data suggest that the reticulon-atlastin interactions occur within the membrane. Consistent with this proposal, DP1 has only short hydrophilic segments outside of its hydrophobic double-hairpin structure, yet can interact with the atlastins.
An intra-membrane interaction is supported by the observation that the C-terminal domains of ATL1-3 (ATL1-TM, ATL2-TM, and ATL3-TM), which each harbor two closely spaced transmembrane segments, are sufficient to bind Rtn3c or DP1 (Figure 2E, lanes 3 and 5, and Figures S4A and S4B). The large N-terminal cytoplasmic domains of the atlastins (cytATLs) did not bind Rtn3c or DP1 (Figure S5). Thus, the two transmembrane segments of the atlastins likely interact with the double-hairpin membrane anchors of the reticulons and DP1. Consistent with this model, the nucleotide state of the atlastins did not affect the interaction; an ATL1 mutant defective in GTP binding (ATL1 K80A; corresponding to K44A in dynamin-1) associated equally as well as wild-type ATL1 with DP1 (Figure 2B, lanes 8 and 10), Rtn4HD (Figure 2C, lanes 8 and 10), and Rtn3c (data not shown).
Atlastins localize to and affect the tubular ER
Because the reticulons and DP1 localize specifically to the tubular ER and are absent from ER sheet structures (Voeltz et al., 2006), we tested whether the atlastins have a similar localization. Indeed, when Myc-ATL1was expressed at low or moderate levels in COS-7 cells, it was mostly found in the tubular ER (Figure 3A). Very little protein was seen in peripheral ER sheets, but in some cells low levels were observed in the nuclear envelope, unlike the reticulons or DP1, which were totally excluded (Voeltz et al., 2006). Similar results were obtained with Myc-ATL2 and Myc-ATL3 (Figure S6). The Myc-tagged C-terminal domains of the atlastins (ATL-TMs) also localized to the tubular ER (shown for Myc-ATL1-TM in Figure S7), indicating that the two closely spaced membrane anchors are sufficient for targeting. Our data indicate that all atlastins localize predominantly to the ER, consistent with their ER retrieval motif at the C-terminus (Rismanchi et al., 2008), although in neuronal cells, atlastin-1 was also found in the cis-Golgi and vesicular tubular clusters (Zhu et al., 2003; Zhu et al., 2006).
Figure 3. ATL1 localizes to the tubular ER and alters ER morphology upon overexpression.
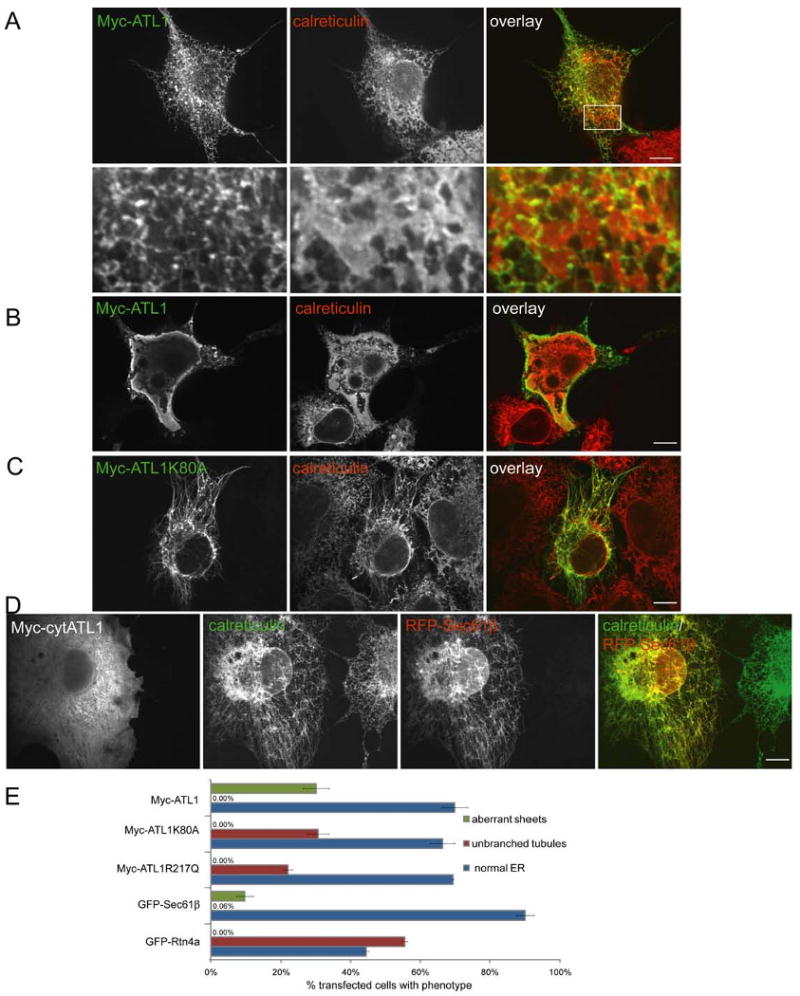
(A) Myc-ATL1 was expressed in COS-7 cells. Its localization was identified with anti-Myc antibodies (green) and compared to that of an endogenous luminal ER protein, calreticulin (red) using indirect immunofluorescence and confocal microscopy. Lower panels show an enlargement of the boxed region centered on ER sheets. Bar, 10 μm.
(B) As in (A), but a cell is shown with particularly high expression of Myc-ATL1. ER sheets are aberrant.
(C) As in (A), but with cells expressing the GTP-binding mutant Myc-ATL1 K80A. Shown is a cell with an unbranched ER tubule phenotype.
(D) As in (A), but with cells expressing a Myc-ATL1 construct that contains only the N-terminal cytoplasmic domain (Myc-cytATL1). Cells also expressed a RFP-tagged version of the ER protein Sec61β and were then stained with Myc- and calreticulin-antibodies. Shown is a cell with an unbranched ER tubule phenotype.
(E) Myc-ATL1, Myc-ATL1 K80A, Myc-ATL1 R217Q, GFP-Sec61β, or GFP-Rtn4a were expressed in COS-7 cells and the percentage of cells with normal ER, with aberrant sheet-like ER structures, or with unbranched tubules was determined. Means ± standard errors were calculated from three different samples of each transfection (200-500 cells per sample).
A role for atlastins in shaping the tubular ER network is suggested by overexpression experiments. When wild-type ATL1 was highly expressed, ER morphology was drastically altered, with aberrant sheet-like structures in essentially all highly expressing cells (Figure 3B, 3E, and S8). In contrast, overexpression of the GTP-binding mutant of ATL1, Myc-ATL1 K80A, led to long, unbranched ER tubules (Figures 3C and 3E); again, the phenotype correlated with the expression level (Figure S8). As in wild type cells, the ER tubules did not co-align completely with microtubules (data not shown). Long, unbranched ER tubules were also observed when a Myc-tagged version of an HSP-causing ATL1 mutant (Myc-ATL1 R217Q) was overexpressed (data not shown). Although the unbranched ER phenotype was previously observed in HeLa cells (Rismanchi et al., 2008), moderate alterations of Golgi morphology were also seen. The latter changes are probably indirect because only slight, if any, alterations of the Golgi were seen in similar experiments in COS-7 cells (Figure S9A). Mitochondrial morphology and distribution were also unchanged (Figure S9B).
The unbranched ER phenotype is similar to that seen upon overexpression of a GFP fusion of Rtn4a (Voeltz et al., 2006) (Figure 3E). The unbranched tubules generated by Myc-ATL1 K80A contain endogenous reticulons (data not shown). In addition, when Myc-ATL1 K80A was co-expressed with GFP-Rtn3c, which by itself does not perturb the tubular ER network, both proteins co-localized to long, unbranched ER tubules (Figure S10). The unbranched tubule phenotype was also observed when the N-terminal cytoplasmic domain of ATL1 or of the K80A mutant was overexpressed (Figures 3D and S11A), as well as when a Myc-tagged version of the membrane-embedded domain of an atlastin was highly expressed (shown for Myc-ATL3-TM in Figure S11B). These results indicate that the atlastins can modulate the structure of the ER network. The experiments with full-length ATL1 further suggest that GTP binding is required to form interconnections of the tubular ER.
The depletion of atlastins also caused defects in the branching of ER tubules. When siRNA was used to deplete atlastin-2 and atlastin-3, the predominant isoforms in HeLa cells (Rismanchi et al., 2008) (Figure 4A), long, unbranched ER tubules were observed (Figure 4B and 4C). The Golgi was somewhat fragmented, as reported previously (Rismanchi et al., 2008), but the morphologies of mitochondria and lysosomes were unaltered (Figure S12). The unbranched ER morphology is reminiscent of that generated upon overexpression of ATL mutants (Figures 3C and 3D). The mutants thus seem to have a dominant-negative effect. Taken together, these results indicate that both overexpression and depletion of the atlastins inhibit the formation of interconnections between ER tubules.
Figure 4. Depletion of the atlastins leads to unbranched ER tubules.
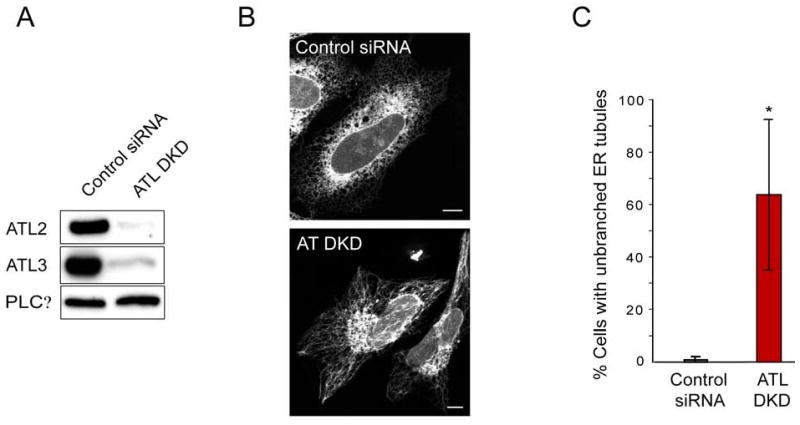
(A) HeLa cells were transfected with siRNA oligonucleotides directed against ATL2 and ATL3 (ATL DKD) or with control siRNA. ATL2 and ATL3 protein levels were then determined by immunoblotting. PLCγ levels were monitored as a loading control.
(B) HeLa cells depleted or not depleted for ATL2 and ATL3 were transfected with GFP-Sec61β and visualized by fluorescence confocal microscopy. Bar, 10 μm.
(C) The number of cells displaying the “unbranched tubule” phenotype (lower panel in (B)) was quantitated as a percentage relative to the total number of cells (means ± SD from three different experiments, with 100 cells per experiment). P<0.02.
Atlastins function in ER network formation in vitro
To test whether the atlastins have a direct role in ER network formation, we used a previously established in vitro assay (Dreier and Rapoport, 2000). When a membrane fraction derived from Xenopus laevis egg extracts is incubated at room temperature in the presence of GTP, small vesicles fuse to generate an elaborate ER network (Dreier and Rapoport, 2000; see also Figure S13A). When these membrane vesicles were preincubated with affinity-purified antibodies that recognize all atlastin isoforms in Xenopus (Figure S13B), network formation was severely inhibited (Figure 5). A similar effect had previously been observed with antibodies against Rtn4a (Voeltz et al., 2006). We therefore conclude that the atlastins function directly in ER network formation, perhaps explaining the GTP requirement for the in vitro assay. These data also indicate that the atlastins affect ER morphology in organisms other than mammals.
Figure 5. Atlastin antibodies inhibit ER network formation in vitro.
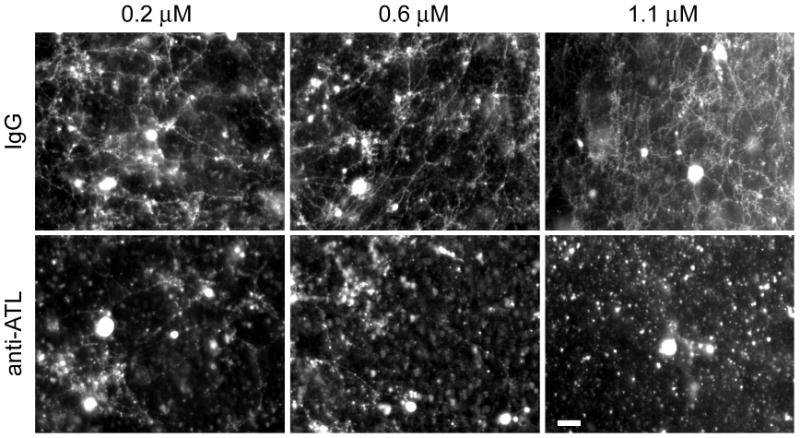
Membranes from Xenopus eggs were preincubated for 60 min with increasing concentrations of affinity-purified, pan-ATL antibodies or with control IgG. GTP was then added, and network formation was visualized by fluorescence microscopy after staining with octadecyl rhodamine. Bar, 20 μm.
A yeast GTPase is similar to the atlastins
Although atlastin homologs appear to exist in all metazoans, including Drosophila and C. elegans, no obvious sequence-related protein could be detected in S. cerevisiae. However, a yeast GTPase, Sey1p, was identified as a putative functional ortholog. While every metazoan has atlastin-related proteins, all other eukaryotes have Sey1p homologs; no organism seems to have both, consistent with the notion that these GTPases have the same function. Like the atlastins, Sey1p is predicted to have two hydrophobic transmembrane segments close to the C-terminus (Figure 6A). These likely form a hairpin structure, with only three residues on the luminal side of the ER membrane. The GTPase domain of Sey1p contains the signature motifs characteristic of dynamin GTPases (G1, G2, and G3; Figure 6B). However, the atlastins, Sey1p and its Arabidopsis ortholog “Root Hair Defective 3” (RHD3), as well as the mammalian guanylate-binding proteins (GBPs) form a distinct subclass that has a G4 motif comprising three hydrophobic residues preceding a Arg-Asp (RD) sequence (Figure 6B). Sey1p was originally identified in genetic screens for mutants that have a synthetic growth defect in conjunction with deletion of Yop1p, the yeast ortholog of the tubule-shaping protein DP1 (hence the name “synthetic enhancer of Yop1p”) (Brands and Ho, 2002). A large-scale screen for genetic interaction partners also demonstrated synthetic effects of yop1 and sey1 deletions (Schuldiner et al., 2005). In our strain background, a sey1Δyop1Δ double-deletion mutant only grew more slowly than the single deletion mutants in high-osmolarity medium (Figure S14). These genetic interactions suggest that, like the atlastins, Sey1p may function in ER network formation in yeast.
Figure 6. Sey1p, a yeast GTPase structurally similar to the atlastins.
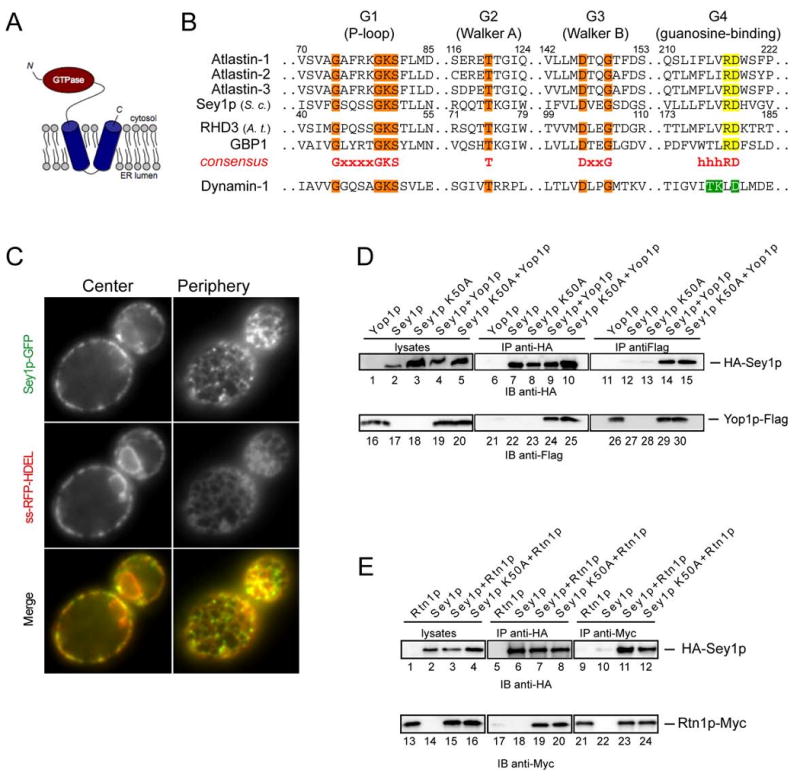
(A) Predicted membrane topology of Sey1p.
(B) Signature motifs of the GTPase domains of the Sey1p/atlastin family. G1, G2, and G3 motifs shared with dynamin-1 are shown in orange, and the divergent G4 motifs of the Sey1p/atlastin family and of dynamin-1 are shown in yellow and green, respectively. The atlastins, GBP1, and dynamin-1 are human forms, Sey1p is from S. cerevisiae (S. c.), and RHD3 is from Arabidopsis thaliana (A. t.). Residue numbers shown are for atlastin-1 and Sey1p.
(C) Sey1p-GFP was expressed under the endogenous promoter from a CEN plasmid together with RFP targeted to the ER lumen (ss-RFP-HDEL). Proteins were visualized by fluorescence microscopy, focusing either at the center or periphery of the cells. Bar, 1 μm.
(D) HA-Sey1p and Yop1p-FLAG were co-expressed individually or together under their endogenous promoters on CEN plasmids in a sey1Δyop1Δ strain. Proteins were immunoprecipitated (IP) from cell lysates using HA or FLAG antibodies, separated by SDS-PAGE, and analyzed by immunoblotting (IB) with HA or FLAG antibodies. Similar experiments were also performed with a Sey1p mutant (Sey1p K50A) that is defective in GTP binding.
(E) As in (D), except that HA-Sey1p and Myc-tagged Rtn1p (Rtn1p-Myc) were expressed individually or together in a sey1Δrtn1Δ strain, and Myc-epitope antibodies were used.
Consistent with the genetic interactions, a Sey1p-GFP fusion, expressed from a CEN plasmid under the endogenous promoter, localized to the ER (Figure 6C). The fusion protein was somewhat enriched in the cortical ER (Figure 6C), although not as much as the reticulons and Yop1p (Voeltz et al., 2006). Interestingly, the protein appeared to be concentrated in punctae along the ER tubules, sometimes at three-way junctions of tubules. When the Sey1p-GFP fusion was expressed from the chromosome, the protein also localized to the ER and showed punctate staining (data not shown), but the labeling was very weak, likely reflecting the low abundance of Sey1p (Ghaemmaghami et al., 2003).
To test whether Sey1p physically interacts with Yop1p, we used a sey1Δyop1Δ yeast strain and expressed epitope-tagged versions of both proteins under endogenous promoters on a CEN plasmid. When HA-Sey1p was precipitated with HA-antibodies, Yop1p-FLAG was co-precipitated (Figure 6D, lane 24). Conversely, when Yop1p-FLAG was precipitated with FLAG-antibodies, HA-Sey1p was co-precipitated (Figure 6D, lane 14). Thus, Sey1p and Yop1p interact with one another, as the atlastins and DP1 do in mammals. Similar results were obtained when the interaction of HA-Sey1p and Rtn1p-Myc was tested in a strain lacking endogenous Sey1p and Rtn1p (Figure 6E, lanes 11 and 19). To determine whether GTP binding to Sey1p is important for its interaction with Yop1p and Rtn1p, we generated a mutant in Sey1p (Sey1p K50A), equivalent to ATL1 K80A. The purified GTPase domain of this mutant was indeed inactive (Figure S15). The full-length Sey1p K50A mutant showed undiminished interaction with Yop1p and Rtn1p (Figure 6D, lanes 15 and 25; Figure 6E, lanes 12 and 20), indicating that, as for the atlastins, GTP binding is not required for interaction with the tubule-shaping proteins.
Together, these results indicate that Sey1p not only shares structural features and ER localization with the atlastins, but also interactions with ER tubule-shaping proteins.
Sey1p and Rtn1p/Yop1p cooperate in maintaining ER morphology
To test whether Sey1p plays a role in maintaining ER morphology, we expressed a GFP fusion to the ER protein Sec63p (Sec63-GFP) in various deletion strains. Fluorescence microcopy was used to visualize the nuclear envelope and cortical ER by focusing the microscope on the center or periphery of the cell, respectively. In yeast lacking only Sey1p the ER resembled that in wild-type cells (Figure 7A versus 6C), comparable to observations made previously for single deletions of Rtn1p or Yop1p (Voeltz et al., 2006). The sheet-like nuclear envelope appeared normal, and the reticular, cortical ER morphology was maintained, although in some cells the network consisted of fewer tubules. In cells lacking both Sey1p and Rtn1p the cortical ER was severely perturbed (Figure 7B); most cells lacked the tubular network and instead showed aberrant structures (Figure 7F). Similar results were obtained with cells lacking Sey1p and Yop1p (Figures 7C and 7F). In this case, the buds of many cells contained only patches of cortical ER, prefiguring a defect in ER inheritance. As expected, expression of wild-type Sey1p from a CEN plasmid rescued the ER morphology defects seen in the double-deletion mutants (Figures 7D and 7F). On the other hand, expression of the GTP-binding mutant Sey1p K50A did not rescue the ER morphology defect (Figures 7E and 7F). Together, these results indicate that Sey1p cooperates with Rtn1p and Yop1p to maintain the structure of the tubular ER. In addition, GTP binding by Sey1p is required for its function.
Figure 7. Sey1p and Rtn1p/Yop1p cooperate in maintaining ER morphology in S. cerevisiae.
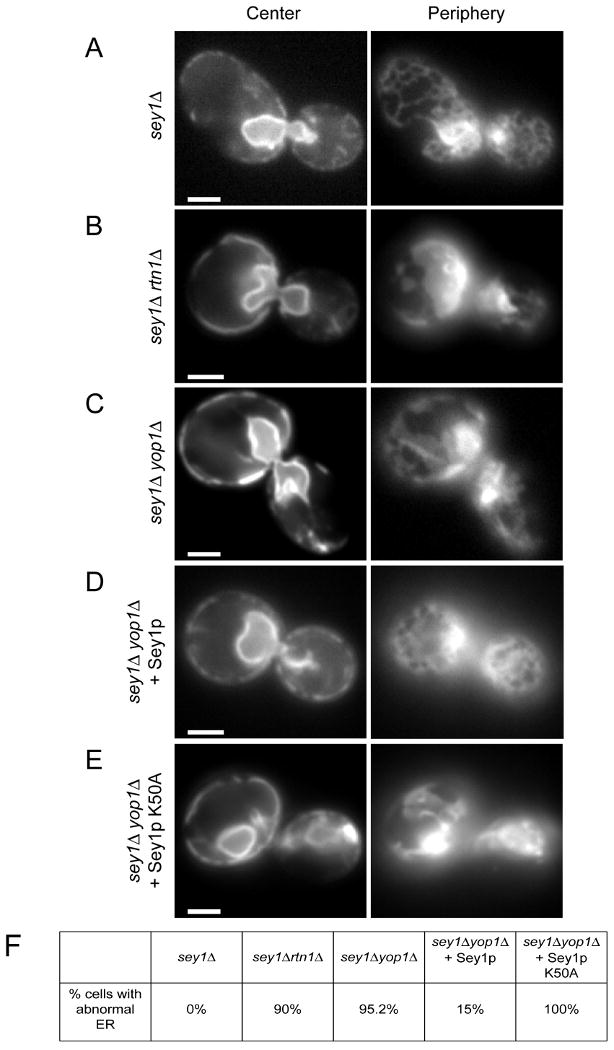
(A) A GFP-fusion of the ER protein Sec63p was expressed in cells lacking Sey1p (sey1Δ). The localization of the protein was determined by fluorescence microscopy, focusing the microscope either at the center or the periphery of the cell. Bar, 1 μm.
(B) As in (A), but with cells lacking both Sey1p and Rtn1p (sey1Δrtn1Δ).
(C) As in (A), but with cells lacking both Sey1p and Yop1p (sey1Δyop1Δ).
(D) As in (C), but with cells expressing wild-type Sey1p from a CEN plasmid.
(E) As in (C), but with cells expressing a GTP-binding mutant of Sey1p (Sey1p K50A) from a CEN plasmid.
(F) The percentage of cells with abnormal ER was determined from 20-40 cells per mutant. The small percentage of cells with abnormal ER in (D) likely reflects loss of the plasmid.
Discussion
Our results provide evidence that a class of dynamin-related, membrane-bound GTPases functions in the formation of the tubular ER network. We show that the mammalian atlastins interact with the previously identified tubule-shaping proteins of the reticulon and DP1 families, and that overexpression or depletion of the atlastins changes the morphology of the tubular ER network. Specifically, both depletion of the atlastins and overexpression of dominant-negative mutants inhibited the formation of tubule interconnections. Experiments using an established in vitro assay confirm that the atlastins play a direct role in ER network formation. We also identified a likely functional ortholog of the atlastins in yeast, Sey1p, which belongs to the same class of dynamin-related GTPases and, like the atlastins, has two closely spaced transmembrane segments at its C-terminus. Sey1p preferentially localizes to the ER, interacts genetically and physically with the tubule-shaping proteins, and cooperates with them to maintain the morphology of the tubular ER. These results indicate that membrane-bound GTPases are involved in shaping the ER network, and they have important implications for the pathogenesis of the HSPs.
Previous work had suggested that the reticulons and DP1/Yop1p proteins play a major role in generating ER tubules (Hu et al., 2008; Shibata et al., 2008; Voeltz et al., 2006). Our new data now indicate that these proteins collaborate with the atlastin/Sey1p GTPases to generate the tubular ER network. Sey1p is much less abundant than Rtn1p or Yop1p in yeast cells (∼2,300 versus 37,000 molecules) (Ghaemmaghami et al., 2003), suggesting that the reticulons and DP1/Yop1p are the main structural components that shape ER tubules. The fact that Sey1p and Rtn1p/Yop1p deletions in yeast have synergistic effects could indicate that these proteins function in parallel pathways of ER tubule formation. However, given the physical interaction of the reticulons and DP1/Yop1p with the atlastins/Sey1p, it appears more likely that they cooperate in the formation of the tubular ER network.
In principle, the atlastins and Sey1p could simply regulate the function of the reticulons and DP1/Yop1p. For example, our observation that the depletion of the atlastins results in the generation of long, unbranched ER tubules might be interpreted as excessive tubulation of ER membranes, which in turn could be caused by excessive oligomerization of the reticulons and DP1. In this scenario, the atlastin and Sey1p GTPases would be required for disassembly of these oligomers. However, this would simply that the tubule-shaping proteins and Sey1p have opposing effects on ER network formation, which does not fit with the genetic synergism seen in yeast. In addition, deletion of Sey1p in yeast does not change the mobility of Rtn1-GFP in fluorescence recovery after photobleaching experiments or the sedimentation behavior of Yop1p-FLAG in sucrose gradient centrifugation experiments (data not shown). Thus, there is currently no evidence that the atlastin/Sey1p GTPases directly regulate the tubule-shaping properties of the reticulons and DP1/Yop1p.
We therefore favor the idea that the atlastins and Sey1p directly affect branching of the ER tubules, likely by promoting their fusion or fission. This idea is supported by analogy to other members of the dynamin family of GTPases. Dynamin-1 causes membrane fission during endocytosis by forming short spirals around the neck of budding vesicles in a GTP-dependent manner (Bashkirov et al., 2008; Marks et al., 2001; Pucadyil and Schmid, 2008; Sweitzer and Hinshaw, 1998; Takei et al., 1995). While this suggests that the atlastin/Sey1p proteins are involved in fission, they are structurally more similar to the mitofusin/Fzo1p proteins. These are dynamin-like GTPases of the outer mitochondrial membrane that contain two closely spaced transmembrane segments and are implicated in the fusion of the tubular mitochondria (Chen et al., 2003; Hermann et al., 1998). A role for the atlastins in the fusion of ER tubules would also be more consistent with the observation that their absence leads to fewer three-way junctions, and the atlastins may be the best candidates to directly mediate homotypic ER fusion. Regardless of whether the atlastins function in fission or fusion, they may generate high local membrane curvature. In fact, the narrow spirals formed by dynamin-1 are thought to cause membrane stress that eventually results in fission (Bashkirov et al., 2008), and the mitofusins/Fzo1p could cause fusion if they generated tubules with exposed, strongly curved tips that are expected to be highly fusogenic (Praefcke and McMahon, 2004; Chernomordik and Kozlov, 2008). We therefore propose that all dynamin family proteins, including the atlastins and Sey1p, deform the lipid bilayer, a general property that is exploited in endocytosis, mitochondrial morphology, and -- as demonstrated here -- ER morphology.
As expected for members of the dynamin family, GTP binding is required for the function of the atlastins and Sey1p. A GTP-binding mutant of ATL1 had a dominant-negative effect on ER tubule connections. Because the atlastins form oligomers (Rismanchi et al., 2008; Zhu et al., 2003), it is likely that the mutant proteins associate with wild-type ATL1 and prevent it from properly functioning. This model could also explain the dominant-negative effect of the cytosolic domains of the atlastins.
Why might the membrane-bound atlastin/Sey1p GTPases physically interact and cooperate with the reticulons and DP1/Yop1p in ER network formation? One possibility is that the reticulons and DP1/Yop1p are responsible for shaping ER tubules, both by generating the high curvature seen in cross-section and by scaffolding the tubules. Thus, the GTPases would associate with the reticulons and DP1/Yop1p at certain points along the tubules. Following GTP-dependent conformational changes, they would further destabilize the bilayer, which in turn could lead to membrane remodeling. A similar model may apply to dynamin-1 during endocytosis, as it also interacts with the curvature-generating proteins sorting nexin 9, amphiphysin, and endophilin (Grabs et al., 1997; Ringstad et al., 1997; Soulet et al., 2005).
The interaction of the atlastins/Sey1p with the reticulons and DP1/Yop1p appears to occur within the membrane. Previous results showed that the reticulon and DP1/Yop1p domains that include the double-hairpin transmembrane segments are responsible for their propensity to form tubules in vitro (Hu et al., 2008), for their oligomerization, and for the localization of these proteins to the tubular ER (Shibata et al., 2008). We now show that these domains are also responsible for the interaction with the membrane-bound atlastin/Sey1p GTPases. These GTPases interact through their two C-terminal transmembrane segments that are predicted to form a hairpin-like structure in the membrane. Because this hairpin structure localizes the atlastins to the high-curvature ER tubules, hairpin-shaped membrane anchors might generally be used to partition proteins to high-curvature membranes. Sey1p is much less abundant than the tubule-shaping proteins in yeast, so it seems possible that the GTPase associates with only a subset of reticulon and DP1/Yop1p oligomers, consistent with its observed punctate staining along ER tubules. The GTPases could then cause fission or fusion of ER tubules at these sites, either directly or in conjunction with other proteins. The activity of the GTPases is probably tightly regulated, as too much or too little fission or fusion would be expected to cause ER morphology defects, consistent with the observed effects of atlastin overexpression and depletion.
While depletion of atlastins in mammalian cells resulted in unbranched ER tubules, the deletion of SEY1 in S. cerevisiae had a subtler phenotype. In some cells fewer tubules were seen, perhaps indicating a branching defect, but a network was still maintained. Thus, Sey1p is not essential for ER network formation, suggesting that yeast cells have additional mechanisms that promote fission and fusion of ER tubules. The situation in plants appears to be more similar to that seen with the mammalian atlastins, since mutants of the atlastin/Sey1p ortholog in Arabidopsis, RHD3, have abnormal ER morphology characterized by unbranched bundles of ER tubules (Zheng et al., 2004). RHD3 is expressed in all plant cells, but the phenotype of mutants is most pronounced in root hairs, which are ER-containing tubular processes of root epidermal cells, similar to neurites in neuronal cells (Wang et al., 1997). RHD3 mutants exhibit short and wavy root hairs (Schiefelbein and Somerville, 1990), reminiscent of the shortened axons in neurons depleted of atlastin-1 (Zhu et al., 2006). These data suggest that RHD3 is involved in the maintenance of the ER network in plants, and that mutants in RHD3 most severely affect cells with long protrusions that likely rely most stringently on proper ER network morphology.
Our results suggest that a defect in ER network formation may underlie some of the most common forms of HSP. Mutations in ATL1 cause the most frequent early-onset HSP, SPG3A. Because many mutations affect the GTPase activity of ATL1 (Zhu et al., 2006), and the overexpression of some disease-causing mutants in cultured cells results in the formation of long, unbranched ER tubules (Rismanchi et al., 2008; and this paper), it is possible that SPG3A is caused by ER morphology defects. SPG3A mutations are autosomal dominant (Zhao et al., 2001), consistent with suggestions that the mutant ATL1 proteins associate with wild-type ATL1 and perturb its function (Rismanchi et al., 2008). This idea is also consistent with our observations that the overexpression of a GTP-binding mutant of ATL1 has the same phenotype as seen upon depletion of the atlastins. Similar to explanations for why RHD3 mutations affect most prominently root hairs in plants, one might expect that ER network integrity is of particular importance for the development or maintenance of corticospinal motor neurons, whose axons can reach a length of one meter. An alternative model in which the SPG3A HSP is caused by defects in axonal vesicle transport appears less likely (Namekawa et al., 2007), because recent experiments show that vesicular traffic in tissue culture cells is not inhibited when atlastins are depleted or when mutant forms are expressed (Rismanchi et al., 2008). Because in these experiments some changes of Golgi morphology were observed, and because some atlastin-1 was localized to the cis-Golgi, it is conceivable that the atlastins also have a role in compartments other than the ER. However, the interactions of the atlastins with the ER resident reticulons and DP1 as well as their prominent role in ER morphology make it more likely that any effect on other organelles is indirect.
Two other proteins whose mutations frequently cause HSP may also affect ER network formation. The SPG4 protein spastin, mutations of which cause about 40% of pure HSP, is a microtubule-severing ATPase (for review, see Salinas et al., 2008). The longer isoform of spastin is enriched in cerebral cortex and spinal cord and localizes to the ER (Connell et al., 2008). It interacts with the atlastins through an N-terminal domain that includes a hydrophobic transmembrane segment (Sanderson et al., 2006; Evans et al., 2006). Interestingly, spastin has also been shown to interact in yeast two-hybrid assays with the reticulons (Mannan et al., 2006), providing a potential direct link to ER-shaping proteins. Mechanistically, spastin mutants could cause defects in microtubule dynamics, which in turn could destabilize ER tubules. Mutations in REEP1 (SPG31) (Beetz et al., 2008), a member of the DP1/Yop1p family of tubule-shaping proteins (Saito et al., 2004; Voeltz et al., 2006), could affect the shape of the ER tubules directly. Although REEP1 has been reported to localize to mitochondria (Züchner et al., 2006), our recent data support an ER localization (data not shown), as for all other members of the family. Since SPG3A, SPG4, and SPG31 collectively account for well over 50% of all HSP cases (Salinas et al., 2008), defects in ER network formation may be the predominant neuropathogenic mechanism underlying these disorders.
Experimental procedures
Mammalian DNA Constructs
HA-DP1, HA-Rtn3c and GFP-Rtn3c constructs were described previously (Shibata et al., 2008; Voeltz et al., 2006). HA-Rtn4HD was modified from GFP-Rtn4HD (Shibata et al., 2008) and inserted into pcDNA3.1D (Invitrogen). For HA-cytRtn4a, the region encoding amino acids 1-960 was PCR-amplified from human Rtn4a-Myc (Voeltz et al., 2006) and inserted into pcDNA3.1D (Invitrogen). GFP-Rtn4a was generated from Rtn4a-Myc in pACGFP-C1 (Clontech). Myc-ATL1, Myc-ATL1K80A, Myc-ATL1R217Q, Myc-ATL2, Myc-ATL3, Myc-ATL1-TM, Myc-ATL2-TM, Myc-ATL3-TM, Myc-cytATL1, Myc-cytATL2, and Myc-cytATL3 were described previously (Rismanchi et al., 2008). Myc-cytATL1 K80A was generated by site-directed mutagenesis from Myc-cytATL1. RFP-and GFP-Sec61β were described previously (Shibata et al., 2008).
Brain Tissue Immunoprecipitations and Mass Spectrometry
Tissue lysates were prepared from adult Sprague-Dawley rats. Dissected spinal cord and brain were homogenized in phosphate-buffered saline (PBS) with 2 mM EDTA, 1 mM EGTA, 0.5% CHAPS and protease inhibitor cocktail (Sigma-Aldrich) and centrifuged at 20,000 × g for 30 min. The supernatant was incubated with 14 μg of anti-atlastin-1 antibodies (#5409; Zhu et al., 2003) or control rabbit IgG and 30 μl of protein A agarose beads at 4°C overnight. Where indicated, the antibody was pre-incubated with immunogenic peptide (0.25 mM). Beads were washed twice in PBS with 2 mM EDTA, 1 mM EGTA and 0.5% CHAPS, and then twice in PBS with 2 mM EDTA and 1 mM EGTA. Proteins were resolved by SDS-PAGE, stained with Coomassie blue, and specific bands were excised and analyzed by MALDI-TOF mass spectrometry. In some experiments, extracts were immunoprecipitated with anti-Rtn4a antibodies. Immunoprecipitated proteins were resolved by SDS-PAGE and immunoblotted with the following antibodies: mouse monoclonal anti-TrkB (clone 47, BD Biosciences), rabbit polyclonal anti-calnexin (StressGen), or mouse monoclonal anti-Rtn4a (clone 17, BD Biosciences).
Mammalian Tissue Culture, Transfections, and Co-immunoprecipitation
Cells were maintained at 37°C with 5% CO2 in DMEM containing 10% fetal bovine serum and passaged every 2-3 days. For co-IP experiments, 60-70% confluent cells were transfected with various expression constructs using Lipofectamine 2000 (Invitrogen), and harvested 24-36 h later in HKM buffer (25 mM Hepes pH 7.4, 150 mM potassium acetate, 2 mM magnesium acetate, and protease inhibitors) containing 1% digitonin (Calbiochem). Extracts were incubated at 4°C for 1 h, and insoluble material was cleared by centrifugation for 10 min at 18,000 × g. Extracts were pre-cleared with protein G-conjugated Sepharose beads (GE Healthcare), incubated with mouse anti-FLAG (clone M2, Sigma-Aldrich), rat anti-HA (clone 3F10, Roche), or mouse anti-Myc (clone 9E10, Sigma-Aldrich) antibodies at 4°C, and protein-antibody complexes were precipitated with protein G beads. Samples were separated by SDS-PAGE and analyzed by immunoblotting. Immunoblots were imaged with a Fuji-LAS 3000 system.
Immunofluorescence and Confocal Microscopy
Indirect immunofluorescence of paraformaldehyde-fixed COS-7 and HeLa cells was described previously (Shibata et al., 2008; Rismanchi et al., 2008). Transfected cells were grown on coverslips, and immunostained using anti-HA, anti-Myc, polyclonal anti-calreticulin (Abcam) primary antibodies, and various Alexa Fluor-conjugated secondary antibodies (Invitrogen). SiRNA studies for ATL2 and ATL3 in HeLa cells were performed as described previously (Rismanchi et al., 2008), except that cells were treated for 72 h with the oligonucleotides (150 nM) and the ER was visualized after re-transfection with GFP-Sec61β 48 h after the siRNA transfection. These cells were immunostained using mouse monoclonal anti-GM130 (clone 35, BD Biosciences), anti-cytochrome c (6H2.B4, BD Biosciences) or anti-LAMP1 (H4A3, SouthernBiotech) antibodies. Images were captured on a Yokogawa spinning disk confocal on a Nikon TE2000U inverted microscope with a 100× Plan Apo NA 1.4 objective lens, a Hamamatsu ORCA ER cooled CCD camera, and MetaMorph 7.0 software or else a Zeiss LSM510 confocal microscope with a 63× 1.4 NA Plan-APOCHROMAT lens, with image acquisition using LSM510 version 3.2 SP2 software (Carl Zeiss Microimaging). Brightness and contrast were adjusted across the entire image using Adobe Photoshop.
In Vitro ER Network Formation
Membrane fractions of Xenopus eggs were prepared as described previously (Dreier and Rapoport, 2000). Network formation was carried at 25°C for 60 min in 50 mM Hepes pH 7.7, 200 mM KCl, 2.5 mM MgCl2, 250 mM sucrose, 1 mM DTT, 1 mM ATP, and 0.5 mM GTP. A pan-atlastin antibody against a region common to all Xenopus atlastins (#6944; Rismanchi et al., 2008) was affinity purified and used in the network formation test. Non-immune rabbit IgG (Sigma-Aldrich) was used as a control. Fluorescence microscopy was performed after octadecyl rhodamine staining.
S. cerevisiae Strains and Constructs
The Sey1p deletion strain (BY4741 sey1Δ∷kanMX4) was purchased from Open Biosystems. The following strains were also used: BY4741 (MATa his3Δ1 leu2Δmet15Δ ura3Δ), BY4742 (MATalpha his3Δ1 leu2Δ lys2Δ ura3Δ), JHY15 (BY4741 SEY1-GFP:kanMX4), JHY1 (BY4742yop1Δ∷HIS3MX6), JHY4 (BY4741 sey1Δ∷kanMX4 yop1Δ∷HIS3MX6), JHY6 (BY4741 sey1Δ∷kanMX4 rtn1Δ∷HIS3MX6). For endogenous level expression of Sey1p, the full coding region of Sey1p plus an N-terminal HA tag and 300 bp upstream and downstream sequences was amplified and inserted into the BamHI/PstI site of pRS315 (a LEU2/CEN plasmid). Similarly, Yop1p-FLAG and Rtn1p-Myc with their endogenous promoters were inserted into the BamHI/XbaI site of pYC2/CT (a URA3/CEN plasmid), with the original GAL promoter removed. The plasmid encoding GFP fused to the C terminus of Sey1p was constructed by replacing the coding region of Sec63p with that of Sey1p plus its endogenous promoter in the plasmid pJK59 (Prinz et al., 2000). Sey1p K50A was generated by site-directed mutagenesis. The plasmids pJK59, encoding Sec63-GFP, and YIplac201/TKC-DsRed-HDEL, encoding ss-RFP-HDEL (a gift from B. Glick), were used to visualize ER morphology as described previously (Hu et al., 2008). Yeast cells were imaged live in growth medium using an Olympus BX61 microscope, UPlanApo 100×/1.35 lens, Qimaging Retiga EX camera, and IVision version 4.0.5 software.
Immunoprecipitation from Yeast Cells
Yeast membranes were prepared as described (Hu et al., 2008) and solubilized in HKM buffer with 1% digitonin for 1 h at 4°C. After removal of insoluble material by centrifugation, extracts were incubated with anti-HA affinity matrix (clone 3F10, Roche), anti-FLAG affinity gel (clone M2, Sigma-Aldrich), or anti-Myc antibody affinity gel (clone 9E10, Sigma-Aldrich) at 4°C for 4 h, and then washed with HKM buffer containing 0.2% digitonin. Bound proteins were eluted with SDS buffer, separated by SDS-PAGE, and immunoblotted with various antibodies.
Supplementary Material
Figure S1. Endogenous ATL1 does not interact with control membrane proteins.
Detergent extracts from mouse brain or spinal cord were incubated with ATL1 antibodies or control IgG. Precipitated proteins were then analyzed by immunoblotting (IB) with Rtn4a, calnexin, or TrkB antibodies. Lanes 1 and 2 (loads) show 10% of the starting material used for immunoprecipitation.
Figure S2. Overexpressed ATL1 and Rtn3c do not interact with control membrane proteins.
(A) Myc-ATL1 and HA-Rtn3c were expressed either individually or together in COS-7 cells. Digitonin-extracts were used for immunoprecipitation (IP) with anti-HA, anti-Myc, or anti-FLAG antibodies. Ten percent of the starting material (load) and of the material not bound to the antibodies (unbound), as well as the precipitates were analyzed by SDS-PAGE and immunoblotting (IB) with anti-HA or anti-Myc antibodies. As a control, the blot was also probed with antibodies to the integral ER membrane protein calnexin.
(B) Co-immunoprecipitation as in (A), but with cells expressing Myc-ATL1 and the HA-tagged integral membrane protein Sec61alpha (HA-Sec61alpha).
(C) Co-immunoprecipitation as in (A), but with cells expressing Myc-ATL1 and the FLAG-tagged integral membrane protein CLIMP63 (FLAG-CLIMP63). The blot was probed with anti-FLAG, instead of HA-antibodies.
(D) Co-immunoprecipitation as in (A), but with cells expressing Myc-Sec61beta and HA-Rtn3c.
Figure S3. ATL2 interacts with Rtn3c and DP1.
(A) Myc-tagged ATL2 (Myc-ATL2) was expressed together with HA-tagged Rtn3c (HA-Rtn3c) in COS-7 cells. Digitonin-solubilized extracts were used for immunoprecipitation (IP) with anti-HA or anti-Myc antibodies. Ten percent of the starting material (load) and of the material not bound to the antibodies (unbound), as well as the material bound to the antibodies were analyzed by SDS-PAGE and immunoblotting (IB) with anti-HA or anti-Myc antibodies.
(B) As in (A), but with HA-tagged DP1 (HA-DP1) instead of HA-Rtn3c.
Figure S4. Interactions of the transmembrane segments of ATL2 and ATL3.
(A) The Myc-tagged C-terminal domains of ATL2 or ATL3 harboring the transmembrane domains (Myc-ATL2-TM or Myc-ATL3-TM) were expressed together with HA-tagged Rtn3c (HA-Rtn3c) in COS-7 cells. Digitonin-solubilized extracts were used for immunoprecipitation (IP) with anti-HA or anti-Myc antibodies. Ten percent of the starting material (load) and of the material not bound to the antibodies (unbound), as well as the material bound to the antibodies were analyzed by SDS-PAGE and immunoblotting (IB) with anti-HA or anti-Myc antibodies.
(B) As in (A), but with Myc-ATL3-TM and HA-DP1.
Figure S5. The cytoplasmic domains of the atlastins do not interact with Rtn3c or DP1.
(A) Myc-tagged versions of the large N-terminal cytoplasmic domains of ATL1, ATL2, or ATL3 (Myc-cytATL1, Myc-cytATL2, or Myc-cytATL3) and HA-tagged Rtn3c (HA-Rtn3c) were co-expressed in COS-7 cells. Digitonin-solubilized extracts were used for immunoprecipitation (IP) with anti-HA or anti-Myc antibodies. Ten percent of the starting material (load), the material not bound to the antibodies (unbound), and the material bound to the antibodies were analyzed by SDS-PAGE and immunoblotting (IB) with anti-HA or anti-Myc antibodies.
(B) As in (A), but with HA-tagged DP1 (HA-DP1) instead of HA-Rtn3c.
Figure S6. ATL2 and ATL3 localize to the tubular ER.
(A) Myc-ATL2 (green) was expressed in COS-7 cells. Its localization was identified with anti-Myc antibodies and compared to that of an endogenous luminal ER protein, calreticulin (red) using indirect immunofluorescence and confocal microscopy. The lower panels show an enlargement of the boxed region centered on ER sheets. Scale bar, 10 μm.
(B) As in (A), but with Myc-ATL3 instead of Myc-ATL2.
Figure S7. The membrane-anchoring domain of the atlastins localizes to the tubular ER network.
Myc-tagged C-terminal region of ATL1 harboring the transmembrane domains (Myc-ATL1-TM) (green) was expressed in COS-7 cells. Its localization was revealed with anti-Myc antibodies and compared to that of an endogenous luminal ER protein, calreticulin (red), using indirect immunofluorescence and confocal microscopy. The lower panels show an enlargement of the boxed region centered on ER sheets. Scale bar, 10 μm.
Figure S8. ER morphology defects correlate with the expression level of ATL1 and ATL1 K80A.
Myc-ATL1 or Myc-ATL1 K80A was expressed in COS-7 cells. The cells were stained with Myc-antibodies and imaged by fluorescence microscopy. For each cell, the ER morphology was visually scored, and the average fluorescence intensity, defined as the total fluorescence in the ER divided by its area, was measured using MetaMorph software. The cells were divided into five groups according to their expression levels. Shown is the percentage of cells with normal and abnormal ER in each group (means ± standard errors from three different transfections; 100 cells per sample).
Figure S9. Expression of ATL1 K80A changes the morphology of the ER, but not that of other organelles.
(A) GFP-Sec61beta or Myc-ATL1 K80A was expressed in COS-7 cells. The localization of the Golgi marker giantin was analyzed using indirect immunofluorescence and confocal microscopy. Bar, 10 μm.
(B) As in (A), but the cells were stained with antibodies to the mitochondrial marker cytochrome c.
Figure S10. Co-localization of a GTP-binding mutant of ATL1 with Rtn3c.
Myc-ATL1 K80A was co-expressed with GFP-Rtn3c in COS-7 cells. The proteins were visualized by indirect immunofluorescence using Myc-antibodies and by GFP fluorescence. Bar, 10 μm.
Figure S11. ER morphology changes caused by overexpression of atlastin mutants.
(A) A Myc-tagged version of the N-terminal, cytoplasmic domain of the GTP-binding mutant of ATL1 (Myc-cytATL1 K80A) was co-expressed in COS-7 cells with an RFP-tagged version of the ER protein Sec61β. The cells were stained with Myc-epitope and calreticulin antibodies and visualized by indirect immunofluorescence and confocal microscopy. Shown is a cell with an unbranched ER tubule phenotype. Bar, 10 μm.
(B) A Myc-tagged version of the membrane-anchoring domain of ATL3 (Myc-ATL3-TM) was expressed in COS-7 cells. The cells were stained with Myc-epitope and calreticulin antibodies and visualized by indirect immunofluorescence and confocal microscopy. Note the unbranched tubular ER morphology.
Figure S12. Effect of atlastin depletion on the morphology of organelles.
(A) HeLa cells were transfected with siRNA oligonucleotides directed against ATL2 and ATL3 (ATL DKD) or else with an equivalent concentration of control siRNA. The cells were transfected with GFP-Sec61β (green) and visualized by fluorescence confocal microscopy after staining for the Golgi marker GM130 (red). Merged images are to the right. Bar, 10 μm.
(B) As in (A), except cells were immunostained for the mitochondrial marker cytochrome c (Cyto c; red). Bar, 10 μm.
(C) As in (A), except cells were immunostained for the lysosomal marker LAMP-1 (red). Bar, 10 μm.
Figure S13. Xenopus egg membranes contain atlastins and form an ER network in vitro
(A) Membranes from Xenopus eggs were incubated with or without GTP for 60 min, stained with octadecyl rhodamine, and visualized by fluorescence microscopy. Bar, 20 μm.
(B) Membrane extracts from Xenopus eggs were separated by SDS-PAGE and analyzed by immunoblotting with anti-ATL antibodies that recognize all Xenopus atlastin isoforms. Molecular mass standards (in kDa) are on the left.
Figure S14. A sey1Δyop1Δ mutant shows a growth defect in high-osmolarity medium
The indicated yeast strains were grown to saturation in YPD media and diluted sequentially ten-fold. Three μl of each dilution were spotted onto YPD plates in the absence or presence of 1 M sorbitol, and the plates were incubated at 30°C for 2 days.
Figure S15. GTPase activity of the cytoplasmic domain of the Sey1p K50A mutant.
The Sey1p GTPase domain (residue 1-351) with an N-terminal 6×His-tag and with or without the K50A mutation was expressed from the pET28a vector (Novagen) in E. coli. Cells were broken in 50 mM Tris pH 7.5, 150 mM NaCl and 5 mM MgCl2 with protease inhibitors and 5 mM 2-mercaptoethanol. The protein was purified from cleared lysates by Ni-NTA chromatography (Qiagen) and gel filtration chromatography (Superdex 200, GE Healthcare). Purified proteins were mixed with 500 μM GTP and 2.5 mM MgCl2, and assayed for generation of phosphate using the PiPer Phosphate Assay (Invitrogen) at 37°C.
Acknowledgments
We thank H. Jaffe (NINDS) for MALDI-TOF analysis, F. Yang (NICHD) for providing Xenopus eggs, L. Dreier and G. Voeltz for advice, and P. Carvalho for generating some yeast deletion strains. We thank S. Schulman, A. Palazzo, and D. Pellman for critical reading of the manuscript. Y.S. was supported by the American Heart Association, and C.B. and W.A.P. by the Intramural Research Programs of the NINDS and NIDDK, respectively, National Institutes of Health. T.A.R. is a Howard Hughes Medical Institute investigator.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Bashkirov PV, Akimov SA, Evseev AI, Schmid SL, Zimmerberg J, Frolov VA. GTPase cycle of dynamin is coupled to membrane squeeze and release, leading to spontaneous fission. Cell. 2008;135:1276–1286. doi: 10.1016/j.cell.2008.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beetz C, Schule R, Deconinck T, Tran-Viet KN, Zhu H, Kremer BP, Frints SG, van Zelst-Stams WA, Byrne P, Otto S, et al. REEP1 mutation spectrum and genotype/phenotype correlation in hereditary spastic paraplegia type 31. Brain. 2008;131:1078–1086. doi: 10.1093/brain/awn026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brands A, Ho TH. Function of a plant stress-induced gene, HVA22. Synthetic enhancement screen with its yeast homolog reveals its role in vesicular traffic. Plant Physiol. 2002;130:1121–1131. doi: 10.1104/pp.007716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H, Detmer SA, Ewald AJ, Griffin EE, Fraser SE, Chan DC. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chernomordik LV, Kozlov MM. Mechanics of membrane fusion. Nat Struct Mol Biol. 2008;15:675–683. doi: 10.1038/nsmb.1455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Connell JW, Lindon C, Luzio JP, Reid E. Spastin couples microtubule severing to membrane traffic in completion of cytokinesis and secretion. Traffic. 2009;10:42–56. doi: 10.1111/j.1600-0854.2008.00847.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dreier L, Rapoport TA. In vitro formation of the endoplasmic reticulum occurs independently of microtubules by a controlled fusion reaction. J Cell Biol. 2000;148:883–898. doi: 10.1083/jcb.148.5.883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Du Y, Ferro-Novick S, Novick P. Dynamics and inheritance of the endoplasmic reticulum. J Cell Sci. 2004;117:2871–2878. doi: 10.1242/jcs.01286. [DOI] [PubMed] [Google Scholar]
- Evans K, Keller C, Pavur K, Glasgow K, Conn B, Lauring B. Interaction of two hereditary spastic paraplegia gene products, spastin and atlastin, suggests a common pathway for axonal maintenance. Proc Natl Acad Sci U S A. 2006;103:10666–10671. doi: 10.1073/pnas.0510863103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghaemmaghami S, Huh WK, Bower K, Howson RW, Belle A, Dephoure N, O'Shea EK, Weissman JS. Global analysis of protein expression in yeast. Nature. 2003;425:737–741. doi: 10.1038/nature02046. [DOI] [PubMed] [Google Scholar]
- Grabs D, Slepnev VI, Songyang Z, David C, Lynch M, Cantley LC, De Camilli P. The SH3 domain of amphiphysin binds the proline-rich domain of dynamin at a single site that defines a new SH3 binding consensus sequence. J Biol Chem. 1997;272:13419–13425. doi: 10.1074/jbc.272.20.13419. [DOI] [PubMed] [Google Scholar]
- Hermann GJ, Thatcher JW, Mills JP, Hales KG, Fuller MT, Nunnari J, Shaw JM. Mitochondrial fusion in yeast requires the transmembrane GTPase Fzo1p. J Cell Biol. 1998;143:359–373. doi: 10.1083/jcb.143.2.359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoppins S, Lackner L, Nunnari J. The machines that divide and fuse mitochondria. Annu Rev Biochem. 2007;76:751–780. doi: 10.1146/annurev.biochem.76.071905.090048. [DOI] [PubMed] [Google Scholar]
- Hu J, Shibata Y, Voss C, Shemesh T, Li Z, Coughlin M, Kozlov MM, Rapoport TA, Prinz WA. Membrane proteins of the endoplasmic reticulum induce high-curvature tubules. Science. 2008;319:1247–1250. doi: 10.1126/science.1153634. [DOI] [PubMed] [Google Scholar]
- Lee C, Chen LB. Dynamic behavior of endoplasmic reticulum in living cells. Cell. 1988;54:37–46. doi: 10.1016/0092-8674(88)90177-8. [DOI] [PubMed] [Google Scholar]
- Mannan AU, Boehm J, Sauter SM, Rauber A, Byrne PC, Neesen J, Engel W. Spastin, the most commonly mutated protein in hereditary spastic paraplegia interacts with Reticulon 1 an endoplasmic reticulum protein. Neurogenetics. 2006;7:93–103. doi: 10.1007/s10048-006-0034-4. [DOI] [PubMed] [Google Scholar]
- Marks B, Stowell MH, Vallis Y, Mills IG, Gibson A, Hopkins CR, McMahon HT. GTPase activity of dynamin and resulting conformation change are essential for endocytosis. Nature. 2001;410:231–235. doi: 10.1038/35065645. [DOI] [PubMed] [Google Scholar]
- Namekawa M, Muriel MP, Janer A, Latouche M, Dauphin A, Debeir T, Martin E, Duyckaerts C, Prigent A, Depienne C, et al. Mutations in the SPG3A gene encoding the GTPase atlastin interfere with vesicle trafficking in the ER/Golgi interface and Golgi morphogenesis. Mol Cell Neurosci. 2007;35:1–13. doi: 10.1016/j.mcn.2007.01.012. [DOI] [PubMed] [Google Scholar]
- Praefcke GJ, McMahon HT. The dynamin superfamily: universal membrane tubulation and fission molecules? Nat Rev Mol Cell Biol. 2004;5:133–147. doi: 10.1038/nrm1313. [DOI] [PubMed] [Google Scholar]
- Prinz WA, Grzyb L, Veenhuis M, Kahana JA, Silver PA, Rapoport TA. Mutants affecting the structure of the cortical endoplasmic reticulum in Saccharomyces cerevisiae. J Cell Biol. 2000;150:461–474. doi: 10.1083/jcb.150.3.461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pucadyil TJ, Schmid SL. Real-time visualization of dynamin-catalyzed membrane fission and vesicle release. Cell. 2008;135:1263–1275. doi: 10.1016/j.cell.2008.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ringstad N, Nemoto Y, De Camilli P. The SH3p4/Sh3p8/SH3p13 protein family: binding partners for synaptojanin and dynamin via a Grb2-like Src homology 3 domain. Proc Natl Acad Sci U S A. 1997;94:8569–8574. doi: 10.1073/pnas.94.16.8569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rismanchi N, Soderblom C, Stadler J, Zhu PP, Blackstone C. Atlastin GTPases are required for Golgi apparatus and ER morphogenesis. Hum Mol Genet. 2008;17:1591–1604. doi: 10.1093/hmg/ddn046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saito H, Kubota M, Roberts RW, Chi Q, Matsunami H. RTP family members induce functional expression of mammalian odorant receptors. Cell. 2004;119:679–691. doi: 10.1016/j.cell.2004.11.021. [DOI] [PubMed] [Google Scholar]
- Salinas S, Proukakis C, Crosby A, Warner TT. Hereditary spastic paraplegia: clinical features and pathogenetic mechanisms. Lancet Neurol. 2008;7:1127–1138. doi: 10.1016/S1474-4422(08)70258-8. [DOI] [PubMed] [Google Scholar]
- Sanderson CM, Connell JW, Edwards TL, Bright NA, Duley S, Thompson A, Luzio JP, Reid E. Spastin and atlastin, two proteins mutated in autosomal-dominant hereditary spastic paraplegia, are binding partners. Hum Mol Genet. 2006;15:307–318. doi: 10.1093/hmg/ddi447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schiefelbein JW, Somerville C. Genetic Control of Root Hair Development in Arabidopsis thaliana. Plant Cell. 1990;2:235–243. doi: 10.1105/tpc.2.3.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuldiner M, Collins SR, Thompson NJ, Denic V, Bhamidipati A, Punna T, Ihmels J, Andrews B, Boone C, Greenblatt JF, et al. Exploration of the function and organization of the yeast early secretory pathway through an epistatic miniarray profile. Cell. 2005;123:507–519. doi: 10.1016/j.cell.2005.08.031. [DOI] [PubMed] [Google Scholar]
- Shibata Y, Voeltz GK, Rapoport TA. Rough sheets and smooth tubules. Cell. 2006;126:435–439. doi: 10.1016/j.cell.2006.07.019. [DOI] [PubMed] [Google Scholar]
- Shibata Y, Voss C, Rist JM, Hu J, Rapoport TA, Prinz WA, Voeltz GK. The reticulon and DP1/Yop1p proteins form immobile oligomers in the tubular endoplasmic reticulum. J Biol Chem. 2008;283:18892–18904. doi: 10.1074/jbc.M800986200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soulet F, Yarar D, Leonard M, Schmid SL. SNX9 regulates dynamin assembly and is required for efficient clathrin-mediated endocytosis. Mol Biol Cell. 2005;16:2058–2067. doi: 10.1091/mbc.E04-11-1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sweitzer SM, Hinshaw JE. Dynamin undergoes a GTP-dependent conformational change causing vesiculation. Cell. 1998;93:1021–1029. doi: 10.1016/s0092-8674(00)81207-6. [DOI] [PubMed] [Google Scholar]
- Takei K, McPherson PS, Schmid SL, De Camilli P. Tubular membrane invaginations coated by dynamin rings are induced by GTP-gamma S in nerve terminals. Nature. 1995;374:186–190. doi: 10.1038/374186a0. [DOI] [PubMed] [Google Scholar]
- Terasaki M, Chen LB, Fujiwara K. Microtubules and the endoplasmic reticulum are highly interdependent structures. J Cell Biol. 1986;103:1557–1568. doi: 10.1083/jcb.103.4.1557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Voeltz GK, Prinz WA, Shibata Y, Rist JM, Rapoport TA. A class of membrane proteins shaping the tubular endoplasmic reticulum. Cell. 2006;124:573–586. doi: 10.1016/j.cell.2005.11.047. [DOI] [PubMed] [Google Scholar]
- Wang H, Lockwood SK, Hoeltzel MF, Schiefelbein JW. The ROOT HAIR DEFECTIVE3 gene encodes an evolutionarily conserved protein with GTP-binding motifs and is required for regulated cell enlargement in Arabidopsis. Genes Dev. 1997;11:799–811. doi: 10.1101/gad.11.6.799. [DOI] [PubMed] [Google Scholar]
- Waterman-Storer CM, Salmon ED. Endoplasmic reticulum membrane tubules are distributed by microtubules in living cells using three distinct mechanisms. Curr Biol. 1998;8:798–806. doi: 10.1016/s0960-9822(98)70321-5. [DOI] [PubMed] [Google Scholar]
- Zhao X, Alvarado D, Rainier S, Lemons R, Hedera P, Weber CH, Tukel T, Apak M, Heiman-Patterson T, Ming L, et al. Mutations in a newly identified GTPase gene cause autosomal dominant hereditary spastic paraplegia. Nat Genet. 2001;29:326–331. doi: 10.1038/ng758. [DOI] [PubMed] [Google Scholar]
- Zheng H, Kunst L, Hawes C, Moore I. A GFP-based assay reveals a role for RHD3 in transport between the endoplasmic reticulum and Golgi apparatus. Plant J. 2004;37:398–414. doi: 10.1046/j.1365-313x.2003.01969.x. [DOI] [PubMed] [Google Scholar]
- Zhu PP, Patterson A, Lavoie B, Stadler J, Shoeb M, Patel R, Blackstone C. Cellular localization, oligomerization, and membrane association of the hereditary spastic paraplegia 3A (SPG3A) protein atlastin. J Biol Chem. 2003;278:49063–49071. doi: 10.1074/jbc.M306702200. [DOI] [PubMed] [Google Scholar]
- Zhu PP, Soderblom C, Tao-Cheng JH, Stadler J, Blackstone C. SPG3A protein atlastin-1 is enriched in growth cones and promotes axon elongation during neuronal development. Hum Mol Genet. 2006;15:1343–1353. doi: 10.1093/hmg/ddl054. [DOI] [PubMed] [Google Scholar]
- Züchner S, Wang G, Tran-Viet KN, Nance MA, Gaskell PC, Vance JM, Ashley-Koch AE, Pericak-Vance MA. Mutations in the novel mitochondrial protein REEP1 cause hereditary spastic paraplegia type 31. Am J Hum Genet. 2006;79:365–369. doi: 10.1086/505361. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Endogenous ATL1 does not interact with control membrane proteins.
Detergent extracts from mouse brain or spinal cord were incubated with ATL1 antibodies or control IgG. Precipitated proteins were then analyzed by immunoblotting (IB) with Rtn4a, calnexin, or TrkB antibodies. Lanes 1 and 2 (loads) show 10% of the starting material used for immunoprecipitation.
Figure S2. Overexpressed ATL1 and Rtn3c do not interact with control membrane proteins.
(A) Myc-ATL1 and HA-Rtn3c were expressed either individually or together in COS-7 cells. Digitonin-extracts were used for immunoprecipitation (IP) with anti-HA, anti-Myc, or anti-FLAG antibodies. Ten percent of the starting material (load) and of the material not bound to the antibodies (unbound), as well as the precipitates were analyzed by SDS-PAGE and immunoblotting (IB) with anti-HA or anti-Myc antibodies. As a control, the blot was also probed with antibodies to the integral ER membrane protein calnexin.
(B) Co-immunoprecipitation as in (A), but with cells expressing Myc-ATL1 and the HA-tagged integral membrane protein Sec61alpha (HA-Sec61alpha).
(C) Co-immunoprecipitation as in (A), but with cells expressing Myc-ATL1 and the FLAG-tagged integral membrane protein CLIMP63 (FLAG-CLIMP63). The blot was probed with anti-FLAG, instead of HA-antibodies.
(D) Co-immunoprecipitation as in (A), but with cells expressing Myc-Sec61beta and HA-Rtn3c.
Figure S3. ATL2 interacts with Rtn3c and DP1.
(A) Myc-tagged ATL2 (Myc-ATL2) was expressed together with HA-tagged Rtn3c (HA-Rtn3c) in COS-7 cells. Digitonin-solubilized extracts were used for immunoprecipitation (IP) with anti-HA or anti-Myc antibodies. Ten percent of the starting material (load) and of the material not bound to the antibodies (unbound), as well as the material bound to the antibodies were analyzed by SDS-PAGE and immunoblotting (IB) with anti-HA or anti-Myc antibodies.
(B) As in (A), but with HA-tagged DP1 (HA-DP1) instead of HA-Rtn3c.
Figure S4. Interactions of the transmembrane segments of ATL2 and ATL3.
(A) The Myc-tagged C-terminal domains of ATL2 or ATL3 harboring the transmembrane domains (Myc-ATL2-TM or Myc-ATL3-TM) were expressed together with HA-tagged Rtn3c (HA-Rtn3c) in COS-7 cells. Digitonin-solubilized extracts were used for immunoprecipitation (IP) with anti-HA or anti-Myc antibodies. Ten percent of the starting material (load) and of the material not bound to the antibodies (unbound), as well as the material bound to the antibodies were analyzed by SDS-PAGE and immunoblotting (IB) with anti-HA or anti-Myc antibodies.
(B) As in (A), but with Myc-ATL3-TM and HA-DP1.
Figure S5. The cytoplasmic domains of the atlastins do not interact with Rtn3c or DP1.
(A) Myc-tagged versions of the large N-terminal cytoplasmic domains of ATL1, ATL2, or ATL3 (Myc-cytATL1, Myc-cytATL2, or Myc-cytATL3) and HA-tagged Rtn3c (HA-Rtn3c) were co-expressed in COS-7 cells. Digitonin-solubilized extracts were used for immunoprecipitation (IP) with anti-HA or anti-Myc antibodies. Ten percent of the starting material (load), the material not bound to the antibodies (unbound), and the material bound to the antibodies were analyzed by SDS-PAGE and immunoblotting (IB) with anti-HA or anti-Myc antibodies.
(B) As in (A), but with HA-tagged DP1 (HA-DP1) instead of HA-Rtn3c.
Figure S6. ATL2 and ATL3 localize to the tubular ER.
(A) Myc-ATL2 (green) was expressed in COS-7 cells. Its localization was identified with anti-Myc antibodies and compared to that of an endogenous luminal ER protein, calreticulin (red) using indirect immunofluorescence and confocal microscopy. The lower panels show an enlargement of the boxed region centered on ER sheets. Scale bar, 10 μm.
(B) As in (A), but with Myc-ATL3 instead of Myc-ATL2.
Figure S7. The membrane-anchoring domain of the atlastins localizes to the tubular ER network.
Myc-tagged C-terminal region of ATL1 harboring the transmembrane domains (Myc-ATL1-TM) (green) was expressed in COS-7 cells. Its localization was revealed with anti-Myc antibodies and compared to that of an endogenous luminal ER protein, calreticulin (red), using indirect immunofluorescence and confocal microscopy. The lower panels show an enlargement of the boxed region centered on ER sheets. Scale bar, 10 μm.
Figure S8. ER morphology defects correlate with the expression level of ATL1 and ATL1 K80A.
Myc-ATL1 or Myc-ATL1 K80A was expressed in COS-7 cells. The cells were stained with Myc-antibodies and imaged by fluorescence microscopy. For each cell, the ER morphology was visually scored, and the average fluorescence intensity, defined as the total fluorescence in the ER divided by its area, was measured using MetaMorph software. The cells were divided into five groups according to their expression levels. Shown is the percentage of cells with normal and abnormal ER in each group (means ± standard errors from three different transfections; 100 cells per sample).
Figure S9. Expression of ATL1 K80A changes the morphology of the ER, but not that of other organelles.
(A) GFP-Sec61beta or Myc-ATL1 K80A was expressed in COS-7 cells. The localization of the Golgi marker giantin was analyzed using indirect immunofluorescence and confocal microscopy. Bar, 10 μm.
(B) As in (A), but the cells were stained with antibodies to the mitochondrial marker cytochrome c.
Figure S10. Co-localization of a GTP-binding mutant of ATL1 with Rtn3c.
Myc-ATL1 K80A was co-expressed with GFP-Rtn3c in COS-7 cells. The proteins were visualized by indirect immunofluorescence using Myc-antibodies and by GFP fluorescence. Bar, 10 μm.
Figure S11. ER morphology changes caused by overexpression of atlastin mutants.
(A) A Myc-tagged version of the N-terminal, cytoplasmic domain of the GTP-binding mutant of ATL1 (Myc-cytATL1 K80A) was co-expressed in COS-7 cells with an RFP-tagged version of the ER protein Sec61β. The cells were stained with Myc-epitope and calreticulin antibodies and visualized by indirect immunofluorescence and confocal microscopy. Shown is a cell with an unbranched ER tubule phenotype. Bar, 10 μm.
(B) A Myc-tagged version of the membrane-anchoring domain of ATL3 (Myc-ATL3-TM) was expressed in COS-7 cells. The cells were stained with Myc-epitope and calreticulin antibodies and visualized by indirect immunofluorescence and confocal microscopy. Note the unbranched tubular ER morphology.
Figure S12. Effect of atlastin depletion on the morphology of organelles.
(A) HeLa cells were transfected with siRNA oligonucleotides directed against ATL2 and ATL3 (ATL DKD) or else with an equivalent concentration of control siRNA. The cells were transfected with GFP-Sec61β (green) and visualized by fluorescence confocal microscopy after staining for the Golgi marker GM130 (red). Merged images are to the right. Bar, 10 μm.
(B) As in (A), except cells were immunostained for the mitochondrial marker cytochrome c (Cyto c; red). Bar, 10 μm.
(C) As in (A), except cells were immunostained for the lysosomal marker LAMP-1 (red). Bar, 10 μm.
Figure S13. Xenopus egg membranes contain atlastins and form an ER network in vitro
(A) Membranes from Xenopus eggs were incubated with or without GTP for 60 min, stained with octadecyl rhodamine, and visualized by fluorescence microscopy. Bar, 20 μm.
(B) Membrane extracts from Xenopus eggs were separated by SDS-PAGE and analyzed by immunoblotting with anti-ATL antibodies that recognize all Xenopus atlastin isoforms. Molecular mass standards (in kDa) are on the left.
Figure S14. A sey1Δyop1Δ mutant shows a growth defect in high-osmolarity medium
The indicated yeast strains were grown to saturation in YPD media and diluted sequentially ten-fold. Three μl of each dilution were spotted onto YPD plates in the absence or presence of 1 M sorbitol, and the plates were incubated at 30°C for 2 days.
Figure S15. GTPase activity of the cytoplasmic domain of the Sey1p K50A mutant.
The Sey1p GTPase domain (residue 1-351) with an N-terminal 6×His-tag and with or without the K50A mutation was expressed from the pET28a vector (Novagen) in E. coli. Cells were broken in 50 mM Tris pH 7.5, 150 mM NaCl and 5 mM MgCl2 with protease inhibitors and 5 mM 2-mercaptoethanol. The protein was purified from cleared lysates by Ni-NTA chromatography (Qiagen) and gel filtration chromatography (Superdex 200, GE Healthcare). Purified proteins were mixed with 500 μM GTP and 2.5 mM MgCl2, and assayed for generation of phosphate using the PiPer Phosphate Assay (Invitrogen) at 37°C.