

Abstract
Artificial ECMs that not only closely mimic the hybrid nature of the natural ECM but also provide tunable material properties and enhanced biological functions are attractive candidates for tissue engineering applications. This review summarizes recent advances in developing multicomponent hybrid hydrogels by integrating modular and heterogeneous building blocks into well-defined, multifunctional hydrogel composites. The individual building blocks can be chemically, morphologically, and functionally diverse, and the hybridization can occur at molecular level or microscopic scale. The modular nature of the designs, combined with the potential synergistic effects of the hybrid systems, has resulted in novel hydrogel matrices with robust structure and defined functions.
Keywords: drug delivery, extracellular matrix, heparin, hyaluronic acid, hybrid hydrogels, microgels, networks, polysaccharide particles, tissue engineering
Introduction
Principles of Tissue Engineering
Tissue engineering aims at regenerating functional tissues or complex organs via the combination of viable cells, biomimetic matrices, spatio-temporal presentation of morphogenic factors, and external biophysical cues.[1–3] It has been demonstrated that the cells, being the key player in tissue engineering, maintain their ability not only to produce their native extracellular matrix (ECM) but also to self-organize into complex tissues on the condition that their native biochemical and biophysical microenvironment can be recapitulated in vitro.[1,4,5] Therefore, in order to harness the regenerative potential of the cells, multifunctional biomimetic matrices that foster the delicate and dynamic interplay among these four components are critically needed. [6–9]
The three dimensional (3D), artificial matrix is designed not only to provide the initial structural support for cells, but also to direct cell differentiation and proliferation, ultimately leading to the assembly of functional tissues.[10] The biochemical as well as the biophysical properties of the matrices are essential to the functional tissue regeneration since cells can display different phenotypes depending on their microenvironment.[11–14] Meanwhile, numerous studies have demonstrated the importance of biological factors, such as growth factors, in stimulating cell proliferation and differentiation.[15,16] Therefore, drug delivery is an integral component of tissue engineering; tissue engineering can be viewed as a special case of controlled drug delivery in which cells are “delivered” and the formation of neotissue can be stimulated by locally released bioactive molecules.[17]
The Need for Hybrid Multicomponent Hydrogels
Among various tissue engineering scaffolds that have been investigated, hydrogels remain the most appealing candidates due to their structural similarity to the natural ECM, inherent biocompatibility, tunable viscoelasticity, high water content and high permeability for oxygen and essential nutrients.[18,19] Their physical properties can be readily manipulated in response to changes of environmental factors and cellular activities.[19–22] Physically or chemically crosslinked hydrogels can be generated in the presence of living cells, allowing in situ encapsulation for tissue engineering. [23–27] A variety of hydrogel materials have been utilized for tissue engineering applications[28,29] including reconstituted ECM components or natural proteins and carbohydrates,[30–33] self-assembling peptides,[34–36] and synthetic hydrogels.[37–39]
Although hydrogel materials have shown promise in tissue repair, critical issues regarding their bioactivities, mechanical strength, and degradation kinetics cannot be neglected. Traditional hydrogels are bulk gels consisting of inert synthetic polymers that are randomly interconnected. They are inherently heterogeneous, containing network defects such as entanglements, chain ends, and phase-separated regions. Uncontrolled heterogeneity at different length scales can dramatically affect the diffusion characteristics, mechanical properties, and cellular functions. More importantly, many traditional hydrogels do not exhibit biological activities, hierarchical organization, and structural integrity that are necessary to facilitate cell infiltration and neovascularization.[4,10,40,41]
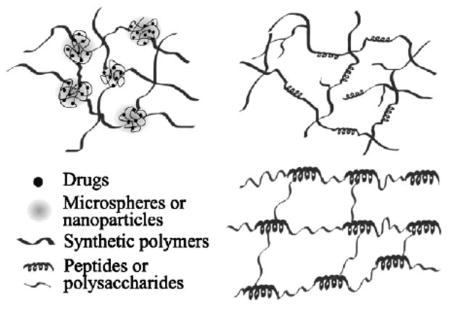
The native ECM can be considered as a hybrid hydrogel containing multiple structural and functional components interdigitated at all length scales.[10,42–44] Recapitulating this hybrid nature into artificial ECMs is likely the most promising approach for scaffold-based tissue engineering. In this review, we summarize recent efforts in engineering hybrid multicomponent hydrogels with enhanced materials properties and controlled biological functions by judicial integration of modular and heterogeneous components into unifying hydrogel constructs (Figure 1). In this context, hybrid multicomponent hydrogels are referred to as hydrogel systems composed of chemically, morphologically, and functionally different building blocks interconnected via chemical or physical means. Depending on the size and the nature of the building blocks, the hybridization can occur at molecular level or microscopic scale. The modular nature of the design, combined with the potential synergistic effect of the hybrid system, has led to novel hydrogels with potential in tissue engineering applications.
Figure 1.
Schematic representation of hybrid hydrogel systems discussed. Left: microscopic level hybridization approaches; right: molecular level hybridization approaches. The components illustrated in the first schematic may be present either separately or together depending on the specific hybrid material. Not drawn to scale.
Molecular Integration of Biological Macromolecules with Synthetic Matrices
Synthetic polymers are attractive candidates for use as tissue engineering matrices because they can be easily synthesized in large quantities with controlled molecular weights, molecular architectures, and microscopic morphologies. Readily accessible functional groups can be introduced to the polymers at defined locales for subsequent crosslinking using well-defined chemistry. [45–47] Their mechanical properties can be easily manipulated at micro- and macroscopic levels by blending, copolymerization, or crosslinking. [48] In most cases, however, the synthetic polymer alone only functions as a passive support material and does not foster active cellular interactions.
On the other hand, natural proteins exhibit distinct primary, secondary, and tertiary structures that are crucial for their functions. In addition to providing structural support for cells, many proteins elicit active cellular responsive, biological recognition, and cell-triggered remodeling. [44,49,50] With current advances in biotechnology, a variety of peptides with specific sequences and secondary structures have been engineered. The expansion of solid-phase peptide synthesis (SPPS) methods and ligation strategies has permitted synthesis of tailored sequences of peptides that capture the most important functions of the intact proteins. [51,52] Taking advantage of recombinant protein technology, researchers have engineered a variety of proteins and polypeptides with unprecedented structural and functional control. Many of these polypeptides have been shown to meet the complex biological requirements of cells, rendering them attractive building blocks for tissue engineering. [53–55]
Owing to the many intriguing potential uses of hybrid peptide/polymer molecular composites in tissue engineering applications, their design and synthesis have been a focus of significant research over the last decade. Hybrid hydrogels have been engineered to mimic the natural proteins in terms of their molecular architectures, dynamic responsiveness, and defined cellular functions, with the added attributes of tunability and processability provided by the synthetic polymer constituents.
Hybrid Hydrogels Mimicking the Natural Structural Proteins
Collagen[56] and elastin[57] are two major structural proteins in natural ECM. In addition to their biological functions in modulating cell/cell interactions and cell/ECM interactions, they provide the tissues with desired tensile strength and elasticity for proper functions. Thus, various approaches have been attempted to recapture the basic functions of collagen and elastin in synthetic hydrogel matrices.
Collagen Mimetic Hybrid Hydrogels
Although collagen isolated from animals can be reconstituted in vitro to form 3D tissue engineering scaffolds, its application is limited due to batch to batch variation, immunogenicity, poor mechanical strength, and heterogeneous chemical composition as well as complex molecular architecture.[50,58] Although hybrid composite hydrogels consisting of synthetic polymer-based, porous scaffolds infused with native collagen have been extensively investigated as bioactive matrices for 3D cell culture and fundamental biology studies,[32,33,59,60] these materials suffer from the same limitations associated with the intact collagen macromolecules.
Collagen molecules, with molecular dimensions of 1.5 nm width and 300 nm length, comprise a triple helix formed from three helically wound α-chains. Each strand has a repeating sequence of XaaYaaGly, with the most abundant triplet being ProHypGly. Individual collagen molecules self-assemble by end-on and side-by-side interactions to generate fibrils, which are approximately cylindrical, with diameters ranging from 20 to 500 nm. They are further strengthened by covalent disulfide crosslinks between collagen chains. [58,61] Theoretical and molecular modeling suggests that the staggered array of ultra-long procollagen molecules in collagen fibrils maximizes the strength and provides large energy dissipation during deformation, thus creating a tough and robust material.[62]
Using molecular self-assembly approaches, several groups have produced collagen-like fibrillar materials through self-assembly. [63–66] Most of these materials, however, are only capable of mimicking the basic level of assembly in collagen although the natural collagen assembles in a coordinated fashion at multiple length scales, ranging from molecular level up to macroscopic scale. In fact, mimicking collagen from the bottom up is extremely daunting, and as of yet not possible. Realizing this difficulty, Yu and coworkers introduced and retained mature collagen in their tissue engineering constructs by taking advantage of both the cellular machinery and the ability of a collagen-mimetic peptide [–(Pro–Hyp–Gly)x–], CMP to bind and associate with cell-produced or externally supplied collagen.[67,68] The binding is believed to occur through a process involving both strand invasion and triple-helix assembly. Hybrid hydrogels based on CMP and poly(ethylene glycol) (PEG) (e.g., as illustrated in Figure 1) were synthesized using a photopolymerizable CMP derivative and PEG diacrylate (PEGDA). In this hybrid system, PEG provides an inert polymeric backbone for CMP conjugation; CMP in turn, reinforces the matrix through physical association and provides biological functions by association with collagen. Chondrocytes encapsulated in the hybrid hydrogels exhibited enhanced cartilage-specific ECM production (87% increase in GAG content and a 103% increase in collagen content) as compared to those encapsulated in an inert PEG gel. However, no comparison with the native cartilage tissues in terms of the biochemical compositions and the mechanical properties was attempted.
Elastin Mimetic Hybrid Hydrogels
Unlike collagen, native elastin is rarely used as a tissue engineering scaffold, and when it is used, only poorly defined preparations have been employed. Major challenges to the use of native elastin result from its extensive covalent crosslinking and substantial insolubility,[69,70] which necessitate multistep purification[71] or high temperature hydrolysis that may lead to undesirable structural alteration.[72] Elastin is rich in mechanically active tissues, such as tendon, bladder, lungs, blood vessels, elastic cartilage, and vocal folds.[73–76] A network of elastic fibers gives the tissue the required strength, flexibility, and resilience so that it can reversibly recoil after transient stretch up to 200%.[77] Thus, engineering elastomeric scaffolds that mimic the mechanical properties of natural elastin may therefore provide useful tissue engineering strategies for repairing mechanically active tissues.
Elastin achieves its excellent mechanical properties through a multiblock copolypeptide structure composed largely of two types of short segments that alternate along the polypeptide chain: highly flexible hydrophobic segments composed of VPGVG repeats, with many transient structures that can easily change their conformation when stretched; and alanine- and lysine-rich α-helical segments, which form crosslinks between adjacent molecules via the action of lysyl oxidase in the ECM.[57,78] The elasticity of the protein has been discussed primarily in terms of dominant entropic components[79,80] and the main models of elastin's elasticity share the perspective that either chain entropy or internal chain dynamics are at least in part at the origin of the elasticity.[81–85] Elastin's ability to modulate a variety of cellular and protein interactions may be a direct result of its extensibility.[86,87] Over the past decade, tremendous research efforts have been devoted to the synthesis of elastin-like (poly)peptides for tissue engineering applications.[54,88–94] Although variations exist in terms of their amino acid compositions and polymer architectures, these materials are not hybrid in the context of this article, thus will not be discussed. The readers are referred to several recent reviews for more information.[95–98]
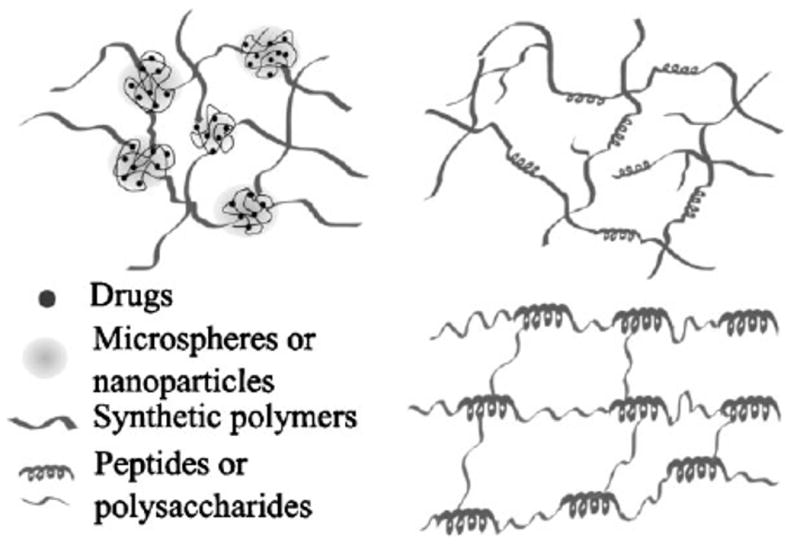
Intrigued by the entropic origin of elastin elasticity, we have been engaged in developing multiblock peptide/polymer hybrid polymers consisting of hydrophobic flexible synthetic polymer alternating with alanine-rich peptides that are the structural component of the hydrophilic crosslinking domain of the natural elastin (Figure 1).[99] Mimicking the hydrophobic domains of natural elastin with synthetic polymeric chains that are molecularly flexible may not compromise the superior elasticity demonstrated by the natural polypeptide, as the polymeric segments will adopt a random coil conformation when the polymer chains are fully relaxed, and will easily extend past one another when exposed to stress, effectively dissipating energy.[100–102] The combination of synthetic polymers and peptides offers chemical versatility and added tunability in molecular weight, structure, and mechanical properties of the hybrid copolymer, considerably expanding design options.
Multiblock elastin mimetic hybrid polymers have been synthesized via step-growth polymerization approaches employing macromonomers that are end-functionalized with mutually reactive functional groups. Copper (I) catalyzed azide-alkyne cycloaddition[46] was employed as a strategy for the production of strictly alternating multiblock architectures. In our initial studies, PEG was chosen as the synthetic block because of its biocompatibility, water solubility, and simplicity in structure. Azide-functionalized telechelic PEG (M̄w = 1 450 g • mol−1) was synthesized by nucleophilic substitution of mesylated hydroxyl end groups with near-quantitative conversion of the end group functionality as confirmed by 1H NMR. Separately, alanine-based, lysine-containing peptides [X(AKAAAKA)2X] based on the hydrophilic crosslinking domains of elastin were synthesized by standard Fmoc SPPS, and alkyne end groups were incorporated by adding the non-natural amino acid l-propargylglycine (X) to both ends of the peptide chain. The peptide was shown to exhibit α-helicity under basic conditions (pH > 10.5), reminiscent of the α-helical structure observed in the crosslinking domains of natural elastin.[103] Hybrid multi-block copolymers with alternating PEG and peptide blocks were formed by step growth polymerization using copper-catalyzed azide-alkyne cycloaddition between the azide end groups of PEG with the alkyne end groups of the peptide. The reaction was conducted in a mixed solvent with CuSO4 as a catalyst and sodium ascorbate as a reducing agent in the presence of the appropriate ligand. The formation of the multiblock product was confirmed collectively by FT-IR, 1H NMR, size exclusion chromatography (SEC), and sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE). Due to the hybrid nature of the multiblock copolymer, its molecular weight cannot be precisely determined. However, an estimation using protein standards suggested an M̄w value of ≈34 000 g · mol−1. The multiblock copolymers were crosslinked using hexamethylene diisocyanate (HMDI) in DMSO, forming covalent crosslinking universally through the lysine side chains of the peptide domain to form urea linkages (Figure 2). The compressive moduli of the dry and hydrated elastin mimetic hybrid hydrogels were calculated to be 0.32 and 0.12 MPa, respectively. With a swelling ratio of 5, the hybrid hydrogels became soft and more compressible, consistent with results observed for natural elastin.[104] For comparison purposes, the compression modulus of a relatively hydrophobic polyurethane elastomer used for tissue engineering applications (Tecoflex™ SG80A)[105] was measured as 0.21 MPa. Live/dead assay (indirect contact method) using primary porcine vocal fold fibroblasts indicate the biocompatibility of the hybrid gels.
Figure 2.
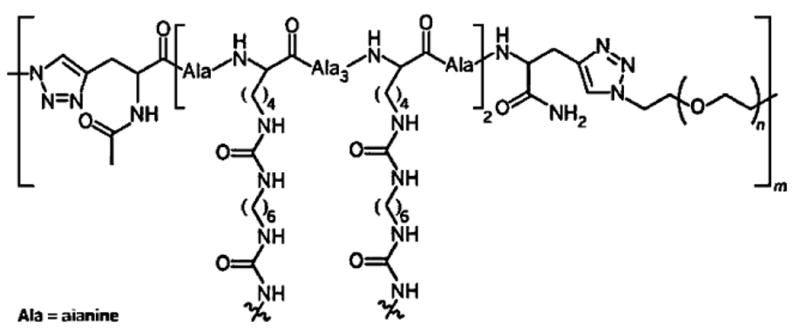
Chemical structure of elastin mimetic hybrid hydrogel composed (1) alternating PEG and peptide along the backbone; and (2) covalent crosslinking through the lysine side chains via the formation of urea bonds. (Reprinted from ref. [193], Copyright (2008) American Chemical Society).
Our on-going effort includes systematic variation of the molecular architectures and chemical compositions of the synthetic block and the specific sequence and the sequence length of the peptide domain. To promote integrin-mediated cell adhesion to these gels, fibronectin-derived GRGDS domains will be included in the peptide building blocks. The modular nature of the design, coupled with the chemical nature of the synthesis, will permit facile adjustment of mechanical, morphological, and biological properties of the resulting polymers.
Hybrid Hydrogels with Unique Responsiveness
As discussed above, collagen and elastin are important structural proteins in natural tissues. Many other native proteins are inherently responsive, changing their molecular conformations in response to minute changes of environmental factors.[106] Studies have shown that proteins exhibit distinctly different biological functions depending on their conformations.[107] By incorporating protein/peptide segments with tailored amino acid sequences into synthetic polymers, the responsiveness, degradability, aggregation properties, and the biological activities of natural proteins can be effectively harnessed.[108–111]
Protein/Peptide-Functionalized Responsive Hydrogels
Hybrid hydrogels that exhibit reversible volume changes to specific antigens were first described by Miyata et al.[111] The initial hybrid semi-interpenetrating (IPN) hydrogel was prepared by grafting the antigen and corresponding antibody to the acrylamide-based polymer network. The antigen/antibody binding within the hydrogel further reinforced network non-covalently. Competitive binding of free antigen triggered a change in gel volume owing to breaking of these non-covalent crosslinks. Similarly, glycoprotein-responsive hybrid hydrogels were synthesized using biomolecular imprinting technique with an acrylamide-based, lightly crosslinked polymer networks as the matrices. The hybrid hydrogels could dynamically recognize tumor-specific marker glycoproteins, resulting in significant volume reduction via the formation of lectin/glycoprotein/antibody complexes.[112]
Novel hybrid hydrogels composed of synthetic polymer and protein-folding motifs were pioneered by Kopecek (Figure 1).[108,113] In these hybrid gels, the water-soluble synthetic polymers serve as the carrier for the peptide, whereas the peptide/protein domains were designed to drive the formation of hydrogel via recognition-directed assembly. In one example,[114] nature-inspired or de novo designed coiled-coil motifs with His-tags were prepared by recombinant technologies. Separately, a linear hydrophilic copolymer of N-(2-hydroxypropyl)methacrylamide (HPMA) and a metal-chelating monomer N-(N′,N′-dicarboxymethyl-aminopropyl) ethacrylamide (DAMA) was prepared using radical polymerization. The synthetic polymers were linked together through the directed assembly of the coiled-coil motifs in conjunction with metal chelating between DAMA and the His-tag on the proteins. Hydrogels thus formed underwent dramatic volume transitions at the melting temperature of the coiled-coil modules. The transition was attributed to the temperature-induced conformational change in the coiled coil domains. Using a similar approach, the same group developed hybrid hydrogels consisting of β-sheet structures based on Ig-like modules from muscle titin.[115] These hydrogels demonstrated positive temperature responsiveness, swelling three times their initial volume at temperatures above the melting temperature of the β-sheet. These hybrid hydrogel systems offer a novel strategy for incorporating and delivering therapeutic proteins.
To improve homogeneity and responsiveness of the gels, hybrid hydrogels based on PHPMA covalently grafted with associating peptides were developed. Two distinct penta-heptad peptides capable of creating dimerization motifs were covalently conjugated to the PHPMA backbone. Equimolar mixtures of the graft copolymers in PBS solution at neutral pH at concentrations as low as 0.1 wt.-% resulted in the formation of self-assembled hydrogels via the formation of antiparallel heterodimeric coiled-coils. The enhanced homogeneity was attributed to the orthogonality of interchain dimerization and the decreased steric hindrance of the polymer backbone on the “in-register” alignment of the peptide heterodimers.[116] The hydrogels exhibit reversible responses in the presence of competing peptides or guanidine hydrochloride.
Structural control of the swelling of hybrid hydrogels has also been demonstrated in the work of Murphy and coworkers[117–119] whereby the function of an actuating protein building block is retained in hybrid hydrogels. Specifically, calmodulin (CaM), produced by molecular biology techniques, was rendered photocrosslinkable by reacting with large excess of PEGDA using thiol-acrylate chemistry. Hydrogels were formed by subjecting PEG-CaM-PEG mixed with PEG to UV irradiation. In the presence of Ca2+, the hydrogels expanded due to the extended dumbbell-shaped conformation of CaM. Upon exposure to trifluoperazine, a small molecule drug that binds CaM, the hydrogel underwent measurable shrinkage as a result of binding induced conformational changes of CaM. This response is reversible and dynamic. Crosslinking synthetic polymers with well-characterized protein modules or whole proteins proves to be a practical strategy for creating new materials with unique environmental responsiveness. Taking advantage of the adaptable nature of the dynamic building block, new materials that are capable of controlled release of therapeutics can be engineered.
One essential and indispensable step in accomplishing aforementioned goals is to link protein/peptide with the synthetic polymers. In Murphy's method, CaM-containing cysteine residues in place of threonine residue at each end of the dumbbell-shaped protein was generated. Instead of using site-specific bioconjugation, Esser-Kahn and Francis have targeted the universal protein termini to generate reactive handles for subsequent conjugation with synthetic polymers.[120] Transamination and protein ligation were used to introduce aldehydes or ketones at the protein termini for subsequent crosslinking with polymers carrying alkoxyamine groups along the backbone. The resulting hydrogels retained the pH/temperature sensitivities and enzymatic degradability of the native protein.
Another important stimulus is the cell itself. As discussed above, tissue engineering scaffolds are designed to serve as provisional matrices for cells to proliferate, organize, and perform normal cellular functions. Thus, cell/matrix interaction is critically important, and ideal synthetic matrices should provide instructive microenvironment that respond to changes of cell cycles.[4] RGD peptides, alone or in conjunction with other cell-binding motifs, have been widely employed as means to improve the cell adhesive properties of hydrogels.[41,121] A further improvement in cell/matrix interaction has been demonstrated by integrating the degradation characteristics of native ECM into the synthetic hydrogels. Hydrogel materials that were not sensitive to hydrolytic cleavage, but susceptible to enzymatic degradation have been developed by Hubbell and coworkers.[4,122] In early studies, peptide-PEG-peptide block triblock copolymers were synthesized and end-functionalized with acrylate groups to allow for UV-initiated photocrosslinking. The peptide sequences incorporated were potential collagenase or plasmin substrates. The resulting hydrogels were degraded under the action of the target enzyme but remained stable in the presence of the other enzyme.[123] With the addition of cell-adhesive peptide (RGD) grafted into the photocrosslinked hydrogel, cell migration into these hydrogels was observed.[124]
Following the same concept, integrin-binding (RGD) sites, and substrates for matrix metalloproteinases (MMP) were conjugated into the PEG-based hydrogels using Michael-type addition reaction. This was achieved by two-step reaction of vinyl sulfone-functionalized four-arm PEG with mono-cysteine adhesion peptides and bis-cysteine MMP substrate peptides. Cells could be entrapped in 3D biomimetic matrices in the presence of these modular building blocks. The matrix was degradable and invasive by cells via cell-secreted MMPs, thus the properties of the matrix is directly controlled by cell cycles (Figure 3). Further, primary human fibroblasts were demonstrated to proteolytically invade these networks, a process that depended on MMP substrate activity, adhesion ligand concentration and network crosslinking density.[125–127] The PEG-based, cell adhesive, MMP-sensitive hydrogels are attractive 3D matrices for cell culture and tissue engineering. By systematic modulation of matrix elasticity, MMP-sensitivity,and the concentration of a matrix-bound RGDSP peptide, mature cardiac cells could be obtained from pluripotent cardiac progenitors.[128]
Figure 3.
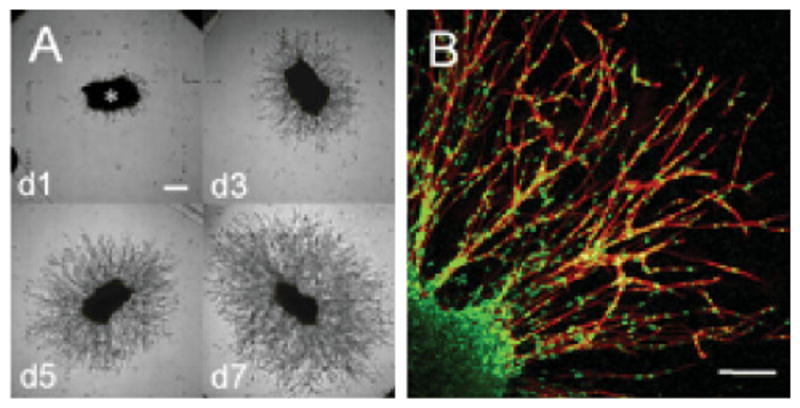
Cell-responsive hybrid hydrogels facilitates fibroblasts migration in 3D. (A) Fibroblasts radially invaded the adhesive and MMP-sensitive synthetic hydrogel matrix (bar=250 μm); (B) migration of spindle-like-shaped fibroblasts occurred in a cohort manner (bar=150 μm). (Reprinted from ref. [127], Copyright (2003) National Academy of Sciences).
It is clear that efficient incorporation of peptide functionalities in hydrogel matrices with a controlled density and spatial distribution is important for directing cellular function in a hydrogel environment. The Anseth group recently studied hydrogel formation using thiol-presenting peptides and PEGDA via a thiol-acrylate mixed mode photopolymerization method. The chain transfer constant of acrylates to thiols was determined to range from 1.5 to 2. Complete conversion of the functional groups occurred within ≈40–60 s. The crosslinking densities of the final gels ranged from 1.5 × 10−2 to 2.0 × 10−2 mol · L−1 when 10 × 10−3m of peptide was reacted with 40 × 10−3m of acrylate groups.[129] Their study has demonstrated that thiol-ene chemistry provides an easy robust, cost-efficient, and cytocompatible reaction scheme for the incorporation of cysteine-containing peptide sequences into PEG hydrogels. However, it is not clear whether amino acid side chain functional groups would interfere with the crosslinking chemistry.
Natural proteins are complex molecules in which multiple domains function synergistically; it is difficult to capture all these complex functions by simple and short peptide sequences. For example, while hydrogels modified with adhesion peptides allow cell adhesion on 2D hydrogel surfaces, adaptation of these hydrogels into 3D culture is not trivial. To further enhance the complexity of the hybrid hydrogel matrices, artificial proteins containing the repeating amino acid sequences based on fibrinogen and antithrombin III, comprising an RGD integrin-binding motif, two plasmin degradation sites, and a heparin-binding site was created by recombinant DNA methods and chemically grafted with PEGDA. Subsequent photopolymerization of the macromers yielded protein-graft-PEG hydrogels. With the initial Young's moduli up to 3.5 kPa, these hydrogels are (i) cell adhesive, both on 2D and in 3D; (ii) cell invasive, via cell-secreted serine proteases; and (iii) capable of binding heparin.[130] Alternatively, researchers resort on the intact protein for enhanced biological functions. PEG-fibrinogen or PEG-fibronectin hydrogels were first described by Seliktar[131] and cowokers and have been demonstrated to support 3D culture of smooth muscle cells (SMCs),[132] chondrocytes,[133] cardiomyocytes,[134] and dorsal root ganglia.[135] PEGylation of fibrinogen or fibronectin gave rise to photocrosslinkable proteins that can be copolymerized with varying amount of PEGDA to form 3D hybrid hydrogels in the presence of UV irradiation.[136] The addition of fibrinogen or fibronectin into this system provided an opportunity for both cell adhesion and cell-mediated degradation and remodeling of the original gel with time. The mechanical properties of PEG-fibrinogen gels can be readily tuned to match that of the targeted tissues.[134] Although hybrid protein-containing hydrogels are promising candidates for use as 3D matrices for tissue engineering, it is difficult to obtain large quantity of pure and well-defined proteins for structure-function assessment, and the chemical modification of the proteins is often heterogeneous and ill-defined. A merge of the advances in protein modification and hydrogel formation will likely address these shortcomings and provide new matrices for tissue engineering.
Carbohydrate-Functionalized Responsive Hydrogels
In addition to proteins, the natural ECM also contains glycosaminoglycans (GAGs) that perform a wide spectrum of biological functions. Their incorporation into the aforementioned synthetic matrices can further enhance the overall bioactivities of the matrix (Figure 1). Contrary to proteins, GAG molecules usually exhibit significant chemical and structural heterogeneity. The direct, controlled synthesis of most of these complex macromolecules is not yet possible, limiting the potential for their controlled incorporation into tissue engineering matrices. Hyaluronic acid (HA), however, represents an exception. It is a simple and homogeneous molecular structure, comprised of β-1,4-linked d-glucuronic acid and β-1,3-N-acetyl-d-glucosamine disaccharide units.[137] Technological advance has permitted facile synthesis of HA with high molecular weight, narrow molecular weight distribution, and high purity by bacterial fermentation.[138] HA is an essential ECM component that not only modulates cellular adhesion, signaling, differentiation, and motility, but also plays a key role in natural wound healing and morphogenesis.[139] HA has recently become one of the most extensively investigated naturally polysaccharides due to its biocompatibility, viscoelasticity, and lack of immunogenicity.[27,140–144]
Prestwich and coworkers have developed a modular and hybrid hydrogels for 3D cell culture, with the goal of replicating the complexity of the native ECM environment with the minimum number of components necessary to allow cells to rebuild and replicate a given tissue. They have developed a set of modular components that can be assembled into biomimetic materials that meet these requirements. These semi-synthetic ECMs are based on HA derivatives as the matrix, modified gelatin for cell attachment, and PEGDA for stability and enhanced crosslinking kinetics. These covalently crosslinked, biodegradable hydrogels are suitable for 3D culture of primary and stem cells in vitro, and for tissue formation in vivo. The synthetic ECMs can be engineered to provide appropriate biological cues needed to recapitulate the complexity of a given ECM environment.[24]
Many investigations on HA-based hydrogels only utilize HA as a passive supporting material without taking advantage of its biological functions. Realizing the crucial involvement of HA in heart morphogenesis and its ability to interact with fibronectin, Anseth anticipated that HA-based scaffolds may simulate important aspects of natural valve development, providing a biomimetic environment that presents signals to encourage formation of a tissue engineered valve. In a 2D model study,[145] valvular interstitial cells (VICs) were found to adhere and spread on collagen and laminin-coated surfaces, but displayed altered morphology and did not proliferate. Further, VICs neither spread on surfaces modified with RGD, nor with other fibronectin-specific peptide sequences such as EILDV and PHSRN. On the other hand, photocrosslinked HA gels were found to facilitate the spreading and proliferation of VICs, forming a confluent monolayer on the gels within 4 d. Modified HA retained its ability to specifically bind FN, allowing for the formation of gels containing both HA and FN. VICs cultured on HA surfaces displayed significantly increased production of ECM, indicating that HA-based scaffolds may provide useful biological cues to stimulate heart valve tissue formation.
Moving these promising results to 3D culture,[141] VICs were encapsulated in enzymatically degradable, crosslinked hydrogels formed from HA and PEG macromers. These hydrogels were shown to have the dual function of permitting the diffusion of ECM elaborated by 3D cultured VICs and promoting the development of a specific matrix composition. Initial cleavage of hydrogel crosslinks, prior to network mass loss, permit the diffusion of collagen, while later stages of degradation promote elastin elaboration and suppress collagen production due to HA fragment release. Exogenous HA delivery through the cell culture media further demonstrated the utility of delivered HA on manipulating the secretory properties of encapsulated VICs. The morphology of VICs adopted within the hydrogel was not mentioned.
Another GAG that has enjoyed increasing application in tissue engineering is heparin, comprising variably sulfated disaccharides of iduronic acid and glucosamine. The highly negatively charged GAG plays an important role not only in coagulation but also in the mediation of binding to many proteins,[146,147] including antithrombin III and growth factors. The biological activity of the heparan sulfates in the ECM, most importantly their protein binding, has motivated the development of heparinized hydrogels for applications in affinity-based protein delivery, with ultimate applications in tissue engineering.[148–154] In these studies, heparin-modified hydrogels have shown useful protein-delivery profiles in support of desirable tissue engineering outcomes such as nerve regeneration and neovascularization. More recently, the direct impact of heparin on modulating cell behavior in human mesenchymal stem cells and porcine VICs has also been established.[155,156]
The use of noncovalent peptide-heparin and protein-heparin interactions has only more recently and less frequently been employed as a way to control both the protein-delivery profiles and mechanical properties of hydrogel matrices. The noncovalent approach offers advantages of reversible gelation on the basis of pH, temperature, changes in shear rate, salt concentration, and ligand concentration. In noncovalently assembled heparinized materials, two main strategies have been employed. In the first approach, described by Panitch and coworkers, vinyl sulfone-functionalized star PEGs were modified with cysteine-equipped, heparin-binding peptides (HBP) to yield PEG-HBP conjugates. Upon incubation of the PEG-HBP conjugates with heparin, viscoelastic materials were formed with mechanical properties that vary as a function of frequency[157] HBPs could be delivered from these matrices at rates that are controlled by the heparin-binding affinity of the peptide. Owing to the mechanical properties of these initial materials, additional, covalent, crosslinks comprising bis-cysteine-functionalized peptides were incorporated into the PEG-based hydrogels.[158] PEG stars carrying eight arms were employed in these studies, to permit some covalent crosslinking concomitant with functionalization of the arms with mono-cysteine-carrying HBPs. With the addition of heparin, these materials showed more rapid rates of gelation than that observed with covalent crosslinking alone, with increased final storage moduli (≈3–5 kPa). The dually crosslinked gels showed temperature- and frequency-dependence of their viscoelastic properties as expected.
Conversely, Kiick and coworkers have employed star PEGs modified with low-molecular-weight heparin (LMWH, M̄r = 3 000) for non-covalent assembly of responsive hydrogels. In this approach, a four-arm star, thiol-functionalized PEG was reacted with maleimide-functionalized LMWH, and the resulting soluble PEG-LMWH has been employed for hydrogel formation with various heparin-binding macromolecules (Figure 4a). Four-arm star PEGs modified with HBPs were initially employed for non-covalent hydrogel assembly, and were produced via the modification of PEG-vinyl-sulfone with cysteine-derivatized HBPs; degrees of substitution ranged from 70 to 100% depending on the HBP employed (based on the heparin-binding domains of antithrombin III, heparin interacting peptide, or human platelet factor 4).[159–161] Self-supporting hydrogels immediately formed upon mixing of solutions of the PEG-LMWH and PEG-HBP, even at reasonably low polymer concentrations (2.5 wt.-%). In contrast, mixing of PEG-HBP with LMWH alone did not result in the formation of hydrogels. In rheological studies, the hydrogels exhibited a bulk storage modulus that exceeds the loss modulus at all frequencies, indicating the formation of a permanently crosslinked network.[159,160] The modulus of the gels could be tuned by tuning the ratio of HBP:LMWH in the network, although an initial elastic response was observed even in the absence of PEG-HBP and was indicated to arise from partial association of LMWH termini.[162] Elastic moduli of several hundred Pa were reproducibly obtained for these hydrogels, regardless of the identity of the HBP, consistent with the properties expected on the basis of the kinetics of binding between LMWH and the HBP.[160] The hydrogels remained competent for binding multiple growth factors, and although growth factor release was mediated by a passive mechanism in these materials, the rate of release could be directly controlled by controlling the rate of erosion of the hydrogel network; the rates of release observed (≈14 d) are potentially useful for the treatment of ischemic conditions.[160,161] It is possible that co-release of PEG-LMWH with the GF may also improve the activities of the released GF.
Figure 4.
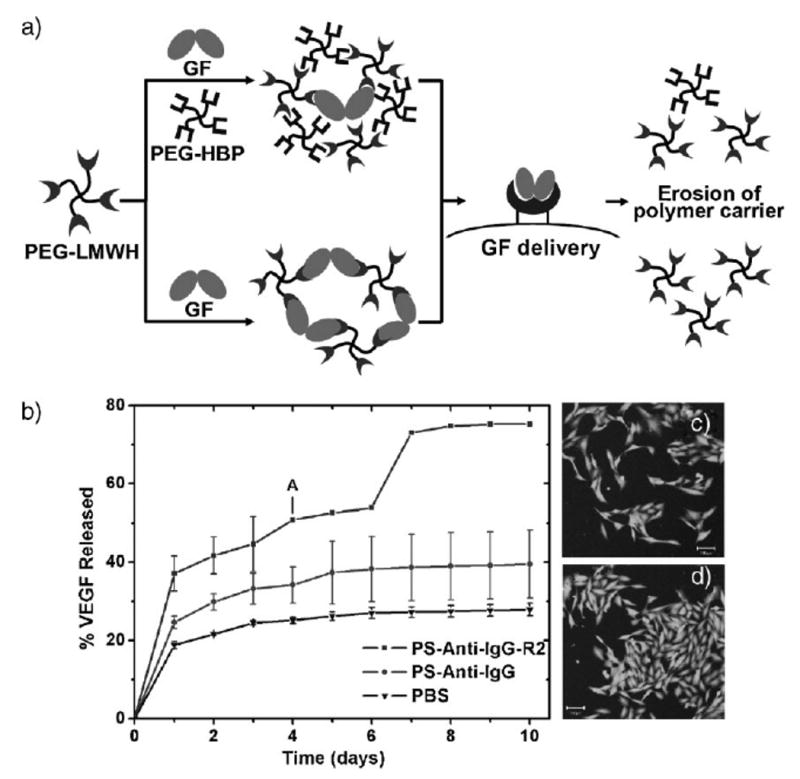
Assembly strategies, erosion data, and cell culture assays for non-covalently assembled PEG-LMWH hydrogels. (a) Schematic of noncovalent hydrogel assembly and erosion strategies employing PEG-LMWH. (b) 125I-VEGF release data for VEGF-crosslinked PEG-LMWH incubated in PBS in the presence of passivated PS particles and VEGFR-2-modified PS particles. Hydrogel erosion is apparent at day 4 (labeled as A). (c and d) Live/dead assays of porcine aortic endothelial cells evaluated in the presence of VEGF crosslinked gels; (c) represents PAE cells incubated with PEG-LMWH; and (d) represents PAE cells incubated in the presence of PEG-LMWH/VEGF hydrogels. Cell proliferation is increased in the presence of VEGF, and the gels do not exhibit toxicity to the cells. (Revised and reprinted from ref. [161, 163]. Copyright (2007) American Chemical Society, Copyright (2008) Royal Society of Chemistry.).
The utility of the PEG-LMWH for hydrogel formation upon binding multivalent PEG-HBP conjugates suggested additional opportunities in the assembly of cell-receptor responsive hydrogels via direct interaction of the PEG-LMWH with dimeric, heparin-binding growth factors. Indeed, mixing of homogeneous, low-viscosity solutions of PEG-LMWH with VEGF (vascular endothelial growth factor), at a final polymer concentration of 4 wt.-%, resulted in the immediate formation of hydrogels.[163] Characterization of the hydrogels via optical tweezer microrheology indicated formation of a weak viscoelastic material with storage moduli in excess of loss moduli at all frequencies. An increase in elastic modulus was observed with increased VEGF concentration (and not with the addition of a non-heparin binding protein, BSA), clearly demonstrating crosslinking by the growth factor. Of most importance for the application of these materials, their selective erosion in the presence of the VEGFR-2 receptor (which plays a primary role in controlling endothelial cell proliferation and migration[164]) has been demonstrated in in vitro experiments.[163] The release of 125I-labeled VEGF from the hydrogels was substantially increased (≈80% release) in the presence of VEGFR-2 modified polystyrene particles, with concomitant visible erosion of the hydrogel network within 4 d (Figure 4b). The released VEGF retained its bioactivity, as indicated by the VEGF-induced proliferation of porcine aortic endothelial cells (Figure 4c and d); importantly, live/dead assays of the PAE cells also indicated the lack of toxicity of the PEG-LMWH/VEGF hydrogels. In contrast, much lower VEGF release (≈40%) was observed in the presence of passivated PS particles, and hydrogels remained intact throughout the experiment (10 d). Similar results have been demonstrated in cell culture assays, and opportunities to employ this receptor-responsiveness in targeted drug delivery and controlled cell migration are underway. While the therapeutic use of these materials has not yet been explored, the opportunities to combine covalent and non-covalent crosslinking, and to employ peptides with specific affinities and therapeutic activities, suggest broad opportunities for these strategies.
Microscopic Integration of Particles into Hydrogel Matrices
As the field of materials science continues to evolve, sophisticated hydrogel scaffolds with hybrid composition, defined bioactivities, and tunable viscoelasticity are being developed. Artificial scaffolds were employed not only to create a 3D structure that mimics the architecture and function of tissue, but also provides a delivery vehicle for cells or to manipulate the environment of cells in vitro.[17] Our discussion on hybrid hydrogels is not limited to matrices containing polymers or macromolecules of different origins, chemical identities, and biological functions. Another distinct and important class of hybrid systems composed of hydrogel matrices with microscopic particles is also of interest to the field of tissue engineering. The particles, irrespective of their chemical nature, hydrophilicity, shape, or sizes, can be engineered as means for physical anchorage and structural reinforcement. In addition, they can be designed as controlled release depots for spatio-temporal presentation of biologically active compounds that ultimately elicit desired cellular activities. To accomplish these goals, the particles have been physically trapped, covalently bound, or coassembled in the matrices.
Hydrogels with Embedded Particles for Structural Control
To engineer mechanically active tissues, synthetic scaffolds that possess appropriate elasticity are required in order to effectively transmit the mechanical forces to the encapsulated cells.[165,166] This requirement is further substantiated by the discovery that cells preferentially differentiate on artificial ECMs that have mechanical stiffness similar to that of their natural tissues.[11,167] Traditional hydrogels are bulk gels consisting of randomly interconnected macromolecules dispersed in a sea of water. They lack hierarchical organizations and structural integrity necessary to facilitate cell infiltration and neovascularization. They exhibit slow response and inferior mechanical properties that pose significant challenges for engineering mechanically active tissues.
Attempts to improve the mechanical properties of traditional hydrogels have been focused mostly on the introduction of hard inorganic particles or hydrophobic particles that can reinforce the matrix.[168,169] However, these particles often aggregate readily and are not compatible with the surrounding hydrogel matrix. More importantly, the ill-defined particle-matrix interface can lead to compromised materials properties.[170] By homogeneously dispersing exfoliated inorganic clay and initiating polymerization from the surface of the clay particles,[171,172] Osada and Gong have constructed nanocomposite (NC) gels with unprecedented mechanical strength. In the NC gel, neighboring clay sheets act as multifunctional crosslinking agents for the polymers in the absence of organic crosslinker. The gel must be formed by initiating polymerization from the clay surface, resulting in the flexible polymer chains with random coil conformation connecting the clay and filling the space between the clay sheets. The preparation of this type of gel is not trivial; the initiator must have strong interactions with the clay sheets, and be stably adsorbed on the surface so that radical polymerization can be initiated from the clay surface and propagated to form the continuous network. This type of NC gel exhibits extraordinary mechanical properties unseen in traditional chemical gels. With a monomer/water ratio of 0.1, the NC gels have an elastic modulus of about 104 Pa, and can be stretched to about ten times their original length. The unique properties were attributed to the reduced fluctuation in the crosslinking density of the NC gels and the cooperativity of the polymer chains connecting the same clays.
Natural bone is a classical example of hybrid composite hydrogel containing organic matrix (collagens, proteoglycans, and cell adhesion proteins) integrated with a mineral phase composed of hydroxyapatite [HAp, Ca3(PO4)2Ca(OH)2].[173] The controlled integration of organic and inorganic components confers natural bone with superior mechanical properties.[174] HAp not only enhances the mechanical integrity of the organic matrix but also has high protein-binding capacity, and has been implied as the natural delivery system for bone morphogenic proteins (BMPs).[175,176] In bone tissue engineering, inorganic materials have been introduced in order to mimic the composite nature of the real bone, with a combination of toughness of a polymer phase with the compressive strength of an inorganic one to generate bioactive materials with improved mechanical properties and degradation profiles.[177]
Stupp and coworkers have reported the formation of composite material using self-assembled peptide-amphiphile (PA) nanofibers as the template.[178] PAs were designed to contain (i) a long alkyl tail that conveys hydrophobicity to the molecule; (ii) four consecutive cysteine residues for stabilization purposes; (iii) a flexible linker of three glycine residues to provide flexibility to the hydrophilic head group; (iv) a single phosphorylated serine residue that is designed to interact strongly with calcium ion and help direct mineralization of HAp; and (v) the cell-adhesive ligand RGD for enhanced cell-matrix interaction. Via the use of pH triggers, the PA self-assembled into a nanostructured fibrous scaffold reminiscent of extracellular matrix. The design of this PA allowed the nanofibers to be reversibly crosslinked via S–S bonds to enhance or decrease their structural integrity. After crosslinking, the fibers were shown to be able to direct mineralization of hydroxyapatite to form a composite material in which the crystallographic c axes of hydroxyapatite were aligned with the long axes of the fibers. This alignment is the same as that observed between collagen fibrils and HAp crystals in bone. This study is intriguing in that multiple self-assembly processes were closely mimicked and coordinated to form a hierarchical structure. The mechanical properties of the resulting hybrid gels were not investigated.
Recently, the same group[179] reported a method to prepare a hybrid bone implant material consisting of a Ti-6Al-4V foam, whose 52% porosity was filled with the PA nanofiber matrix. The Ti-6Al-4V was used presumably to improve the mechanical properties of PA/HAp. The PA molecules self-assembled into a nanofiber matrix within the pores of the metallic foam, fully occupying the foam's interconnected porosity. Furthermore, the method allows the encapsulation of cells within the bioactive matrix, and under appropriate conditions the nanofibers can nucleate mineralization of calcium phosphate phases with a Ca/P ratio that corresponds to that of HAp. The crystal structure and alignment of HAp was not discussed. Preliminary results demonstrate de novo bone formation around and inside the implant, vascularization around the implant, as well as the absence of a cytotoxic response. The PA-Ti hybrid strategy could be potentially tailored to initiate mineralization and direct a cellular response from the host tissue into porous implants to form new bone and thereby improve fixation, osteointegration, and long-term stability of implants.
Mann and coworkers[180] showed that supramolecular hydrogels with hybrid properties can be prepared by in situ calcium phosphate mineralization. Supramolecular hydrogels produced by enzymatic dephosphorylation of N-fluorenylmethyloxycarbonyltyrosine phosphate (I) have been used as self-assembled matrices for calcium phosphate mineralization. Dephosphorylation of N-fluorenylmethyloxycarbonyltyrosine phosphate (I) in water produced a compound that spontaneously assembles into nanofilaments due to π-stacking of the fluorenyl end groups, which adopt a helical orientation due to superhelical arrangements of the amino acid residues arising from hydrogen-bonding interactions between the terminal tyrosine groups. By controlling the levels of calcium and phosphate ions associated with the supramolecular structure, the researchers demonstrated that mineralized hydrogels of variable hardness can be prepared in the form of hybrid composites comprising interconnecting networks of calcified nanofilaments. The hybrid composites can be prepared as supramolecular gels, thin films, as well as hard macroporous monoliths, suggesting that they could be developed toward specific applications as novel biomaterials for tissue engineering, wound treatment, and drug release.
Hybrid Hydrogels with Embedded Particles for Drug Delivery
The development of scaffolding materials with controlled release potential may lead to novel multifunctional platforms able to control and guide the tissue regeneration processes.[17] Particle-based systems have been widely used for drug delivery because the release rate and bioavailability can be tailored by engineering particles with different composition, sizes, surface functionality, and particle morphology to tailor their release rate for specific applications.[9,181,182] Bulk hydrogels have also been widely used as release depots for protein drugs as well as small molecule therapeutics.[142,183,184] Generally, drug diffusion from the gels is not significantly hindered if they are simply dispersed within the bulk gel matrix without any covalent linkage or other specific physical interactions.[185] Thus, micro- or nanostructured matrices capable of sequestering and delivering bioactive compounds in a highly specific manner can be engineered by formulating particulate systems into the hydrogel matrix to form composite or “plum pudding” hydrogel networks (Figure 1).[184] This integration effectively overcomes the inherent pharmacological limitations of hydrogels and toxicity associated with particles alone, providing defined biological and biophysical cues for tissue repair, functional angiogenesis and stem cell differentiation. The hydrogel matrix and the dispersed particles may synergistically alter the drug release profile, eliminate the burst release, and extend duration of release.[186]
In one study, the release kinetics for dexamethasone (DX) and/or VEGF from PLGA microspheres and hydrogel alone was compared to that from PLGA-embedded hydrogels. The hydrogels were based on copolymers of 2-hydroxyethyl methacrylate (HEMA), 1-vinyl-2-pyrrolidinone (VP), and PEG. In vitro VEGF release from hydrogels was much greater than from VEGF-loaded microspheres embedded in hydrogels. DX release from microspheres embedded in hydrogels lacked the initial burst associated with DX-loaded hydrogels; however, the sustained release characteristics of DX release were similar from both embedded microspheres and hydrogels. Concurrent release of VEGF and DX was determined to be best from either VEGF/DX-loaded hydrogels or VEGF-loaded hydrogels with embedded DX microspheres.
In another study[187] the in vitro and in vivo biological activities of BMP-2 released from four sustained delivery vehicles for bone regeneration was systematically evaluated. BMP-2 was incorporated into (i) a gelatin hydrogel, (ii) PLGA microspheres embedded in a gelatin hydrogel, (iii) microspheres embedded in a poly(propylene fumarate) (PPF) scaffold, and (iv) microspheres embedded in a PPF scaffold surrounded by a gelatin hydrogel. The four implant types showed different in vitro release profiles over the 12-week period, which changed significantly upon implantation. The alkaline phosphatase (AP) induction by BMP-2 released from gelatin implants showed a loss in bioactivity after 6 weeks in culture, while the BMP-2 released from the other implants continued to show bioactivity over the full 12-week period. Significantly more bone formation after 6 weeks of implantation was observed in the microsphere/PPF scaffold composites. After 12 weeks, the amount of newly formed bone in the microsphere/PPF scaffolds remained significantly higher than that in the gelatin and microsphere/gelatin hydrogels, however, there was no statistical difference compared to the microsphere/PPF/gelatin composite.
Using confocal microscopy, researchers studied the distribution of drug-loaded PLGA microspheres within physically crosslinked poly(vinyl alcohol) (PVA) gels.[188] Due to the hydrophilicity mis-match (immiscibility) between the gel matrix and the microspheres, microparticles did not distribute evenly within the gels. The hydrogel composite system appeared to provide better control of hydrocortisone release than the microparticles and hydrogels alone. However, the inherent inhomogeneity of the composite hydrogels may prevent overall control in the pharmacological kinetics. Replacing the hydrophobic microspheres embedded in hydrogel matrices with hydrogel microparticles (or microgels) may overcome aforementioned difficulties.
Mikos and coworkers have developed composite hydrogels with growth factor-laden microgels embedded in a secondary hydrogel matrix for cartilage tissue engineering.[39] In most cases, the matrix is based on radically crosslinked oligo[poly(ethylene glycol) fumarate] (OPF) that is hydrolytically degradable, while the microgels (≈100 μm) are based on glutaraldehyde-crosslinked gelatin microparticles that are enzymatically degradable.[189] Their encapsulation procedure allows for a uniform distribution of microgels within the secondary hydrogel matrix.[190] Compared to growth factor release directly from the OPF matrix, incorporation of TGF-loaded gelatin microgels in the OPF matrix significantly attenuated both the burst release and the cumulative release. The equilibrium swelling ratio of the microgels was shown to be greater than that of the OPF component. Thus, the release kinetics can be controlled by adjusting OPF formulation and microparticle loading, factors affecting the swelling behavior of these composites. This technology was extended to the dual delivery of IGF-1 (insulin-like) and TGF-β1 by loading these growth factors into either the OPF hydrogel phase or gelatin microparticle phase of composites. Release profiles were successfully manipulated by altering the phase of growth factor loading and extent of microparticle crosslinking.[191] When bovine chondrocytes were embedded in composite hydrogels and coencapsulated with gelatin microgels loaded with TGF-β1[190], they attached to the surface of the microgels and formed cell aggregates, which were not observed in the absence of encapsulated microgels. There was a statistically significant increase in cellular proliferation for samples containing gelatin MPs as compared with samples without MPs. The release of TGF-β1 further increased cell construct cellularity. Over the same time period, GAG content per cell remained constant for all formulations, suggesting that the dramatic increase in cell number for samples with TGF-β1-loaded MPs was accompanied by maintenance of the cell phenotype. Overall, these data indicate the potential of OPF gelatin composites containing embedded chondrocytes and TGF-β1-loaded gelatin MPs as a novel strategy for cartilage tissue engineering.
Our discussion so far has been limited to composite hydrogel systems with chemically and functionally distinct dispersed phases (microspheres) and matrix phases. The absence of specific linkages between the two phases may give rise to a depleted interphase that can result in inferior mechanical properties and limit the tunability of the overall release profile for the composite gels. It is known that nature modulates the mechanical and biological properties of tissue by subtle adjustments of its composition, which yield perceivable alterations of its micro- or nanoscale organization. In an effort to develop injectable hydrogels for vocal fold tissue regeneration, Jia and coworkers have created a new class of HA-based hydrogel materials with HA hydrogel particles (HGPs) embedded in and covalently crosslinked to a secondary network that is also HA-based (Figure 5).[143,192] The critical biochemical and biomechanical roles HA plays in vocal folds suggest its relevance as a scaffolding material for vocal fold tissue engineering. Two types of HGPs with varying sizes and crosslinking chemistry were synthesized using different inverse emulsion systems and crosslinking chemistry. In situ crosslinking of HA derivatives carrying hydrazide (HAADH) and aldehyde (HAALD) functionalities within the inverse emulsion droplets stabilized by span 80 afforded spherical HA HGPs with an average particle size of ≈10 μm (HGP10).[143] Alternatively, HA HGPs with an average diameter of ≈0.9 μm (HGP0.9) were synthesized by chemical crosslinking of HA with divinylsulfone (DVS) using a sodium bis(2-ethylhexyl) sulfosuccinate (AOT)/isooctane reverse micelle system in the presence of 1-heptanol.[192] Both types of HGPs are more resistant to enzymatic degradation as compared to the bulk gels formed using the same crosslinking chemistry. While HGP10 exhibit residual functional groups (aldehyde and hydrazide) after the synthesis, HGP0.9 lacks readily accessible function groups for bioconjugation and subsequent covalent crosslinking. Thus, HGP0.9 were rendered crosslinkable by introducing aldehyde groups via sodium periodate oxidation (oxHGP). The aldehyde groups on both types of microgels were used as reactive handles for covalent crosslinking with HAADH or HAADH/HAALD, forming doubly crosslinked networks (DXNs) with structural integrity and tailored viscoelasticity. The resulting macroscopic gels contain two distinct hierarchical networks (DXNs): one within individual particles and another among different particles.
Figure 5.
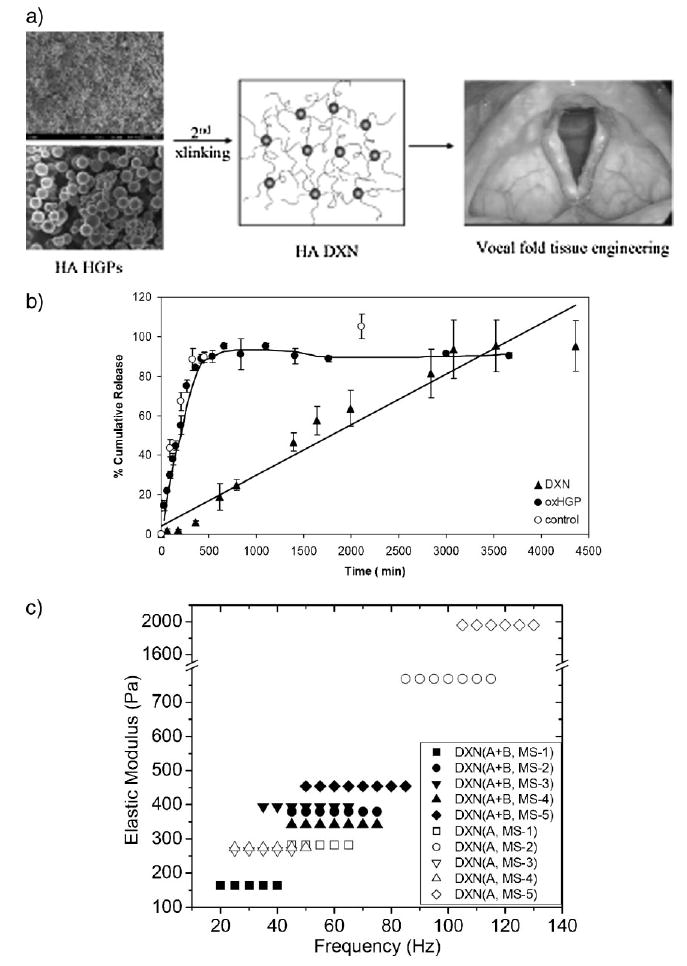
Design principles and basic materials properties of HA-based hydrogel particles (HGPs) and doubly crosslinked networks (DXNs). (a) HA-based HGPs can be linked together via a secondary crosslinker to form DXNs, which are targeted for vocal fold tissue engineering. Not drawn to scale. (b) Rhodamine 6G (R6G) release from HGP0.9 (filled triangles) and DXNs generated from HGP0.9 (filled circles). (c) Tunability of DXN viscoelasticity as determined by torsional wave apparatus at frequencies close to human phonation. (Revised and reprinted from ref. [143, 192]. Copyright (2006) American Chemical Society, Copyright (2008) Koninklijke Brill NV).
The HA-based HGPs are intended not just as inert filler materials, but instead as smart entities that can actively remodel the scar tissue. For this purpose, controlled release of pharmaceutically active compounds (such as growth factors and anti-inflammatory molecules) can be achieved through their anchorage at predetermined locales of the DXN system. In our preliminary studies, a model drug, rhodamine 6G (R6G), was physically trapped inside oxHGP0.9 and HGP10 by the electrostatic interaction between R6G and HA. It was determined that ≈84% of entrapped R6G was liberated from oxHGP0.9 at a rate of 0.24% · min−1 in the first 6 h, while the remaining R6G was gradually released during the next 2 d (Figure 5b). Similarly, approximately ≈80% of entrapped R6G was liberated from HGP10 at a rate of 0.22% · min−1 in the first 7 h, while the remaining R6G was gradually released during the next 1.5 d. The high release rate can be attributed to the rapid diffusion of R6G from oxHGP0.9 and HGP10 during hydration and swelling of the HGP. DXNs were prepared using the R6G-loaded HGP following the procedure described above. A rough estimation shows that R6G was released from HGP0.9-based DXN at a steady rate of 0.03% · min−1 for over 3 d. Faster release kinetics were observed for HGP10-based DXN (0.06% · min−1 for over 3 d). The secondary network introduced by HAADH may have resulted in a reinforced layer at the particle/matrix interface which is conducive to reaching a stable and slow release pattern of R6G.
Recently Jia and coworkers conducted an in-depth structural analysis of DXN made from HGP0.9 and HAADH. The average mesh size of the HGPs was approximately 5.5–7.0 nm determined by a protein uptake experiment. The hierarchical structure with the densely crosslinked HGPs embedded in and covalently linked to a loose secondary network was confirmed locally by cryo-SEM and globally by SANS/USANS. The presence of two levels of crosslinking allows the viscoelasticity of DXN to be readily adjusted by altering (i) the relative concentration of HGPs and the secondary crosslinker; (ii) the particle size and size distribution; (iii) the density of functional groups on the particles; (iv) intraparticles; and (v) interparticle crosslinking. Mechanical measurements using a custom-built torsional wave apparatus indicated that HA-based DXNs exhibit tunable elastic moduli (200–2 000 Pa) that are similar to those of vocal fold lamina propria at frequencies close to the range of human phonation (20–200 Hz) (Figure 5c). DXN exhibit statistically higher tan (δ) values than that of the bulk gels, indicating more damping capability. Derivatives with mutually reactive groups were included for comparison. Although both bulk gels (formed by direct mixing of HAALD and HAADH) and DXNs are stable, elastic gels that become stiffer at higher frequencies. DXNs are less sensitive to the frequency changes than bulk gels. The HA-based DXN offers unique structural hierarchy, mechanical properties, and potential biological functions that are suitable for vocal fold tissue engineering.
Conclusion
In this article, we have discussed recent advances in the engineering of hybrid multicomponent hydrogels consisting of chemically, morphologically, and functionally distinct modules interconnected via chemical or physical means. Depending on the size and the nature of the building blocks, the hybridization can occur at molecular level or microscopic scale. Most of the systems described here were inspired by the hybrid, hierarchical, and multifunctional nature of the native ECM. At the molecular level, integrating peptides/proteins and/or polysaccharides into a synthetic polymer hydrogel can impart biological functions and modulated responsiveness to the system. The molecular level hybridization also offers the flexibility and tunability in molecular architecture, chemical composition, and mechanical properties, considerably expanding design opportunities. At the microscopic scale, incorporation of micro- or nanoparticles into existing hydrogels may improve their mechanical properties as well as their biological functions. The embedded particles not only provide physical anchorage and structural reinforcement but also allow for spatio-temperal release of biologically active compounds capable of stimulating desired cell functions locally within the hybrid hydrogel matrix.
Hybrid approaches offer unprecedented opportunities in mimicking the biological tissues the materials are intended to replace, and thus efforts to overcome the inherent challenges of these approaches will continue. These materials are inherently heterogeneous, exhibiting chemical and structural variation at different length scales, which can complicate efforts to controllably engineer the materials as tissue engineering scaffolds. Stability and consistency are additional challenges to be addressed as the desired biological complexity of hybrid materials has increased. Continued advances in methods to maintain protein structure and function upon chemical modification will expand the use of proteins as structurally and biologically active components. New methods for particle synthesis and modification will enable the design of well-integrated hybrid materials with improved structural stability. The responsiveness demonstrated in some of these systems, although novel in its own right, will require tailoring for the application in drug delivery and tissue engineering. Finally, the immunogenicity related to the new materials will need to be addressed concomitant with materials design. Nevertheless, continued advances in materials science and engineering, translation of these advances to address biological challenges, and their integration with cell biology offer many promising approaches for the application of these hybrid materials in the engineering of new tissues.
Acknowledgments
Work in the authors' laboratories in these areas has been funded by grants from the National Institutes of Health [R01 DC008965 (X. J.); 1 R01 EB00317201 (K. L. K.)], the National Science Foundation [DMR-0643226 (X. J.); DBE-0221651(K. L. K.)], and the Arnold and Mabel Beckman Foundation (K. L. K). We thank Shuang Liu for her help with the schematic illustration of the hybrid hydrogel systems.
Biographies

Xinqiao Jia received her B.S. in Applied Chemistry from Fudan University in China in 1995 and her Ph.D. in Polymer Science and Engineering from the University of Massachusetts Amherst in 2002. She carried out her postdoctoral training with Professor Robert Langer at Massachusetts Institute of Technology prior to joining the Materials Science and Engineering Department at the University of Delaware in 2005. She is an affiliated faculty with several centers and institutes at UD that include the Center for Translational Cancer Research, Delaware Biotechnology Institute, Integrative Graduate Education & Research Traineeship Program, and Graduate Program at the Chemistry/Biology Interface. She received the NSF CAREER Award in 2006 to develop mechano-responsive biomaterials. Xinqiao Jia's current research is focused on the design, synthesis and characterization of biomimetic materials with controlled architectures and functionalities for biomedical applications. Her research activities are currently supported by National Science Foundation and National Institutes of Health.

Kristi L. Kiick is an Associate Professor of Materials Science and Engineering at the University of Delaware. She earned her B.S. degree in Chemistry (summa cum laude) from the University of Delaware, and an M.S. degree in Bioinorganic Chemistry from the University of Georgia as a National Science Foundation Graduate Fellow. Kiick began a doctoral program in Polymer Science and Engineering at the University of Massachusetts Amherst in 1996, funded by a National Defense Science and Engineering Graduate Fellowship. Her doctoral research, conducted at the California Institute of Technology under the direction of David A. Tirrell, pioneered new strategies for incorporating non-natural amino acids into protein-based materials. Dr. Kiick joined the UD faculty in the Department of Materials Science and Engineering as an Assistant Professor in August 2001 and was promoted to the rank of Associate Professor in 2007. Her research programs have been funded in part by a Camille and Henry Dreyfus Foundation New Faculty Award, a Beckman Young Investigator Award, an NSF CAREER Award, and a DuPont Young Professor Award; she has served as a project leader on an NIH Center of Biomedical Research Excellence awarded to the University of Delaware in 2002 and renewed in 2008. Her current research programs combine biosynthetic techniques, chemical methods, and bioinspired assembly strategies for producing novel materials with long-term applications in medicine and/or device technologies.
References
- 1.Freed LE, Vunjak-Novakovic G. Adv Drug Delivery Rev. 1998;33:15. doi: 10.1016/s0169-409x(98)00017-9. [DOI] [PubMed] [Google Scholar]
- 2.Cima LG, Langer R. Chem Eng Prog. 1993;6:46. [Google Scholar]
- 3.Lavik E, Langer R. Appl Microbiol Biotechnol. 2004;65:1. doi: 10.1007/s00253-004-1580-z. [DOI] [PubMed] [Google Scholar]
- 4.Lutolf MP, Hubbell JA. Nat Biotechnol. 2005;23:47. doi: 10.1038/nbt1055. [DOI] [PubMed] [Google Scholar]
- 5.Stevens MM, George JH. Science. 2005;310:1135. doi: 10.1126/science.1106587. [DOI] [PubMed] [Google Scholar]
- 6.Ma PX. Adv Drug Deliv Rev. 2008;60:184. doi: 10.1016/j.addr.2007.08.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ratner BD, Bryant SJ. Annu Rev Biomed Eng. 2004;6:41. doi: 10.1146/annurev.bioeng.6.040803.140027. [DOI] [PubMed] [Google Scholar]
- 8.Shin H, Jo S, Mikos AG. Biomaterials. 2003;24:4353. doi: 10.1016/s0142-9612(03)00339-9. [DOI] [PubMed] [Google Scholar]
- 9.Goldberg M, Langer R, Jia XQ. J Biomater Sci Polym Ed. 2007;18:241. doi: 10.1163/156856207779996931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Zagris N. Micron. 2001;32:427. doi: 10.1016/s0968-4328(00)00011-1. [DOI] [PubMed] [Google Scholar]
- 11.Discher DE, Janmey P, Wang YL. Science. 2005;310:1139. doi: 10.1126/science.1116995. [DOI] [PubMed] [Google Scholar]
- 12.Ghosh K, Ingber DE. Adv Drug Delivery Rev. 2007;59:1306. doi: 10.1016/j.addr.2007.08.014. [DOI] [PubMed] [Google Scholar]
- 13.Griffith LG, Swartz MA. Nat Rev Mol Cell Biol. 2006;7:211. doi: 10.1038/nrm1858. [DOI] [PubMed] [Google Scholar]
- 14.Nerem RM. Tissue Eng. 2006;12:1143. doi: 10.1089/ten.2006.12.1143. [DOI] [PubMed] [Google Scholar]
- 15.Hwang NS, Varghese S, Elisseeff J. Adv Drug Deliv Rev. 2008;60:199. doi: 10.1016/j.addr.2007.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cao L, Mooney DJ. Adv Drug Deliv Rev. 2007;59:1340. doi: 10.1016/j.addr.2007.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Biondi M, Ungaro F, Quaglia F, Netti PA. Adv Drug Delivery Rev. 2008;60:229. doi: 10.1016/j.addr.2007.08.038. [DOI] [PubMed] [Google Scholar]
- 18.Lee KY, Mooney DJ. Chem Rev. 2001;101:1869. doi: 10.1021/cr000108x. [DOI] [PubMed] [Google Scholar]
- 19.Peppas NA. Curr Opin Colloid Interface Sci. 1997;2:531. [Google Scholar]
- 20.Ulijn RV, Bibi N, Jayawarna V, Thornton PD, Todd SJ, Mart RJ, Smith AM, Gough JE. Mater Today. 2007;10:40. [Google Scholar]
- 21.Gil ES, Hudson SA. Prog Polym Sci. 2004;29:1173. [Google Scholar]
- 22.Osada Y, Gong JP. Adv Mater. 1998;10:827. [Google Scholar]
- 23.Mart RJ, Osborne RD, Stevens MM, Ulijn RV. Soft Matter. 2006;2:822. doi: 10.1039/b607706d. [DOI] [PubMed] [Google Scholar]
- 24.Serban MA, Prestwich GD. Methods. 2008;45:93. doi: 10.1016/j.ymeth.2008.01.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ifkovits JL, Burdick JA. Tissue Eng Part A. 2007;13:2369. doi: 10.1089/ten.2007.0093. [DOI] [PubMed] [Google Scholar]
- 26.Van Tomme SR, Storm G, Hennink WE. Int J Pharm. 2008;355:1. doi: 10.1016/j.ijpharm.2008.01.057. [DOI] [PubMed] [Google Scholar]
- 27.Jia XQ, Burdick JA, Kobler J, Clifton RJ, Rosowski JJ, Zeitels SM, Langer R. Macromolecules. 2004;37:3239. [Google Scholar]
- 28.Ladet S, David L, Domard A. Nature. 2008;452:76. doi: 10.1038/nature06619. [DOI] [PubMed] [Google Scholar]
- 29.Anderson DG, Burdick JA, Langer R. Science. 2004;305:1923. doi: 10.1126/science.1099987. [DOI] [PubMed] [Google Scholar]
- 30.Robinson PS, Johnson SL, Evans MC, Barocas VH, Tranquillo RT. Tissue Eng Part A. 2008;14:83. doi: 10.1089/ten.a.2007.0148. [DOI] [PubMed] [Google Scholar]
- 31.Long JL, Tranquillo RT. Matrix Biol. 2003;22:339. doi: 10.1016/s0945-053x(03)00052-0. [DOI] [PubMed] [Google Scholar]
- 32.Wallace DG, Rosenblatt J. Adv Drug Delivery Rev. 2003;55:1631. doi: 10.1016/j.addr.2003.08.004. [DOI] [PubMed] [Google Scholar]
- 33.Zhong SP, Teo WE, Zhu X, Beuerman R, Ramakrishna S, Yung LYL. Biomacromolecules. 2005;6:2998. doi: 10.1021/bm050318p. [DOI] [PubMed] [Google Scholar]
- 34.Zhang S, Zhao X. J Mater Chem. 2004;14:2082. [Google Scholar]
- 35.Stupp SI, Donners J, Li LS, Mata A. MRS Bull. 2005;30:864. [Google Scholar]
- 36.Schneider JP, Pochan DJ, Ozbas B, Rajagopal K, Pakstis L, Kretsinger J. J Am Chem Soc. 2002;124:15030. doi: 10.1021/ja027993g. [DOI] [PubMed] [Google Scholar]
- 37.Mahoney MJ, Anseth KS. Biomaterials. 2006;27:2265. doi: 10.1016/j.biomaterials.2005.11.007. [DOI] [PubMed] [Google Scholar]
- 38.Nicodemus GD, Bryant SJ. Tissue Eng Part B: Rev. 2008;14:149. doi: 10.1089/ten.teb.2007.0332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Holland TA, Bodde EWH, Baggett LS, Tabata Y, Mikos AG, Jansen JA. Biomed J Mater Res, Part A. 2005;75A:156. doi: 10.1002/jbm.a.30379. [DOI] [PubMed] [Google Scholar]
- 40.Aumailley M, Gayraud B. J Mol Med. 1998;76:253. doi: 10.1007/s001090050215. [DOI] [PubMed] [Google Scholar]
- 41.Gullberg D, Ekblom P. Int J Dev Biol. 1995;39:845. [PubMed] [Google Scholar]
- 42.Birk DE, Yurchenco PD, Mecham RP. Extracellular Matrix Assembly and Structure. Academic Press; San Diego: 1994. [Google Scholar]
- 43.Daley WP, Peters SB, Larsen M. J Cell Sci. 2008;121:255. doi: 10.1242/jcs.006064. [DOI] [PubMed] [Google Scholar]
- 44.Bosman FT, Stamenkovic I. J Pathol. 2003;200:423. doi: 10.1002/path.1437. [DOI] [PubMed] [Google Scholar]
- 45.Coessens V, Pintauer T, Matyjaszewski K. Prog Polym Sci. 2001;26:337. [Google Scholar]
- 46.Kolb HC, Finn MG, Sharpless KB. Angew Chem, Int Ed. 2001;40:2004. doi: 10.1002/1521-3773(20010601)40:11<2004::AID-ANIE2004>3.0.CO;2-5. [DOI] [PubMed] [Google Scholar]
- 47.Hawker CJ, Wooley KL. Science. 2005;309:1200. doi: 10.1126/science.1109778. [DOI] [PubMed] [Google Scholar]
- 48.Sperling LH. Introduction to Physical Polymer Science. Wiley Interscience; Hoboken: 2006. [Google Scholar]
- 49.Creighton TE. Proteins: Structures and Molecular Properties. W.H. Freeman and Company; New York: 1993. [Google Scholar]
- 50.Yurchenco PD, Birk DE, Mecham RP. Extracellular Matrix Assembly and Structure. Academic; San Diego: 1994. [Google Scholar]
- 51.Chan WC, White PD. Fmoc Solid Phase Peptide Synthesis: A Practical Approach. Oxford University Press; New York: 2000. [Google Scholar]
- 52.Macmillan D. Angew Chem, Int Ed. 2006;45:7668. doi: 10.1002/anie.200602945. [DOI] [PubMed] [Google Scholar]
- 53.Maskarinec SA, Tirrell DA. Curr Opin Biotechnol. 2005;16:422. doi: 10.1016/j.copbio.2005.06.009. [DOI] [PubMed] [Google Scholar]
- 54.Bellingham CM, Lillie MA, Gosline JM, Wright GM, Starcher BC, Bailey AJ, Woodhouse KA, Keeley FW. Biomaterials. 2003;70:445. doi: 10.1002/bip.10512. [DOI] [PubMed] [Google Scholar]
- 55.Langer R, Tirrell DA. Nature. 2004;428:487. doi: 10.1038/nature02388. [DOI] [PubMed] [Google Scholar]
- 56.Ottani V, Martini D, Franchi M, Ruggeri A, Raspanti M. Micron. 2002;33:587. doi: 10.1016/s0968-4328(02)00033-1. [DOI] [PubMed] [Google Scholar]
- 57.Debelle L, Tamburro AM. Int J Biochem Cell Biol. 1999;31:261. doi: 10.1016/s1357-2725(98)00098-3. [DOI] [PubMed] [Google Scholar]
- 58.Aigner T, Stove J. Adv Drug Deliv Rev. 2003;55:1569. doi: 10.1016/j.addr.2003.08.009. [DOI] [PubMed] [Google Scholar]
- 59.Nam K, Kimura T, Kishida A. Biomaterials. 2007;28:1. doi: 10.1016/j.biomaterials.2006.08.002. [DOI] [PubMed] [Google Scholar]
- 60.Stachowiak AN, Irvine DJ. J Biomed Mater Res, Part A. 2008;85A:815. doi: 10.1002/jbm.a.31661. [DOI] [PubMed] [Google Scholar]
- 61.Fratzl P. Collagen: Structure and Mechanics. Springer; Heidelberg: 2008. [Google Scholar]
- 62.Buehler MJ. Proc Natl Acad Sci USA. 2006;103:12285. doi: 10.1073/pnas.0603216103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Woolfson DN, Ryadnov MG. Curr Opin Chem Biol. 2006;10:559. doi: 10.1016/j.cbpa.2006.09.019. [DOI] [PubMed] [Google Scholar]
- 64.Goodman M, Bhumralkar M, Jefferson EA, Kwak J, Locardi E. Biopolymers. 1998;47:127. doi: 10.1002/(SICI)1097-0282(1998)47:2<127::AID-BIP2>3.0.CO;2-W. [DOI] [PubMed] [Google Scholar]
- 65.Kotch FW, Raines RT. Proc Natl Acad Sci USA. 2006;103:3028. doi: 10.1073/pnas.0508783103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Cejas MA, Kinnney WA, Chen C, Vinter JG, Almond HR, Balss KM, Maryanoff CA, Schmidt U, Breslav M, Mahan A, Lacy E, Maryanoff BE. Proc Natl Acad Sci USA. 2008;105:8513. doi: 10.1073/pnas.0800291105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Lee HJ, Lee JS, Chansakul T, Yu C, Elisseeff JH, Yu SM. Biomaterials. 2006;27:5268. doi: 10.1016/j.biomaterials.2006.06.001. [DOI] [PubMed] [Google Scholar]
- 68.Wang AY, Foss CA, Leong S, Mo X, Pomper MG, Yu SM. Biomacromolecules. 2008;9:1755. doi: 10.1021/bm701378k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Starcher BC, Galione MJ. Anal Biochem. 1976;74:441. doi: 10.1016/0003-2697(76)90224-4. [DOI] [PubMed] [Google Scholar]
- 70.Daamen WF, Hafmans T, Weerkamp JH, van Kuppevelt TH. Biomaterials. 2001;22:1997. doi: 10.1016/s0142-9612(00)00383-5. [DOI] [PubMed] [Google Scholar]
- 71.Smith DW, Brown DM, Carnes WH. J Biol Chem. 1972;247:2427. [PubMed] [Google Scholar]
- 72.Singla A, Lee CH. J Biomed Mater Res. 2002;60:368. doi: 10.1002/jbm.10077. [DOI] [PubMed] [Google Scholar]
- 73.Hammond TH, Gray SD, Butler J, Zhou R, Hammond E. Otolaryngol Head Neck Surg. 1998;119:314. doi: 10.1016/S0194-5998(98)70071-3. [DOI] [PubMed] [Google Scholar]
- 74.Hammond TH, Zhou R, Hammond EH, Pawlak A, Gray SD. J Voice. 1997;11:59. doi: 10.1016/s0892-1997(97)80024-0. [DOI] [PubMed] [Google Scholar]
- 75.Sandberg L. Int Rev Connect Tissue. 1976;6:160. doi: 10.1016/b978-0-12-363707-9.50010-1. [DOI] [PubMed] [Google Scholar]
- 76.Sandberg LB, Soskel NT, Leslie JG. New Engl J Med. 1981;304:566. doi: 10.1056/NEJM198103053041004. [DOI] [PubMed] [Google Scholar]
- 77.Alberts B, Johnson A, Lewis J, Raff M, Roberts K, Walter P. Molecular Biology of the Cell. 4th. Garland Science; New York; 2002. [Google Scholar]
- 78.Vrhovski B, Weiss AS. Eur J Biochem. 1998;258:1. doi: 10.1046/j.1432-1327.1998.2580001.x. [DOI] [PubMed] [Google Scholar]
- 79.Aaron BB, Gosline JM. Biopolymers. 1981;20:1247. [Google Scholar]
- 80.Fleming WW, Sullivan CE, Torchia DA. Biopolymers. 1980;19:597. doi: 10.1002/bip.1980.360190311. [DOI] [PubMed] [Google Scholar]
- 81.Martino M, Oerri T, Tamburro A. Macromol Biosci. 2002;2:319. [Google Scholar]
- 82.Urry DW. J Protein Chem. 1988;7:1. doi: 10.1007/BF01025411. [DOI] [PubMed] [Google Scholar]
- 83.Tamburro AM, Guantieri V. In: Classical and Fractal Description of Elastin Structure. Rossi C, Tiezzi E, editors. Elsevier; Amsterdam: 1991. p. 391. [Google Scholar]
- 84.Tamburro AM, Guantieri V, Pandolfo L, Scopa A. Biopolymers. 1990;29:855. doi: 10.1002/bip.360290419. [DOI] [PubMed] [Google Scholar]
- 85.Gosline JM. Biopolymers. 1978;17:677. doi: 10.1002/bip.1978.360170311. [DOI] [PubMed] [Google Scholar]
- 86.Li DY, Brooke B, Davis EC, Mecham RP, Sorensen LK, Boak BB, Eichwald E, Keating MT. Nature. 1998;393:276. doi: 10.1038/30522. [DOI] [PubMed] [Google Scholar]
- 87.Faury G, Garnier S, Weiss AS, Wallach J, Fulop T, Jacob MP, Mecham RP, Robert L, Verdetti J. Circ Res. 1998;82:328. doi: 10.1161/01.res.82.3.328. [DOI] [PubMed] [Google Scholar]
- 88.Wright ER, Conticello VP. Adv Drug Deliv Rev. 2002;54:1057. doi: 10.1016/s0169-409x(02)00059-5. [DOI] [PubMed] [Google Scholar]
- 89.Wright ER, McMillan RA, Cooper A, Apkarian RP, Conticello VP. Adv Funct Mater. 2002;12:149. [Google Scholar]
- 90.Chilkoti A, Dreher MR, Meyer DE, Raucher D. Adv Drug Deliv Rev. 2002;54:613. doi: 10.1016/s0169-409x(02)00041-8. [DOI] [PubMed] [Google Scholar]
- 91.Lee J, Macosko CW, Urry DW. Biomacromolecules. 2001;2:170. doi: 10.1021/bm0000900. [DOI] [PubMed] [Google Scholar]
- 92.Reiersen H, Clarke AR, Rees AR. J Mol Biol. 1998;283:255. doi: 10.1006/jmbi.1998.2067. [DOI] [PubMed] [Google Scholar]
- 93.Schreiner E, Nicolini C, Ludolph B, Ravindra R, Otte N, Kohlmeyer A, Rousseau R, Winter R, Marx D. Phys Rev Lett. 2004;92:148101. doi: 10.1103/PhysRevLett.92.148101. [DOI] [PubMed] [Google Scholar]
- 94.Heilshorn SC, DiZio KA, Welsh ER, Tirrell DA. Biomaterials. 2003;24:4245. doi: 10.1016/s0142-9612(03)00294-1. [DOI] [PubMed] [Google Scholar]
- 95.Daamen WF, Veerkamp JH, van Hest JCM, van Kuppevelt TH. Biomaterials. 2007;28:4378. doi: 10.1016/j.biomaterials.2007.06.025. [DOI] [PubMed] [Google Scholar]
- 96.Kiick KL. Polym Rev. 2007;47:1. [Google Scholar]
- 97.Kim W, Conticello VP. Polym Rev. 2007;47:93. [Google Scholar]
- 98.Simnick AJ, Lim DW, Chow D, Chilkoti A. Polym Rev. 2007;47:121. [Google Scholar]
- 99.Grieshaber SE, Farran AJE, Lin-Gibson S, Kiick KL, Jia X. doi: 10.1021/ma802791z. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Flory JP. Principle of Polymer Chemistry. Cornell University Press; Ithaca: 1953. [Google Scholar]
- 101.Flory JP. Statistical Mechanics of Chain Molecules. New York. Wiley-Interscience; 1969. [Google Scholar]
- 102.Flory PJ. Polymer. 1979;20:1317. [Google Scholar]
- 103.Miao M, Cirulis JT, Lee S, Keeley FW. Biochemistry. 2005;44:14367. doi: 10.1021/bi0510173. [DOI] [PubMed] [Google Scholar]
- 104.Lillie MA, Gosline JM. Biopolymers. 1990;29:1147. doi: 10.1002/bip.360290805. [DOI] [PubMed] [Google Scholar]
- 105.Dennes TJ, Schwartz J. Soft Matter. 2008;4:86. doi: 10.1039/b714947f. [DOI] [PubMed] [Google Scholar]
- 106.Vogel V, Sheetz M. Nat Rev Mol Cell Biol. 2006;7:265. doi: 10.1038/nrm1890. [DOI] [PubMed] [Google Scholar]
- 107.Herczenik E, Gebbink M. FASEB J. 2008;22:2115. doi: 10.1096/fj.07-099671. [DOI] [PubMed] [Google Scholar]
- 108.Kopecek J. Eur J Pharm Sci. 2003;20:1. doi: 10.1016/s0928-0987(03)00164-7. [DOI] [PubMed] [Google Scholar]
- 109.Ulbrich K, Strohalm J, Kopecek J. Biomaterials. 1982;3:150. doi: 10.1016/0142-9612(82)90004-7. [DOI] [PubMed] [Google Scholar]
- 110.Obaidat AA, Park K. Pharm Res. 1996;13:989. doi: 10.1023/a:1016090103979. [DOI] [PubMed] [Google Scholar]
- 111.Miyata T, Asami N, Uragami T. Nature. 1999;399:766. doi: 10.1038/21619. [DOI] [PubMed] [Google Scholar]
- 112.Miyata T, Jige M, Nakaminami T, Uragami T. Proc Natl Acad Sci USA. 2006;103:1190. doi: 10.1073/pnas.0506786103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kopecek J. Biomaterials. 2007;28:5185. doi: 10.1016/j.biomaterials.2007.07.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang C, Stewart RJ, Kopecek J. Nature. 1999;397:417. doi: 10.1038/17092. [DOI] [PubMed] [Google Scholar]
- 115.Chen L, Kopecek J, Stewart RJ. Bioconjugate Chem. 2000;11:734. doi: 10.1021/bc000046h. [DOI] [PubMed] [Google Scholar]
- 116.Yang JY, Xu CY, Wang C, Kopecek J. Biomacromolecules. 2006;7:1187. doi: 10.1021/bm051002k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Murphy WL, Dillmore WS, Modica J, Mrksich M. Angew Chem, Int Ed. 2007;46:3066. doi: 10.1002/anie.200604808. [DOI] [PubMed] [Google Scholar]
- 118.Sui ZJ, King WJ, Murphy WL. Adv Mater. 2007;19:3377. [Google Scholar]
- 119.Sui ZJ, King WJ, Murphy WL. Adv Funct Mater. 2008;18:1824. doi: 10.1002/adfm.200800218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Esser-Kahn AP, Francis MB. Angew Chem, Int Ed. 2008;47:3751. doi: 10.1002/anie.200705564. [DOI] [PubMed] [Google Scholar]
- 121.Hersel U, Dahmen C, Kessler H. Biomaterials. 2003;24:4385. doi: 10.1016/s0142-9612(03)00343-0. [DOI] [PubMed] [Google Scholar]
- 122.Sakiyama-Elbert SE, Hubbell JA. Annu Rev Mater Res. 2001;31:183. [Google Scholar]
- 123.West JL, Hubbell JA. Macromolecules. 1999;32:241. [Google Scholar]
- 124.Mann BK, Gobin AS, Tsai AT, Schmedlen RH, West JL. Biomaterials. 2001;22:3045. doi: 10.1016/s0142-9612(01)00051-5. [DOI] [PubMed] [Google Scholar]
- 125.Lutolf MP, Hubbell JA. Biomacromolecules. 2003;4:713. doi: 10.1021/bm025744e. [DOI] [PubMed] [Google Scholar]
- 126.Lutolf MP, Raeber GP, Zisch AH, Tirelli N, Hubbell JA. Adv Mater. 2003;15:888. [Google Scholar]
- 127.Lutolf MP, Lauer-Fields JL, Schmoekel HG, Metters AT, Weber FE, Fields GB, Hubbell JA. Proc Natl Acad Sci USA. 2003;100:5413. doi: 10.1073/pnas.0737381100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kraehenbuehl TP, Zammaretti P, Van der Vlies AJ, Schoenmakers RG, Lutolf MP, Jaconi ME, Hubbell JA. Biomaterials. 2008;29:2757. doi: 10.1016/j.biomaterials.2008.03.016. [DOI] [PubMed] [Google Scholar]
- 129.Salinas CN, Anseth KS. Macromolecules. 2008;41:6019. [Google Scholar]
- 130.Halstenberg S, Panitch A, Rizzi S, Hall H, Hubbell JA. Biomacromolecules. 2002;3:710. doi: 10.1021/bm015629o. [DOI] [PubMed] [Google Scholar]
- 131.Gonen-Wadmany M, Oss-Ronen L, Seliktar D. Biomaterials. 2007;28:3876. doi: 10.1016/j.biomaterials.2007.05.005. [DOI] [PubMed] [Google Scholar]
- 132.Peyton SR, Kim PD, Ghajar CM, Seliktar D, Putnam AJ. Biomaterials. 2008;29:2597. doi: 10.1016/j.biomaterials.2008.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Schmidt O, Mizrahi J, Elisseeff J, Seliktar D. Biotechnol Bioeng. 2006;95:1061. doi: 10.1002/bit.21072. [DOI] [PubMed] [Google Scholar]
- 134.Shapira-Schweitzer K, Seliktar D. Acta Biomater. 2007;3:33. doi: 10.1016/j.actbio.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 135.Sarig-Nadir O, Seliktar D. Tissue Eng Part A. 2008;14:401. doi: 10.1089/tea.2007.0029. [DOI] [PubMed] [Google Scholar]
- 136.Dikovsky D, Bianco-Peled H, Seliktar D. Biomaterials. 2006;27:1496. doi: 10.1016/j.biomaterials.2005.09.038. [DOI] [PubMed] [Google Scholar]
- 137.Garg HG, Hales CA. Chemistry and Biology of Hyaluronan. 1st. Elsevier Ltd; Oxford: 2004. [Google Scholar]
- 138.Chong BF, Blank LM, McLaughlin R, Nielsen LK. Appl Microbiol Biotechnol. 2005;66:341. doi: 10.1007/s00253-004-1774-4. [DOI] [PubMed] [Google Scholar]
- 139.Laurent TCE. The Chemistry, Biology, and Medical Applications of Hyaluronan and Its Derivatives. Portland Press; Miami: 1998. [Google Scholar]
- 140.Chung C, Erickson IE, Mauck RL, Burdick JA. Tissue Eng Part A. 2008;14:1121. doi: 10.1089/ten.tea.2007.0291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Shah DN, Recktenwall-Work SM, Anseth KS. Biomaterials. 2008;29:2060. doi: 10.1016/j.biomaterials.2008.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Jia XQ, Colombo G, Padera R, Langer R, Kohane DS. Biomaterials. 2004;25:4797. doi: 10.1016/j.biomaterials.2003.12.012. [DOI] [PubMed] [Google Scholar]
- 143.Jia XQ, Yeo Y, Clifton RJ, Jiao T, Kohane DS, Kobler JB, Zeitels SM, Langer R. Biomacromolecules. 2006;7:3336. doi: 10.1021/bm0604956. [DOI] [PubMed] [Google Scholar]
- 144.Leach JB, Schmidt CE. Biomaterials. 2005;26:125. doi: 10.1016/j.biomaterials.2004.02.018. [DOI] [PubMed] [Google Scholar]
- 145.Masters KS, Shah DN, Walker G, Leinwand LA, Anseth KS. J Biomed Mater Res, Part A. 2004;71A:172. doi: 10.1002/jbm.a.30149. [DOI] [PubMed] [Google Scholar]
- 146.Capila I, Linhardt RJ. Angew Chem, Int Ed. 2002;41:390. doi: 10.1002/1521-3773(20020201)41:3<390::aid-anie390>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 147.Fromm JR, Hileman RE, Caldwell EEO, Weiler JM, Linhardt RJ. Arch Biochem Biophys. 1997;343:92. doi: 10.1006/abbi.1997.0147. [DOI] [PubMed] [Google Scholar]
- 148.Sakiyama-Elbert SE, Hubbell JA. J Controlled Release. 2000;65:389. doi: 10.1016/s0168-3659(99)00221-7. [DOI] [PubMed] [Google Scholar]
- 149.Cai S, Liu Y, Zheng Shu X, Prestwich GD. Biomaterials. 2005;26:6054. doi: 10.1016/j.biomaterials.2005.03.012. [DOI] [PubMed] [Google Scholar]
- 150.Tae GY, Scatena M, Stayton P, Hoffman AS. J Biomater Sci Polym Ed. 2006;17:187. doi: 10.1163/156856206774879090. [DOI] [PubMed] [Google Scholar]
- 151.Wissink MJB, Beernink R, Poot AA, Engbers GHM, Beugeling T, van Aken WG, Feijen J. J Controlled Release. 2000;64:103. doi: 10.1016/s0168-3659(99)00145-5. [DOI] [PubMed] [Google Scholar]
- 152.Nakamura S, Ishihara M, Obara K, Masuoka K, Ishizuka T, Kanatani Y, Takase B, Matsui T, Hattori H, Sato T, Kariya Y, Maehara T. J Biomed Mater Res, Part A. 2006;78A:364. doi: 10.1002/jbm.a.30688. [DOI] [PubMed] [Google Scholar]
- 153.Liu LS, Ng CK, Thompson AY, Poser JW, Spiro RC. J Biomed Mater Res. 2002;62:128. doi: 10.1002/jbm.10238. [DOI] [PubMed] [Google Scholar]
- 154.Chinen N, Tanihara M, Nakagawa M, Shinozaki K, Yamamoto E, Mizushima Y, Suzuki Y. J Biomed Mater Res, Part A. 2003;67A:61. doi: 10.1002/jbm.a.10061. [DOI] [PubMed] [Google Scholar]
- 155.Benoit DSW, Anseth KS. Acta Biomater. 2005;1:461. doi: 10.1016/j.actbio.2005.03.002. [DOI] [PubMed] [Google Scholar]
- 156.Cushing MC, Liao JT, Jaeggli MP, Anseth KS. Biomaterials. 2007;28:3378. doi: 10.1016/j.biomaterials.2007.04.005. [DOI] [PubMed] [Google Scholar]
- 157.Seal BL, Panitch A. Biomacromolecules. 2003;4:1572. doi: 10.1021/bm0342032. [DOI] [PubMed] [Google Scholar]
- 158.Seal BL, Panitch A. Macromolecules. 2006;39:2268. [Google Scholar]
- 159.Yamaguchi N, Chae BS, Zhang L, Kiick KL, Furst EM. Biomacromolecules. 2005;6:1931. doi: 10.1021/bm0500042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Zhang L, Furst EM, Kiick KL. J Controlled Release. 2006;114:130. doi: 10.1016/j.jconrel.2006.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Kiick KL. Soft Matter. 2008;4:29. doi: 10.1039/b711319f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Spinelli FJ, Kiick KL, Furst EM. Biomaterials. 2007 doi: 10.1016/j.biomaterials.2007.11.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Yamaguchi N, Zhang L, Chae BS, Palla CS, Furst EM, Kiick KL. J Am Chem Soc. 2007;129:3040. doi: 10.1021/ja0680358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Neufeld G, Cohen T, Gengrinovitch S, Poltorak Z. FASEB J. 1999;13:9. [PubMed] [Google Scholar]
- 165.Ingber DE. FASEB J. 2006;20:811. doi: 10.1096/fj.05-5424rev. [DOI] [PubMed] [Google Scholar]
- 166.Kung C. Nature. 2005;436:647. doi: 10.1038/nature03896. [DOI] [PubMed] [Google Scholar]
- 167.Engler AJ, Griffin MA, Sen S, Bonnetnann CG, Sweeney HL, Discher DE. J Cell Biol. 2004;166:877. doi: 10.1083/jcb.200405004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Malkoch M, Vestberg R, Gupta M, Mespouille L, Dubois P, Mason AF, Hedrick JL, Liao Q, Frank CW, Kingsburg K, Hawker CJ. Chem Commun. 2006:2774. doi: 10.1039/b603438a. [DOI] [PubMed] [Google Scholar]
- 169.Haraguchi K, Farnworth R, Ohbayashi A, Takehisa T. Macromolecules. 2003;36:5732. [Google Scholar]
- 170.Nayak S, Lyon LA. Angew Chem, Int Ed. 2005;44:7686. doi: 10.1002/anie.200501321. [DOI] [PubMed] [Google Scholar]
- 171.Haraguchi K, Takehisa T. Adv Mater. 2002;14:1120. [Google Scholar]
- 172.Haraguchi K, Takehisa T, Fan S. Macromolecules. 2002;35:10162. [Google Scholar]
- 173.Ripamonti U. Biomaterials. 2006;27:807. doi: 10.1016/j.biomaterials.2005.09.021. [DOI] [PubMed] [Google Scholar]
- 174.Fantner GE, Hassenkam T, Kindt JH, Weaver JC, Birkedal H, Pechenik L, Cutroni JA, Cidade GAG, Stucky G, Morse DE, Hansma PK. Nat Mater. 2005;4:612. doi: 10.1038/nmat1428. [DOI] [PubMed] [Google Scholar]
- 175.Olszta MJ, Cheng XG, Jee SS, Kumar R, Kim YY, Kaufman MJ, Douglas EP, Gower LB. Mater Sci Eng R-Rep. 2007;58:77. [Google Scholar]
- 176.Habibovic P, de Groot K. J Tissue Eng Regen Med. 2007;1:25. doi: 10.1002/term.5. [DOI] [PubMed] [Google Scholar]
- 177.Stevens B, Yang YZ, Mohanda A, Stucker SB, Nguyen KT. J Biomed Mater Res, Part B. 2008;85B:573. doi: 10.1002/jbm.b.30962. [DOI] [PubMed] [Google Scholar]
- 178.Hartgerink JD, Beniash E, Stupp SI. Science. 2001;294:1684. doi: 10.1126/science.1063187. [DOI] [PubMed] [Google Scholar]
- 179.Sargeant TD, Guler MO, Oppenheimer SM, Mata A, Satcher RL, Dunand DC, Stupp SI. Biomaterials. 2008;29:161. doi: 10.1016/j.biomaterials.2007.09.012. [DOI] [PubMed] [Google Scholar]
- 180.Schnepp ZAC, Gonzalez-McQuire R, Mann S. Adv Mater. 2006;18:1869. [Google Scholar]
- 181.Oh JK, Drumright R, Siegwart DJ, Matyjaszewski K. Prog Polym Sci. 2008;33:448. doi: 10.1016/j.progpolymsci.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Davis ME, Chen Z, Shin DM. Nat Rev Drug Discov. 2008;7:771. doi: 10.1038/nrd2614. [DOI] [PubMed] [Google Scholar]
- 183.Yu L, Ding JD. Chem Soc Rev. 2008;37:1473. doi: 10.1039/b713009k. [DOI] [PubMed] [Google Scholar]
- 184.Hoare TR, Kohane DS. Polymer. 2008;49:1993. [Google Scholar]
- 185.Siepmann J, Peppas NA. Adv Drug Deliv Rev. 2001;48:139. doi: 10.1016/s0169-409x(01)00112-0. [DOI] [PubMed] [Google Scholar]
- 186.Norton LW, Tegnell E, Toporek SS, Reichert WM. Biomaterials. 2005;26:3285. doi: 10.1016/j.biomaterials.2004.07.069. [DOI] [PubMed] [Google Scholar]
- 187.Kempen DHR, Lu L, Hefferan TE, Creemers LB, Maran A, Classic KL, Dhert WJA, Yaszemski MJ. Biomaterials. 2008;29:3245. doi: 10.1016/j.biomaterials.2008.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Liu X, Nakamura K, Lowman AM. Soft Matter. 2003;1:393. [Google Scholar]
- 189.Holland TA, Tabata Y, Mikos AG. J Controlled Release. 2003;91:299. doi: 10.1016/s0168-3659(03)00258-x. [DOI] [PubMed] [Google Scholar]
- 190.Park H, Temenoff JS, Holland TA, Tabata Y, Mikos AG. Biomaterials. 2005;26:7095. doi: 10.1016/j.biomaterials.2005.05.083. [DOI] [PubMed] [Google Scholar]
- 191.Holland TA, Tabata Y, Mikos AG. J Controlled Release. 2005;101:111. doi: 10.1016/j.jconrel.2004.07.004. [DOI] [PubMed] [Google Scholar]
- 192.Sahiner N, Jha AK, Nguyen D, Jia XQ. J Biomater Sci, Polym Ed. 2008;19:223. doi: 10.1163/156856208783432462. [DOI] [PubMed] [Google Scholar]
- 193.Drahl C. Chemical & Engineering News. American Chemical Society; Washington DC: 2008. Hybrid Polymers for Healing Voices; p. 86.p. 78. [Google Scholar]