

Summary
Although protein-protein interactions are involved in nearly all cellular processes, general rules for describing affinity and selectivity in protein-protein complexes are lacking, primarily because correlations between changes in protein structure and binding energetics have not been well determined. Here, we establish the structural basis of affinity maturation for a protein-protein interaction system that we had previously characterized energetically. This model system exhibits a 1500-fold affinity increase. Also, its affinity maturation is restricted by negative intramolecular cooperativity. With three complex and six unliganded variant X-ray crystal structures, we provide molecular snapshots of protein interface remodeling events that span the breadth of the affinity maturation process and present a comprehensive structural view of affinity maturation. Correlating crystallographically observed structural changes with measured energetic changes reveals molecular bases for affinity maturation, intramolecular cooperativity, and context-dependent binding.
Introduction
Protein-protein interactions are essential for most cellular processes, including signal transduction, gene regulation, and immune responses (Gascoigne and Zal, 2004; Pawson and Nash, 2000; Warren, 2002); thus, understanding the physiochemical principles that govern these interactions would significantly improve our understanding of biological systems. Although the biophysical factors that contribute to protein complex formation, such as van der Waals interactions, hydrogen bonding, the hydrophobic effect, shape and charge complementarity, allostery, plasticity, and cooperativity (Bogan and Thorn, 1998; Conte et al., 1999; DeLano, 2002b; Ma et al., 2001; Nooren and Thornton, 2003; Sheinerman et al., 2000; Wodak and Janin, 2002), have been studied intensively, predicting binding energies of protein complexes based on protein structures alone remains elusive. Notwithstanding the significant progress that has been made recently in developing computational methods for quantitative predictions of protein-protein interactions (Guerois et al., 2002; Huo et al., 2002; Kortemme and Baker, 2002; Massova and Kollman, 1999; Sharp, 1998), errors in predicted binding free energy changes upon mutation of even single residues remain relatively large (at least 1 kcal/mol) and are comparable to the standard cutoff value for distinguishing hot spot and neutral residues in a binding interface.
One strategy for defining the molecular basis of protein recognition is to determine how changes in protein structure affect changes in binding energetics within a single protein complex. Within a given protein-protein interaction, X-ray crystallographic or NMR analyses can define the structural epitopes, those residues that make intermolecular contacts. Functional epitopes, consisting of residues that contribute energetically to the interaction, can be described by using mutagenesis approaches. Combining these two techniques in the analysis of a molecular system can reveal the relationship between the structural and functional epitopes at atomic resolution, and can thus provide quantitative correlations between structural and energetic changes.
While the contributions of some biophysical factors, such as the hydrophobic effect, can be probed successfully by comparing structures and energetic parameters of protein complexes containing single-site mutations (Li et al., 2005; Sundberg et al., 2000), others require the concerted effects of multiple residues. Cooperativity, or nonadditive binding energies resulting from multiple simultaneous mutations, is one such biophysical factor that is now known to commonly contribute to complex formation between proteins, as shown by a variety of methods on diverse molecular systems (Albeck et al., 2000; Pal et al., 2005; Teufel et al., 2003; Yang et al., 2003; Yang and Schultz, 1999). Cooperativity can occur between residues from the same protein of a complex (intramolecular), between residues from both proteins in a complex (intermolecular), or between entire interfaces within a higher-order multiprotein complex (interfacial). The structural basis of cooperativity, in any of these forms, is unclear at present.
The affinity maturation process, by which proteins evolve to bind with increased specificity and affinity, provides an opportunity to dissect cooperative versus additive binding energetics in a protein-protein interaction. This process can be induced in a protein complex by using in vitro-directed evolution techniques such as phage (Lowman, 1997; Winter et al., 1994) or yeast (Boder and Wittrup, 1997) display in order to generate a series of variants that individually represent distinct stages of molecular evolution and together define an affinity maturation pathway. Such a pathway can also be analyzed in reverse (i.e., the affinity reversion pathway), and it will often reveal significant context-dependent energetic differences for mutations at a given position (Yang et al., 2003). These variants act as molecular snapshots within the evolution process from which energetic and structural properties can be derived and correlated to provide insight into how certain mutations, and combinations thereof, contribute to affinity maturation and cooperativity.
A limited number of crystallographic analyses of affinity-matured molecular interaction systems exist presently, and these include both protein-protein and protein-hapten systems. The protein-protein systems studied to date (De Genst et al., 2004; Li et al., 2003; Sundberg et al., 2003) are all characterized by relatively modest affinity maturations (up to ~50-fold). These binding increases are largely attributable to augmentation of the hydrophobic, buried surface area, improvements in shape complementarity, and reduced enthalpic penalties for desolvation, but not by large entropic effects. Generally, no additional hydrogen bonds, significant structural changes, or detectable cooperative binding energetics exist in these systems. Conversely, the protein-hapten systems characterized (Alzari et al., 1990; Midelfort et al., 2004; Mizutani et al., 1995; Wedemayer et al., 1997; Yuhasz et al., 1995) exhibit large affinity changes (up to ~30,000-fold) and significant positively cooperative binding energetics. The structural bases of several examples of these affinity-matured interactions are dependent on large conformational changes and preorganization of the binding site, as well as an increase in the overall number of hydrogen bonds and both electrostatic and van der Waals interactions. In contrast, in another affinity-matured protein-hapten system, relatively modest changes in structure were observed, implying that affinity maturation was due to the cumulative effects of multiple small structural modifications (Midelfort et al., 2004). Clearly, the physiochemical properties of haptens, and consequently their interactions with proteins, are markedly different than those of proteins. Likewise, the affinity maturation of protein-hapten and protein-protein systems may require distinct sets of structural modifications, as the systems characterized presently suggest.
We have described previously the intramolecular cooperative and additive binding energetics in the affinity maturation pathway of murine T cell receptor (TCR) Vβ8.2 domain (Vβ) variants binding the superantigen staphylococcal enterotoxin C3 (SEC3) (Yang et al., 2003), which had been generated by yeast display mutagenesis (Kieke et al., 2001). This maturation pathway exhibits a 1500-fold increase in affinity, significantly greater than any other affinity-matured protein-protein interaction for which structural analysis has been performed. Here, we present the X-ray crystallographic analysis of the Vβ-SEC3 affinity maturation pathway. Together, the energetic and structural analyses elucidate the molecular basis of affinity maturation and intramolecular cooperativity in this protein-protein interaction.
Results
A Model System for Assessing Affinity Maturation and Intramolecular Cooperativity
Our previous study (Yang et al., 2003) assessed the energetic consequences of the mutations that constitute the affinity maturation pathway from wild-type Vβ to L2CM, the highest affinity Vβ variant, in binding to SEC3. This analysis revealed that four of the nine mutated Vβ residues identified during affinity maturation were energetically significant in terms of improved binding to SEC3. These residues constitute the functional epitope that is responsible for increasing affinity from KD = 8 μM to KD = 5 nM, for the low- and high-affinity end point variants, respectively, and include the A52VVβ, S54NVβ, K66EVβ, and Q72HVβ mutations. One additional mutation, E80VVβ, while not energetically significant by itself, interacts with the side chain of K66Vβ and is thus likely to play a role in the differential electrostatic dependence on binding for the K66EVβ (maturation) and E66K-rVβ (reversion) mutations observed previously (Yang et al., 2003). The relative positions of these Vβ variant residues in the Vβ-SEC3 complex are shown in Figure 1A.
Figure 1. Relative Positions of Vβ Variant Residues and Energetic Consequences of Their Variation.
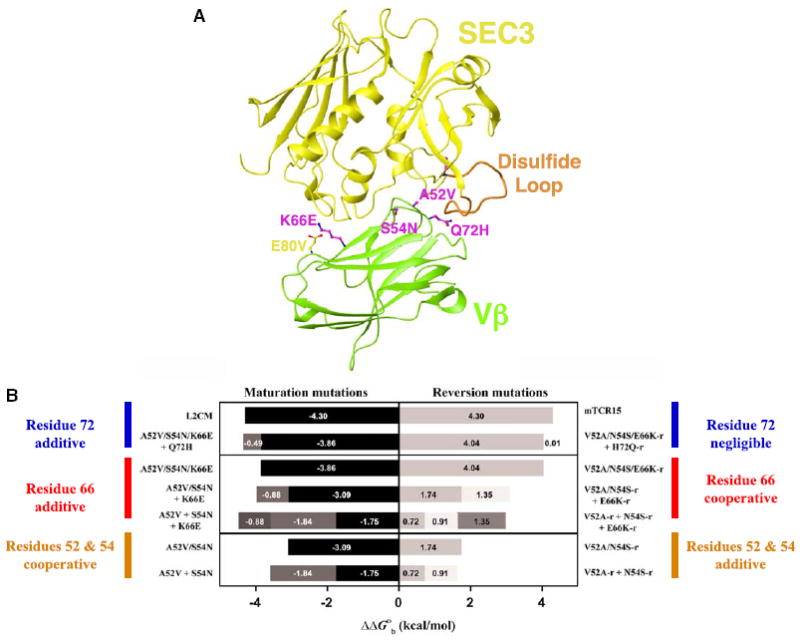
(A) Overview of the Vβ-SEC3 complex and location of the Vβ variant residues. Side chains of mutations in Vβ that are energetically significant in the affinity maturation pathway are shown in magenta; others are in yellow. Vβ is in green, SEC3 is in yellow, and the flexible SEC3 disulfide loop is in orange.
(B) Dissection of cooperative and additive binding energetics. Relative changes in binding free energies of variant Vβ-SEC3 complexes in which point or combinations of mutations were made reveal the context-dependent cooperative versus additive energetic nature of the variant residues. All measurements were made by surface plasmon resonance (SPR) analysis at pH 7.4. Bars are shaded to distinguish binding free energy changes attributable to each of the mutants listed to the side of the graph. Data for this figure are from Yang et al. (2003).
We also found (Yang et al., 2003) that mutations at positions 52 and 54 were energetically cooperative, while mutations at positions 66 and 72 were simply additive when they existed as maturation mutations. As reversion mutations, however, variations at positions 52 and 54 were additive, variations at position 66 were cooperative, and variations at position 72 resulted in no relative change in binding free energy. The relative changes in binding free energy of these variant residues alone or in various combinations, and as maturation and reversion mutations, are summarized in Figure 1B.
In order to elucidate the structural basis for affinity maturation and reversion, as well as for energetic cooperativity between residues, in the Vβ-SEC3 protein-protein interface, we determined the X-ray crystal structures of three variant Vβ-SEC3 complexes (wild-type Vβ-SEC3, A52V/S54N/K66E:SEC3-1D3, and H72Q-r: SEC3-1A4). SEC3-1D3 and SEC3-1A4 are phage display variants of SEC3 that have altered sequences in their disordered disulfide loop regions (Andersen et al., 2001) that do not affect the binding interface at the Vβ variant positions. Both wild-type and variant SEC3 molecules are hereafter referred to generically as SEC3. We also determined the crystal structures of six variant Vβ molecules in their unliganded (apo) states (including: maturation variants S54N and A52V/S54N/K66E; the highest affinity variant, L2CM; and reversion variants H72Q-r, V52A-r, and E66K-r). The variants in the complex and apo structures define distinct stages along the affinity maturation and reversion pathways, providing crystallographic snapshots of the specific protein-protein interface remodeling events that modulate affinity in this molecular interaction (Figure 2). All of the structures were determined to a nominal resolution of at least 2.3 Å. Crystallographic data collection and refinement statistics for the apo and complex structures are shown in Tables 1 and 2, respectively.
Figure 2. Overview of the Crystallographic Analysis of the Affinity Maturation and Reversion Pathways in the Vβ-SEC3 Interaction.
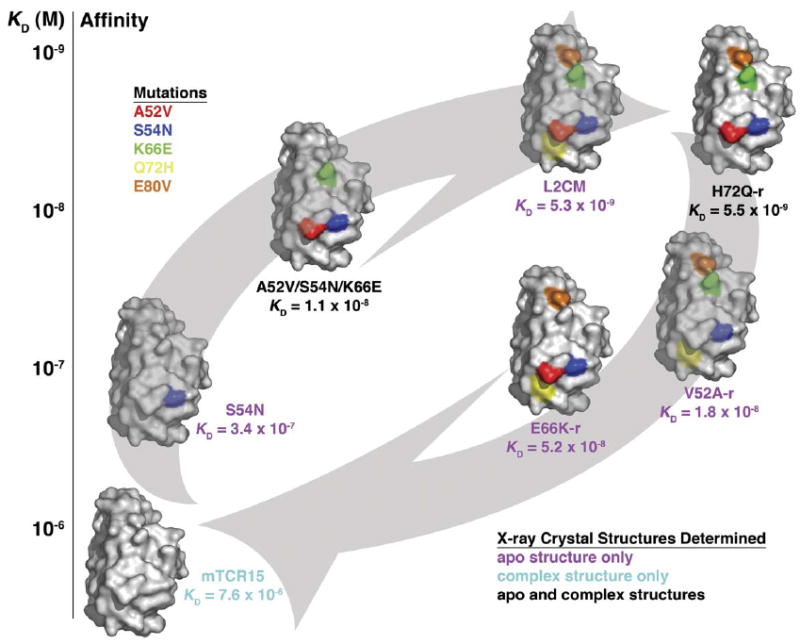
Vβ variants are shown as molecular surface representations; only residues that are in their mutated stated highlighted by the following colors: A52V, blue; S54N, red; K66E, green; Q72H, yellow; and E80V, orange. Arrows indicate the affinity maturation pathway (from wild-type toward L2CM, bottom left to top right) and the affinity reversion pathway (from L2CM to wild-type, from top right to bottom left). Vβ variants are plotted according to their relative affinities for SEC3 (vertical axis) and are placed on the appropriate affinity modulation pathway. Labels of each Vβ variant are color coded to reveal which crystal structures were determined as follows: apo Vβ crystal structure only, magenta; Vβ-SEC3 complex crystal structure only, cyan; both apo Vβ and Vβ-SEC3 complex crystal structures, black. In our naming convention of this affinity maturation pathway, wild-type Vβ and L2CM (highest affinity/fully matured Vβ variant) define the energetic start and end points of the pathway, respectively. Intermediate Vβ variants are described by the particular mutation or combination of mutations found in the variant, with a -r label distinguishing reversion mutations from their maturation counterparts.
Table 1.
Data Collection and Refinement Statistics for Vβ Variants
| S54N | A52V/S54N/K66E | V52A-r | E66K-r | H72Q-r | L2CM | |
|---|---|---|---|---|---|---|
| Data Collection | ||||||
| Space group | I212121 | I212121 | C2 | I212121 | I212121 | I212121 |
| Unit cell dimensions | ||||||
| a (Å), b (Å), c (Å) | 32.502, 74.834, 113.530 | 31.608, 74.708, 113.819 | 79.423, 116.295, 34.769; γ (°) = 110.17 | 32.502, 74.696, 113.591 | 32.521, 74.552, 113.045 | 32.528, 74.575, 113.083 |
| Molecules per asymmetric unit | 1 | 1 | 2 | 1 | 1 | 1 |
| Resolution (Å) | 30.00–1.80 | 30.00–1.80 | 40.00–2.00 | 30.00–1.90 | 30.00–2.00 | 30.00–1.80 |
| Observations | 67,564 | 54,289 | 85,921 | 63,425 | 49,693 | 76,325 |
| Unique reflections | 10,932 (651) | 11,549 (660) | 17,516 (922) | 10,640 (771) | 9,148 (648) | 12,398 (881) |
| Completeness (%) | 86.6 (73.2)a | 94.1 (73.8) | 92.36 (65.37) | 99.1 (99.6) | 99.1 (97.5) | 99.03 (97.9) |
| Rmerge (%)b | 6.8 (29.7) | 8.9 (35.4) | 5.3 (24.6) | 7.8 (30.3) | 4.8 (27.1) | 6.6 (23.6) |
| Refinement | ||||||
| Rcryst (%)c | 19.1 (35.5) | 19.7 (25.0) | 17.6 (28.3) | 18.9 (32.5) | 19.9 (21.2) | 19.0 (32.6) |
| Rfree (%)d | 25.8 (45.7) | 26.3 (28.1) | 21.3 (30.9) | 23.0 (35.7) | 23.9 (30.5) | 23.1 (27.1) |
| Protein residues | 110 | 112 | 218 | 109 | 109 | 109 |
| Water molecules | 144 | 151 | 102 | 162 | 110 | 190 |
| Average B factors (Å2) | ||||||
| Vβ | 17.2 | 26.0 | 38.8 | 17.9 | 15.1 | 16.3 |
| Water | 30.8 | 40.4 | 55.3 | 32.3 | 26.8 | 34.6 |
| Rms deviations | ||||||
| Bonds (Å) | 0.015 | 0.015 | 0.018 | 0.018 | 0.017 | 0.015 |
| Angles (°) | 1.341 | 1.526 | 1.878 | 1.507 | 1.639 | 1.353 |
| Ramachandran plot statistics | ||||||
| Core (%) | 91.2 | 90.3 | 91.8 | 91.2 | 91.2 | 90.1 |
| Allowed (%) | 8.8 | 9.7 | 8.2 | 8.8 | 8.8 | 9.9 |
| Generous (%) | 0 | 0 | 0 | 0 | 0 | 0 |
| Disallowed (%) | 0 | 0 | 0 | 0 | 0 | 0 |
Values in parentheses correspond to the highest-resolution shell: S54N (1.85–1.80 Å); A52V/S54N/K66E (1.85–1.80 Å); V52A-r (2.05–2.00 Å); E66K-r (1.95–1.90 Å); H72Q-r (2.05–2.00 Å); L2CM (1.85–1.80 Å).
Rmerge (I) = (Σ|I[i] − <I[h]>|/ΣI[i]), where I[i] is the ith observation of the intensity of the hkl reflection and <I> is the mean intensity from multiple measurements of the hkl reflection.
Rcryst (F) = Σh||Fobs(h)| − |Fcalc(h)||/Σh. |Fobs(h)| and |Fcalc(h)| are the observed and calculated, respectively, structure factor amplitudes for the hkl reflection.
Rfree is calculated over reflections in a test set not included in atomic refinement: S54N, 561 reflections, 4.9%; A52V/S54N/K66E, 630 reflections, 5.2%; V52A-r, 960 reflections, 5.2%; E66K-r, 574 reflections, 5.1%; H72Q-r, 465 reflections, 4.8%; L2CM, 682 reflections, 5.2%.
Table 2.
Data Collection and Refinement Statistics for Vβ-SEC3 Variant Complexes
| Vβ-SEC3 | H72Q-r:SEC3-1A4 | A52V/S54N/K66E:SEC3-1D3 | |
|---|---|---|---|
| Data Collection | |||
| Space group | P1 | P1 | P65 |
| Unit cell dimensions | |||
| a (Å), b (Å), c (Å), α (°), β (°), γ (°) | 64.160, 70.460, 98.372, 74.18, 75.76, 88.40 | 63.200, 70.186, 98.403, 74.79, 75.05, 88.54 | 96.537, 96.537, 92.182, 90, 90, 120 |
| Molecules per asymmetric unit | 4× SEC3/4× Vβ | 4× SEC3/4× Vβ | 1× SEC3/1× Vβ |
| Resolution (Å) | 40.00–2.30 | 35.00–2.10 | 40.00–1.80 |
| Observations | 272,478 | 747,266 | 297,523 |
| Unique reflections | 63,758 (3,699)a | 83,749 (5,084) | 42,633 (2,922) |
| Completeness (%) | 94.21 (73.66) | 95.98 (79.96) | 99.1 (93.0) |
| Rmerge (%)b | 6.8 (27.3) | 4.4 (25.6) | 5.2 (29.1) |
| Refinement | |||
| Rcryst (%)c | 21.2 (28.6) | 18.8 (24.9) | 18.8 (24.7) |
| Rfree (%)d | 27.1 (34.4) | 24.3 (33.7) | 21.6 (29.7) |
| Protein residues | 1,380 | 1,380 | 344 |
| Water molecules | 198 | 590 | 235 |
| Zinc ions | 0 | 0 | 1 |
| Average B factors (Å2) | |||
| SEC3 | 47.5 | 47.6 | 26.5 |
| Vβ | 40.8 | 39.2 | 34.0 |
| Water | 40.1 | 45.5 | 39.8 |
| Rms deviations | |||
| Bonds (Å) | 0.032 | 0.024 | 0.014 |
| Angles (°) | 2.531 | 2.016 | 1.386 |
| Ramachandran plot statistics | |||
| Core (%) | 80.8 | 86.9 | 89.2 |
| Allowed (%) | 17.5 | 11.7 | 10.8 |
| Generous (%) | 1.4 | 1.1 | 0 |
| Disallowed (%) | 0.2 | 0.3 | 0 |
Values in parentheses correspond to the highest-resolution shell: Vβ-SEC3 (2.36–2.30 Å); H72Q-r:SEC3-1A4 (2.15–2.10 Å); A52V/S54N/K66E:SEC3-1D3 (1.86–1.80 Å).
Rmerge (I) = (Σ|I[i] − <I[h]>|/ΣI[i]), where I[i] is the ith observation of the intensity of the hkl reflection, and <I> is the mean intensity from multiple measurements of the hkl reflection.
Rcryst (F) = Σh||Fobs(h)| − |Fcalc(h)||/Σh. |Fobs(h)| and |Fcalc(h)| are the observed and calculated, respectively, structure factor amplitudes for the hkl reflection.
Rfree is calculated over reflections in a test set not included in atomic refinement: Vβ-SEC3, 3394 reflections, 5.1%; H72Q-r:SEC3-1A4, 4468 reflections, 5.1%; A52V/S54N/K66E:SEC3-1D3, 2257 reflections, 5.0%.
Multiple Structural Consequences of the A52VVβ Mutation
The alanine to valine mutation at position 52Vβ results in the addition of two methyl groups, the Cγ1 and Cγ2 atoms, beyond the single methyl group side chain of the wild-type residue (Figure 3). Each of these two methyl groups appears to serve a distinct function: one increases the buried hydrophobic surface with SEC3, and another induces intramolecular conformational changes in neighboring Vβ domain regions. This mutation thereby contributes not only to the maturation and reversion of affinity to SEC3, but also to intramolecular energetic cooperativity (compare ΔΔGb(A52V/S54N) to Σ[ΔΔGb(A52V) + ΔΔGb(S54N)] values in Figure 1B) and context-dependent binding in this protein-protein interaction (for example, compare ΔΔGb(Q72H) and ΔΔGb(H72Q-r) values in Figure 1B).
Figure 3. Multiple Structural Consequences of the A52VVβ Mutation.
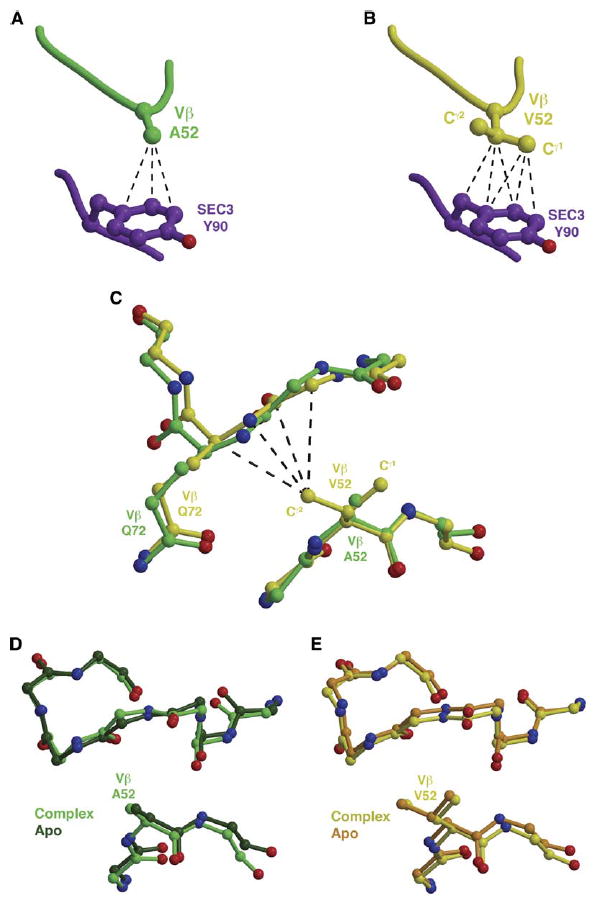
(A) The wild-type residue A52Vβ makes several van der Waals interactions with Y90SEC3.
(B) The Cγ1 atom of the mutant residue V52Vβ makes additional intermolecular contacts with Y90SEC3.
(C–E) (C) The Cγ2 atom of the mutant residue V52Vβ affects the relative conformation of the HV4 loop, in which another variant residue, Q72Vβ, resides. This conformational change is a result of the A52VVβ mutation and not of SEC3 binding, as seen by comparison of apo Vβ and Vβ-SEC3 complex structures in which the residue at position 52Vβ is either the (D) wild-type alanine or the (E) mutant valine. Intermolecular van der Waals contacts are shown as black, dashed lines.
In the wild-type Vβ-SEC3 complex, residue A52Vβ makes several van der Waals contacts with Y90SEC3 (Figure 3A). The A52VVβ mutation, and specifically the Cγ1 atom of the valine, increases significantly the number of van der Waals interactions with Y90SEC3 (Figure 3B). This results in an increased hydrophobic contact area between Vβ and SEC3 of 25.4 Å2 (average of two Vβ-SEC3 complex structures containing the A52VVβ mutation), relative to the wild-type alanine residue (Figure 3B). Using a quantitative estimation of the hydrophobic effect at central regions of protein-protein interfaces (Li et al., 2005), the predicted change in the relative free energies of binding for the A52VVβ mutation is −1.2 kcal/mol. The change in relative binding free energies for the alanine to valine mutation of residue 52Vβ as measured by surface plasmon resonance analysis, however, is −1.8 kcal/mol (Yang et al., 2003); thus, the remaining binding free energy should be ascribable to other effects caused by this mutation.
The other additional methyl group of the valine residue at position 52Vβ, the Cγ2 atom, points toward the HV4 loop of Vβ. This results in a relative displacement of the Cα atom of one of the energetically significant Vβ residues, Q72Vβ, by 0.8 Å (Figure 3C). This conformational change is induced intramolecularly by the A52VVβ mutation, not by complex formation with SEC3, as is shown by comparison of the relevant regions of both wild-type (A52Vβ, Figure 3D) and mutant (V52Vβ, Figure 3E) structures in both apo and complex forms. These intramolecular structural modulations resulting from variation at position 52Vβ are also influenced by the variant residue 72Vβ, and they modulate the conformations of both the CDR1 and CDR2 (in which position 52Vβ resides) loops, with further ramifications for SEC3 binding (see below).
The S54NVβ Mutation Recruits Bridging Water Molecules to Modulate Affinity
The hydroxyl group of the wild-type serine residue at position 54Vβ forms a hydrogen bond with the Oε1 side chain atom of residue E56Vβ, and, as a result, S54Vβ forms no intermolecular contacts with SEC3, as seen in the wild-type Vβ-SEC3 complex structure (Figure 4A). No ordered water molecules are found in this region of the Vβ-SEC3 complex that could bridge the two molecules, even though it resides at the periphery of the binding interface, and waters should therefore be accessible to this site. While the conformation of the side chains of residues S54Vβ and E56Vβ are not wholly conserved in the only comparable apo Vβ crystal structure available (the mouse TCR Vβ8.2 chain determined to a resolution of 1.7 Å, PDB accession code 1BEC [Bentley et al., 1995]), the S54Vβ side chain in this structure points back toward the N-terminal portion of the CDR2 loop and away from the binding interface with SEC3 (Figure 4B). This may be due, in part, to the presence of residue V91SEC3, whose side chain contacts the Cβ atoms of residues at position 54Vβ, in the complex structures.
Figure 4. The S54NVβ Mutation Modulates the Conformation of the Invariant Residue Glu56 to Facilitate an Extensive Water-Mediated Hydrogen Bonding Network with SEC3.
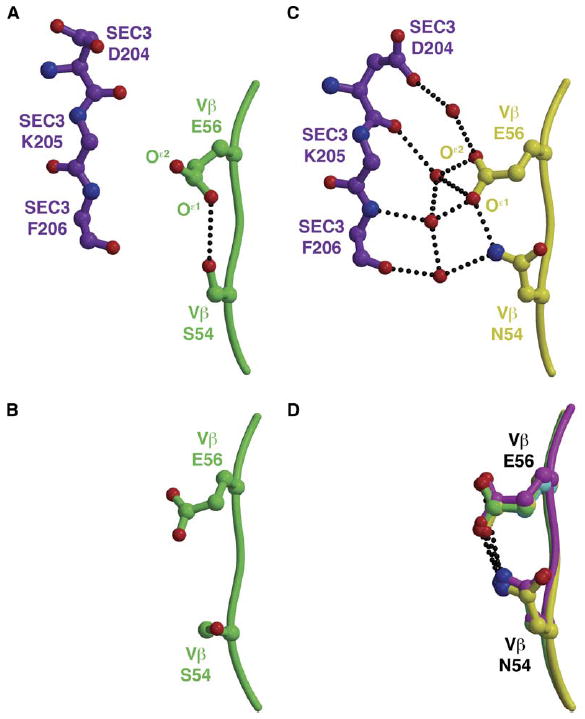
(A) An intramolecular hydrogen bonding interaction between S54Vβ and E56Vβ prevents interactions and intermolecular contacts with SEC3.
(B) The analogous region of the CDR2 loop of wild-type apo Vβ.
(C) The mutation S54NVβ extends both N54Vβ and E56Vβ side chains toward SEC3, resulting in the recruitment of numerous ordered water molecules to the molecular interface.
(D) The relative conformations of the N54Vβ and E56Vβ side chains are ordered prior to binding SEC3, as seen in all apo Vβ variants containing the A52VVβ mutation for which we have determined crystal structure. Hydrogen bonds are shown as black, dotted lines.
When mutated to an asparagine residue, however, the intramolecular hydrogen bonding interaction between the residues at positions 54Vβ and E56Vβ observed in the wild-type complex structure is extended farther from the backbone of the CDR2 loop and toward SEC3, as observed in the A52V/S54N/K66E:SEC3 complex structure (Figure 4C). An additional hydrogen bond formed between the Oδ1 atom of the N54Vβ side chain and the main chain N atom of Y50Vβ, but absent when the wild-type serine residue is present at position 54Vβ, may also contribute to the spatial arrangement of the N54Vβ and E56Vβ side chains. Numerous water molecules are observed in the affinity-matured Vβ-SEC3 interface. This water network bridges the N54Vβ and E56Vβ side chains to main chain atoms of F204SEC3 and K204SEC3, as well as one of the side chain oxygen atoms of D204SEC3, in the affinity-matured N54Vβ variant. The ordering of water molecules is likely the reason for both the high enthalpic favorability of this mutation (ΔΔHb(S54N-WT) = −12.1 kcal/mol), as well as its compensating entropic cost (ΔΔSb(S54N-WT) = −3.9 kcal/mol) (Yang et al., 2003).
All of the apo Vβ crystal structures that we determined that contain the S54NVβ mutation (including S54N, A52V/S54N/K66E, L2CM, V52A-r, E66K-r, and H72Q-r [refer to Figure 2]) reveal an identical relative conformation of the N54Vβ and E56Vβ residues (Figure 3D). Thus, the entropic cost of this mutation must come from the ordering of the water molecules and not through rigidification of side chains upon complex formation.
Interplay between Variant Residues at Positions 66Vβ and 80Vβ
Positions 66Vβ and 80Vβ are located adjacent to one another, and the side chains of residues at these two positions make intramolecular contacts regardless of whether they are in their wild-type or mutant forms. Side chains of residue 66Vβ also invariably make intermolecular contacts with F176SEC3, although the quantity and quality of these contacts vary depending on the residues found at positions 66Vβ and 80Vβ. While residues at position 80Vβ do not make direct intermolecular contacts with any residues in SEC3, they do affect the position and the electrostatic environment of residues at position 66Vβ, and, thus, their interaction with SEC3 (Figure 5).
Figure 5. Molecular Interplay between Variant Residues at Positions 66Vβ and 80Vβ.
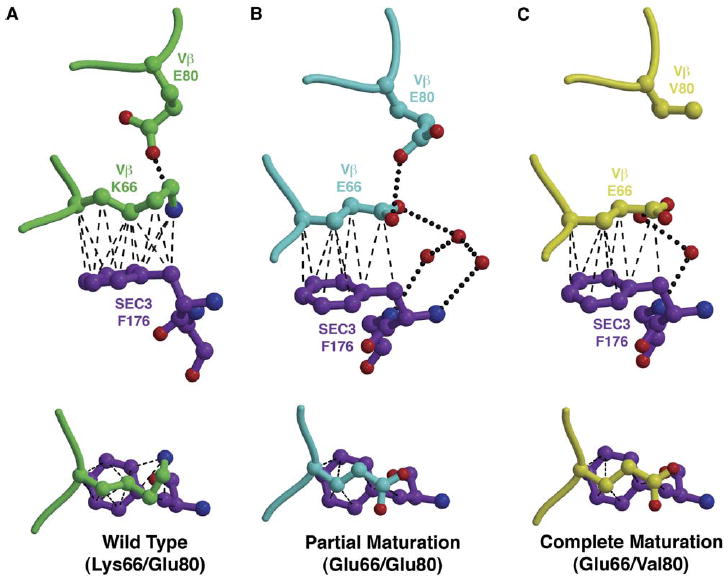
(A–C) Intramolecular and intermolecular contacts change as a result of mutations at positions 66Vβ and 80Vβ, as shown from two orientations for the Vβ-SEC3 complex structures that contain (A) both wild-type residues K66Vβ and E80Vβ, (B) the mutation K66E and the wild-type E80Vβ, or (C) both mutated residues E66Vβ and V80Vβ. Hydrogen bonds are shown as black, dotted lines; van der Waals interactions are shown by black, dashed lines.
In the wild-type Vβ-SEC3 cocrystal structure, the amino group of K66Vβ is held in place via a hydrogen bond to the Oε2 atom of E80Vβ and by numerous van der Waals interactions between the aliphatic portion of K66Vβ and the phenyl ring of F176SEC3 (Figure 5A). Thus, pinned between these two side chains, the K66Vβ side chain adopts a highly constrained conformation.
In the A52V/S54N/K66E:SEC3 complex, in which partial affinity maturation has occurred in this region of the interface (K66EVβ/E80Vβ), many of the hydrophobic intermolecular contacts between residues E66Vβ and F176SEC3 are maintained (Figure 5B), relative to the wild-type complex. Accordingly, there is no significant relative change in the buried surface area attributable to the K66E Vβ mutation. The side chain of E66Vβ adopts an extended, and presumably less constrained, side chain conformation than does wild-type K66Vβ, concomitant with a loss of some van der Waals interactions. This is reflected in the energetic parameters of binding of the K66EVβ variant relative to wild-type Vβ, for which we measured a significantly unfavorable change in the enthalpy of binding (ΔΔHb(K66E-WT) = 5.0 kcal/mol). This enthalpic cost was outweighed by a highly favorable entropic change (ΔTΔSb(K66E-WT) = 5.4 kcal/mol) and results in a higher-affinity complex (Yang et al., 2003).
When the additional maturation mutation, E80VVβ, is present, as it is in the Q72H-r:SEC3 complex, the side chain conformation of E66Vβ (Figure 5C) is unchanged relative to that of the partially matured Vβ (Figure 5B). With a valine side chain at position 66Vβ, no electrostatic interactions can be made between the mutated residues at positions 66Vβ and 80Vβ. Although mutation at position 80Vβ does not by itself contribute significantly to the energetics of binding, when combined with variation at position 66Vβ, the change in electrostatic properties of the region resulting from the E80VVβ mutation has a significant effect on both the association kinetics and binding sensitivity to the ionic strength of the solute (Yang et al., 2003). The association rate (ka) of the E66K-r variant (which includes the E80VVβ maturation mutation) is more than five-fold faster than that of the K66E variant (in which the residue at position 80Vβ is the wild-type Asp), by far the largest difference in association kinetics for any pair of maturation and reversion mutations at the same position. We note that some caution need be taken with this interpretation on account of the relatively high concentration of protein required to extract kinetic data for the E66K-r:SEC3 interaction by SPR analysis. Additionally, the K66E maturation variant exhibits a decrease in affinity of ~110-fold over a NaCl concentration range of 50–500 mM, while the E66K-r variant exhibits an affinity decrease of only ~15-fold over the same NaCl concentration range.
Amino Acid Variations in the Vβ CDR2 and HV4 Loops Affect the Invariant CDR1 Loop
Residue 72Vβ resides in the HV4 loop of Vβ and is located between the variant residue 52Vβ of the CDR2 loop and the invariant residues of the CDR1 loop, including N28Vβ and N30Vβ (Figure 6). These residues of the HV4 and CDR1 loops are thought to form a binding site for the highly flexible SEC3 disulfide loop (Fields et al., 1996). Indeed, the CDR1 loop residues N28Vβ and N30Vβ were both shown to be hot spots for SEC3 binding, with the latter being relatively more important energetically, by alanine scanning mutagenesis (Churchill et al., 2000). In the wild-type Vβ-SEC3 complex crystal structure, the SEC3 disulfide loop is disordered, but this does not necessarily preclude the possibility of transient interactions in solution. These interactions would likely be affected by relative positional changes of the hot spot residues in the CDR1 loop. We present here an analysis of the CDR2/HV4/CDR1 region of the binding interface of the relevant apo Vβ molecules, for which we have crystal structures of all possible combinations of wild-type and mutated residues at positions 52Vβ and 72Vβ.
Figure 6. Molecular Interplay between Variant Residues at Positions 52Vβ and 72Vβ Affects the Vβ CDR1 Loop.
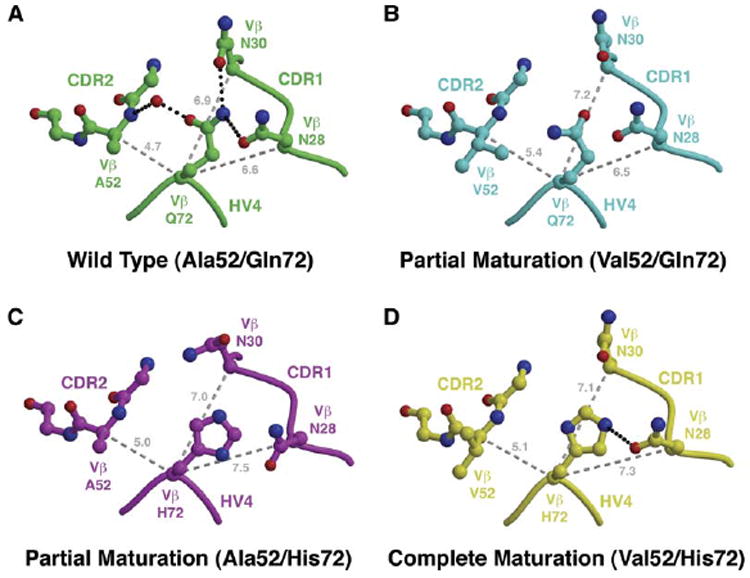
(A–D) Comparison of CDR1/CDR2/HV4 loop arrangements in apo Vβ structures containing (A) both wild-type A52Vβ and Q72Vβ residues, (B) the A52VVβ mutation and the wild-type Q72Vβ residue, (C) the wild-type A52Vβ residue and the Q72HVβ mutation, and (D) both A52VVβ and Q72HVβ mutations. Hydrogen bonds are shown as black, dotted lines. Distances (in Å) between the Cα atom of position 72Vβ and the Cα atoms of positions 28Vβ, 30Vβ, and 52Vβ are shown as gray, dashed lines.
In the wild-type Vβ structure, Q72Vβ bridges the CDR1 and CDR2 loops in a compact arrangement (Figure 6A). A water-mediated hydrogen bond between Oε1 of Q72Vβ and the main chain nitrogen of residue A52Vβ, as well as two hydrogen bonds between Nε2 of Q72Vβ and Oδ1 atoms of both N28Vβ and N30Vβ, maintain the relatively closed conformation of the CDR loops. When partial Vβ maturation occurs via the A52VVβ mutation (Figure 6B), the relative bulkiness of the valine side chain pushes residue Q72Vβ and the HV4 loop significantly away from the CDR2 loop (as in Figures 3C and 3E). This movement breaks entirely the hydrogen bonding network that bridges the CDR1 and CDR2 loops observed in the wild-type structure. This effect is propagated to the CDR1 loop, predominantly to residue N30Vβ, and it is moved slightly away from the CDR2 and HV4 loops, resulting in a more open arrangement of these hypervariable regions. In an alternate partial Vβ maturation scenario, the Q72HVβ mutation also breaks the CDR-bridging hydrogen bonding network of the wild-type (Figure 6C), likewise opening up the CDR2/HV4/CDR1 loop arrangement primarily through movement of residue N28Vβ. When both the A52VVβ and Q72HVβ maturation mutations are present in the same Vβ molecule (Figure 6D), the arrangement of the CDR1, HV4, and CDR2 loops appears as a hybrid of the two partially matured Vβ variants.
The primary energetic significance of these concerted movements between the CDR1, CDR2, and HV4 loops caused by various combinations of mutations at positions 52Vβ and 72Vβ is that the wild-type residue A52Vβ is pushed toward SEC3 to accommodate greater intermolecular contacts and increased affinity when the Q72HVβ maturation mutation alone is present (ΔΔGb(Q72H-r-WT) = −0.5 kcal/mol; Figure 1B). When the H72Q-rVβ reversion mutation is made, however, the presence of the valine residue at position 52Vβ has already induced an opening of the CDR1/CDR2/HV4 loop arrangement, explaining why the energetic effect of this reversion mutation is negligible (ΔΔGb(H72Q-r-L2CM) = 0.0 kcal/mol; Figure 1B). Accordingly, the energetic consequences of mutations of residue 72Vβ are context dependent.
CDR2 Plasticity Allows for Pathway-Dependent Energetic Cooperativity and Additivity
SEC3 induces a conformational change in the CDR2 loop upon binding wild-type Vβ, effectively tilting the apical end, and primarily residues 52Vβ and 53Vβ, of the loop toward the SEC3 molecular surface (to the right in Figure 7A). The flexibility of this loop is likely due to two invariant glycine residues at positions 51Vβ and 53Vβ, interspersed about the variant residues at positions 52Vβ and 54Vβ. Although the entirety of the CDR2 loop was subjected to randomization in the yeast display affinity maturation of this Vβ, no variation in these two glycine residues was ever observed (Kieke et al., 2001), indicating that flexibility of the CDR2 loop may be of functional significance.
Figure 7. Flexibility of the Vβ CDR2 Loop.
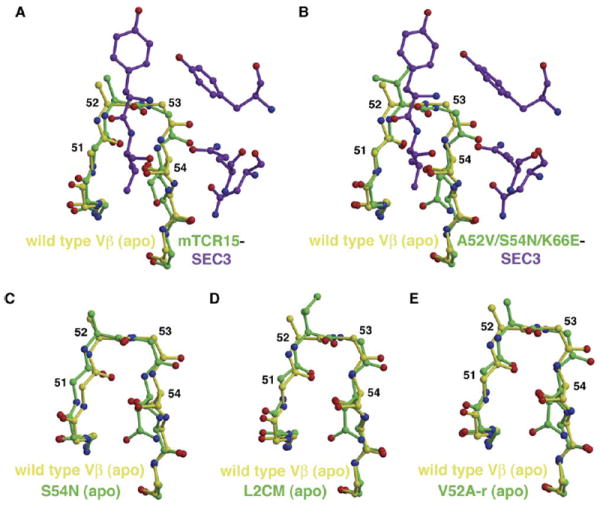
(A and B) Comparison of the CDR2 loop and neighboring residues in the wild-type apo Vβ structure (yellow) with the same region of (A) the wild-type Vβ-SEC3 and (B) the A52V/S54N/K66E:SEC3 complex structures (Vβ residues are green; SEC3 residues are purple).
(C–E) Comparison of the wild-type apo Vβ structure (yellow) with apo Vβ variant structures (green) of (C) S54N, (D) L2CM, and (E) V52A-r.
The conformational shift of the CDR2 loop induced by SEC3 in the wild-type Vβ-SEC3 complex (Figure 7A) is also observed in SEC3 complexes with the H72Q-r and A52V/S54N/K66E (Figure 7B) Vβ variants. From the analysis of the variant apo Vβ crystal structures, however, it appears that these structural changes in the variant complexes are not induced upon complex formation, but instead are arranged prior to SEC3 binding. This may be a contributing factor to the overall affinity maturation pathway, which is driven significantly more by favorable entropic changes (ΔTΔSb(L2CM-WT) = 3.5 kcal/mol) than by favorable enthalpic changes (ΔΔHb(L2CM-WT) = −0.8 kcal/mol) (Yang et al., 2003).
The S54NVβ affinity maturation mutation results in a shift of the CDR2 loop in the opposite direction as observed for those induced by SEC3 binding, primarily for residues G51Vβ and N54Vβ (to the left in Figure 7C). The increase in affinity for this mutant likely comes entirely from the formation of the extensive water-bridging hydrogen bonding network between residues N54Vβ, E56Vβ, D204SEC3, K205SEC3, and F206SEC3 (Figure 4C). This S54NVβ CDR2 loop shift is nullified, however, in all Vβ variants that contain either or both of the maturation mutations A52VVβ or Q72HVβ, for instance in the A52V/S54N/K66E (A52VVβ mutation), L2CM (both A52VVβ and Q72HVβ mutations; Figure 7D), V52A-r (Q72HVβ mutation, Figure 7E), and H72Q-r (A52VVβ mutation) apo structures. The conformations of the CDR2 loops in these variants more closely approximate the conformation that is induced upon binding SEC3. Thus, just as the combination of mutations at positions 52Vβ and 72Vβ affects the structure of the neighboring CDR1 loop (Figure 6), they also affect the structure of the neighboring region (the C-terminal portion) of the CDR2 loop.
This modulation of the position of the CDR2 loop also provides a structural explanation for the negatively cooperative and additive binding energetics measured for the mutations at positions 52Vβ and 54Vβ, as maturation and reversion mutations, respectively (Yang et al., 2003). On the maturation pathway, mutations A52VVβ and S54NVβ exert opposite structural effects on the conformation of the CDR2 loop, and, thus, when combined as a double mutant, the loop is both pushed toward and away from SEC3. In the absence of any overriding influence from the wild-type residue Q72Vβ, this results in negative cooperativity. On the reversion pathway, however, mutations V52A-rVβ and N54S-rVβ are additive due to the presence of the histidine residue at position 72Vβ, which moderates the opposing CDR2 movements caused by these two mutations, and they are thus energetically additive.
Discussion
Our previous energetic analysis (Yang et al., 2003) of the Vβ-SEC3 affinity maturation pathway, isolated by random mutagenesis and yeast display (Kieke et al., 2001), revealed those mutations that were significant energetic contributors to affinity maturation. This study also identified those mutations and/or reversions that affected binding energetics in a context-dependent manner and that acted cooperatively or additively relative to other mutations. With the comprehensive structural analysis of the affinity maturation and reversion pathways that we present here, it is possible to assign structural explanations for these observed energetic effects and to broaden our understanding of some of the underlying relationships between structural and energetic changes in protein-protein interactions generally. This analysis has practical implications for the superantigen system described here, as the high-affinity, soluble Vβ receptors have been shown to have neutralizing potential and thus may be of some therapeutic use (Burnett et al., 2005; Kieke et al., 2001).
Not surprisingly, with the myriad of energetic effects, we observe numerous ways in which the protein-protein interface is remodeled to achieve improvements in binding. Only one of the structural changes (increased van der Waals interactions between A52VVβ and Y90SEC3; Figures 3A and 3B) might have been predicted by thorough examination of a high-resolution wild-type complex crystal structure, and it reflects changes observed in moderately affinity-matured protein-protein systems (De Genst et al., 2004; Li et al., 2003; Sundberg et al., 2003). The majority of the remodeling events, however, appear to play more complex roles in affinity modulation, as in highly affinity-matured protein-hapten systems (Alzari et al., 1990; Midelfort et al., 2004; Mizutani et al., 1995; Wedemayer et al., 1997; Yuhasz et al., 1995).
The protein plasticity exhibited by the Vβ CDR2 loop is similar to, although not as extensive as, that seen for other TCR CDR loops, especially CDR3, upon interaction with peptide-major histocompatibility complexes, as has been shown by both structural (Ding et al., 1999; Garcia et al., 1998; Reiser et al., 2002, 2003) and thermodynamic (Anikeeva et al., 2003; Boniface et al., 1999; Willcox et al., 1999) studies. This protein plasticity is also not nearly as dramatic as that seen in some protein-hapten affinity maturation systems (Wedemayer et al., 1997). Flexibility may be important for SEC3 binding, though, as the two glycine residues of the CDR2 loop at positions 51Vβ and 53Vβ, and which are adjacent in sequence to the two most energetically significant variant residues in the affinity maturation pathway, those at positions 52Vβ and 54Vβ, were absolutely invariant throughout the yeast display process. This is the case even though other glycine residues outside of the Vβ-SEC3 interface varied, presumably to increase stability of the expressed murine protein on the yeast surface (Kieke et al., 2001). Additionally, the CDR2 loop conformational change seen in the formation of the wild-type Vβ-SEC3 complex is mimicked by all of the apo Vβ variants containing the A52VVβ mutation. A conformational change pushing the apical region of the CDR2 loop in the opposite direction is seen in the apo S54NVβ variant structure. Thus, not only is the flexibility of the CDR2 loop important in SEC3 binding and affinity maturation, but it is also the cause of the negative cooperativity between the A52VVβ and S54NVβ maturation mutations on account of the opposing forces that they each exert on this loop. In effect, even though both of these mutations significantly improve affinity in the Vβ-SEC3 complex individually, they partially negate the positive (in terms of binding) impact of one another.
Subtle conformational changes play a role in the energetic significance of residue 72Vβ, neighboring residue 52Vβ on the opposite side from residue 54Vβ. The Q72HVβ mutation is a minor contributor to affinity maturation and induces a more open arrangement of the CDR1, CDR2, and HV4 loops, thereby affecting how residue 52Vβ and residues of the CDR1 loop can interact with SEC3. Mutation at residue 52Vβ, however, can override the effect of the Q72HVβ mutation, by inducing a similar restructuring of the hypervariable loops. This dominant effect of the A52VVβ mutation in relation to a mutation at position 72Vβ is the basis of context-dependent energetic effects of the latter (e.g., a minor but significant contributor to affinity maturation; it is energetically negligible in affinity reversion).
Our structural analysis reveals two seemingly distinct regions of cooperativity in the Vβ-SEC3 molecular interface: one at the nexus of the CDR1, CDR2, and HV4 loops and including residues 28Vβ, 30Vβ, 52Vβ, 54Vβ, and 72Vβ, and the other involving residues 66Vβ and 80Vβ from the nearby framework of β strands. Recent studies have suggested that within distinct regions of protein-protein interfaces packed densely with energetically significant residues, termed hot regions or modules, mutations are energetically cooperative, while mutations in separate regions or modules are energetically additive (Keskin et al., 2005; Reichmann et al., 2005). Whether cooperativity in our model system requires residues to be adjacent to one another is not entirely clear, as reversion mutations at positions 52Vβ, 54Vβ, and 66Vβ appear to be energetically coupled (compare Σ[ΔΔGb(A52V/S54N-r) + ΔΔGb(E66K-r) to ΔΔGb(A52VV/S54N/E66K-r)] in Figure 1B). F176SEC3, the residue that contacts the side chains of the wild-type and mutant residues at position 66Vβ, also contacts residue T55Vβ and may affect the conformation of the CDR2 loop. Further crystallographic analysis of the Vβ-SEC3 affinity maturation pathway will be required to fully elucidate the structural basis of the cooperative energetics measured between residues 52Vβ, 54Vβ, and 66Vβ.
It has been argued that the single-base changes that are commonly introduced and accumulated over many generations during natural molecular evolution disfavor cooperativity, while in vitro-directed evolution techniques artificially bias molecular complexes to utilize positively cooperative binding energetics to more rapidly improve affinity (Bernat et al., 2004). This is not the case for our Vβ-SEC3 model system because the major observed energetic cooperativity in this affinity maturation pathway, occurring between residues at positions 52Vβ and 54Vβ, is negative (Yang et al., 2003), and because many of the mutations that significantly affect one another energetically and structurally (including those in the CDR2 and HV4 loops) arose from distinct rounds of mutagenesis and selection during the yeast display process (only the K66E, Q72H, and E80V mutations were clearly coevolved) (Kieke et al., 2001). It may be that the Vβ-SEC3 protein-protein interaction, and the affinity modulation thereof, is representative of other protein complexes and in vivo affinity maturation processes generally.
The range and types of structural changes invoked along the Vβ-SEC3 affinity maturation pathway are reminiscent of highly affinity-matured protein-hapten systems (Alzari et al., 1990; Midelfort et al., 2004; Mizutani et al., 1995; Wedemayer et al., 1997; Yuhasz et al., 1995). These structural changes include not only increased van der Waals interactions, hydrophobic surface burial, and improved shape complementarity, as seen in other affinity-matured protein-protein interactions (De Genst et al., 2004; Li et al., 2003; Sundberg et al., 2003), but also cooperative conformational changes, context-dependent binding, and structural preorganization of the binding site. One significant difference is the direction of energetic cooperativity in the Vβ-SEC3 (negative cooperativity) and protein-hapten (positive cooperativity) systems.
On account of the complexities of protein-protein interactions, the evolution of high affinity in protein-protein interactions can, and perhaps must, be induced by a wide diversity of structural changes. Indeed, the ~1500-fold affinity increase between the wild-type Vβ-SEC3 complex and that involving the highest affinity variant, L2CM, is brought about through numerous structural changes. In order for this degree of affinity maturation to take place due simply to a change in a single biophysical factor, it would, for example, require an increase in the buried hydrophobic surface area of at least 100 Å2, according to our estimates of the hydrophobic effect (Li et al., 2005; Sundberg et al., 2000). Instead, we actually observe a decrease in the buried hydrophobic surface area between the wild-type Vβ-SEC3 complex and the H72Q-r:SEC3 complex (which exhibits an affinity equivalent to the L2CM-SEC3 complex). Likewise, the molecular interface of the wild-type complex is already highly complementary, as indicated by an Sc value of 0.61. Shape complementarity is increased only to an Sc value of 0.66 in the H72Q-r:SEC3 complex, well short of Sc increases we have observed previously in much less affinity-matured protein-protein interactions (Sundberg et al., 2003). Thus, the majority of the affinity increase in this molecular system is due to a combination of other types of structural changes, the resulting interface remodeling events of which we have documented in this study.
It is likely that the complexities of protein-protein interactions will restrict our understanding of the fundamental rules that govern protein recognition for some time. This is especially the case as difficult-to-quantify biophysical factors, such as cooperativity and plasticity, are routinely involved in controlling the affinities and specificities of these interactions. Correlating the structural and energetic changes in an affinity maturation pathway, however, as we have done for the Vβ-SEC3 model system, is a critical step in understanding and developing predictive algorithms for protein-protein interactions.
Experimental Procedures
Crystallization
Vβ variants were expressed in E. coli, refolded from inclusion bodies, and purified as described previously (Yang et al., 2003). Crystals of Vβ variants alone were grown at room temperature by hanging drop vapor diffusion by mixing 1 μl concentrated protein solution (12 mg/ml) with an equal volume of reservoir buffer containing 2.0 M sodium malonate (pH 7.0), 0.2% dioxane. Prismatic crystals as large as 0.2 × 0.2 × 0.5 mm were obtained within 1 week.
SEC3 and variants thereof were expressed in E. coli and purified from periplasmic fractions as described previously (Andersen et al., 2001). Vβ and SEC3 proteins were mixed in a molar ratio of 1.2:1, respectively, and incubated for 24 hours at 4°C. Protein mixtures were further purified by size exclusion chromatography with a Superdex 75 HR 10/30 column (Amersham Biosciences) equilibrated with 50 mM Tris-HCl (pH 8.5). Cocrystals of wild-type Vβ-SEC3 and H72Q-r:SEC3-1A4 were grown at room temperature by hanging drop vapor diffusion by mixing 1 μl concentrated protein solution (8 mg/ml) and an equal volume of reservoir buffer containing 20% PEG3350, 0.2 M tri-ammonium citrate (pH 7.0), 0.3% dioxane. Showers of thin, needle-like crystals were formed within a week. These crystals gradually dissolved, and prismatic crystals as large as 0.05 × 0.1 × 0.3 mm were obtained after ~1 month.
A52V/S54N/K66E:SEC3-1D3 crystals were grown at room temperature by hanging drop vapor diffusion by mixing 1 μl concentrated protein solution and an equal volume of reservoir buffer containing 2.0 M (NH4)2SO4, 0.1 M Tris (pH 7.0), 0.3% 1,6-diaminohexane. Hexagonal crystals as large as 0.05 × 0.05 × 0.1 mm were obtained after 2 months.
Data Collection
For the apo Vβ crystals, saturated Li2SO4 was used as cryoprotectant. Diffraction data were collected from frozen crystals at 100 K on an R-axis IV++ image plate detector (Rigaku). The crystals belonged to space group I212121 (except the A52V-r variant), with one molecule in the asymmetric unit. The calculated Matthew’s co-efficient (Vm) was 2.5 Å3/Da, and the solvent content was 51.3%. The collected data were processed by using d*TREK incorporated in the Crystal Clear v1.35 software suite (Molecular Structure Corporation).
Vβ-SEC3 cocrystals were transferred to mother liquor containing 10% (v/v) sucrose and were frozen in liquid nitrogen. Diffraction data were collected by using synchrotron radiation at beamline X-25 of the National Synchrotron Light Source and were processed by using the programs DENZO and SCALEPACK (Otwinowski and Minor, 1997).
Structure Determination and Refinement
The apo Vβ and Vβ-SEC3 complex structures were determined by molecular replacement by using the program Molrep in the CCP4 program suite (CCP4, 1994) with components or the entirety of the wild-type Vβ-SEC3 complex crystal structure (PDB accession code 1JCK) (Fields et al., 1996), respectively, as search models. Initial refinement was performed with CNS (Brunger et al., 1998) with positional, simulated annealing, and individual B factor refinement. Manual model rebuilding was carried out iteratively in XtalView (McRee, 1999) by using σA-weighted 2Fo − Fc maps. After CNS refinement converged, further refinement was carried out with Refmac5 (Murshudov et al., 1997), during which solvent molecules were placed in >2σ peaks in the σA-weighted 2Fo − Fc maps with regard to potential interactions with hydrogen bonding partners. The solvent model was further checked comprehensively during refinement by omitting water molecules that exhibited high B factors (> 60 Å2) or poor hydrogen bonding distances or geometries.
Figures were generated with PyMOL (DeLano, 2002a), Molscript (Kraulis, 1991), and Raster3D (Merritt and Bacon, 1997).
Acknowledgments
Data for this study were measured in part at beamline X-25 of the National Synchrotron Light Source, financial support for which comes principally from the Offices of Biological and Environmental Research and of Basic Energy Sciences of the U.S. Department of Energy and from the National Center for Research Resources of the National Institutes of Health. In particular, we wish to thank Dr. Howard Robinson from the Mail-in Data Collection team for data collection and processing, and Drs. Michael K. Gilson and Roberto Dominguez for critical reading of, and comment on, the manuscript. This work was supported in part by National Institutes of Health grants GM55767 and AI064611 (to D.M.K.), GM52801 and AI49564 (to R.A.M.), and AI55882 (to E.J.S.).
Footnotes
Accession Numbers
Atomic coordinates and structure factors for S54N, A52V/S54N/K66E, V52A-r, E66K-r, H72Q-r, L2CM, mTCR15-SEC3, H72Q-r: SEC3-1A4, and A52V/S54N/K66E:SEC3-1D3 have been deposited into the Protein Data Bank with PDB ID codes 2APB, 2APF, 2APT, 2APV, 2APW, 2APX, 2AQ3, 2AQ1, and 2AQ2, respectively.
References
- Albeck S, Unger R, Schreiber G. Evaluation of direct and cooperative contributions towards the strength of buried hydrogen bonds and salt bridges. J Mol Biol. 2000;298:503–520. doi: 10.1006/jmbi.2000.3656. [DOI] [PubMed] [Google Scholar]
- Alzari PM, Spinelli S, Mariuzza RA, Boulot G, Poljak RJ, Jarvis JM, Milstein C. Three-dimensional structure determination of an anti-2-phenyloxazolone antibody: the role of somatic mutation and heavy/light chain pairing in the maturation of an immune response. EMBO J. 1990;9:3807–3814. doi: 10.1002/j.1460-2075.1990.tb07598.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andersen PS, Geisler C, Buus S, Mariuzza RA, Karjalainen K. Role of the T cell receptor ligand affinity in T cell activation by bacterial superantigens. J Biol Chem. 2001;276:33452–33457. doi: 10.1074/jbc.M103750200. [DOI] [PubMed] [Google Scholar]
- Anikeeva N, Lebedeva T, Krogsgaard M, Tetin SY, Martinez-Hackert E, Kalams SA, Davis MM, Sykulev Y. Distinct molecular mechanisms account for the specificity of two different T-cell receptors. Biochemistry. 2003;42:4709–4716. doi: 10.1021/bi026864+. [DOI] [PubMed] [Google Scholar]
- Bentley GA, Boulot G, Karjalainen K, Mariuzza RA. Crystal structure of the β chain of a T cell antigen receptor. Science. 1995;267:1984–1987. doi: 10.1126/science.7701320. [DOI] [PubMed] [Google Scholar]
- Bernat B, Sun M, Dwyer M, Feldkamp M, Kossiakoff AA. Dissecting the binding energy epitope of a high-affinity variant of human growth hormone: cooperative and additive effects from combining mutations from independently selected phage display mutagenesis libraries. Biochemistry. 2004;43:6076–6084. doi: 10.1021/bi036069b. [DOI] [PubMed] [Google Scholar]
- Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol. 1997;15:553–557. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- Bogan AA, Thorn KS. Anatomy of hot spots in protein interfaces. J Mol Biol. 1998;280:1–9. doi: 10.1006/jmbi.1998.1843. [DOI] [PubMed] [Google Scholar]
- Boniface JJ, Reich Z, Lyons DS, Davis MM. Thermodynamics of T cell receptor binding to peptide-MHC: evidence for a general mechanism of molecular scanning. Proc Natl Acad Sci USA. 1999;96:11446–11451. doi: 10.1073/pnas.96.20.11446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brunger AT, Adams PD, Clore GM, DeLano WL, Gros P, Grosse-Kunstleve RW, Jiang JS, Kuszewski J, Nilges M, Pannu NS, et al. Crystallography and NMR system: a new software suite for macromolecular structure determination. Acta Crystallogr D Biol Crystallogr. 1998;54:905–921. doi: 10.1107/s0907444998003254. [DOI] [PubMed] [Google Scholar]
- Burnett JC, Henchal EA, Schmaljohn AL, Bavari S. The evolving field of biodefence: therapeutic developments and diagnostics. Nat Rev Drug Discov. 2005;4:281–297. doi: 10.1038/nrd1694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- CCP4 (Collaborative Computational Project, Number 4) The CCP4 suite: programs for protein crystallography. Acta Crystallogr D Biol Crystallogr. 1994;50:760–763. doi: 10.1107/S0907444994003112. [DOI] [PubMed] [Google Scholar]
- Churchill HR, Andersen PS, Parke EA, Mariuzza RA, Kranz DM. Mapping the energy of superantigen Staphylococcus enterotoxin C3 recognition of an α/β T cell receptor using alanine scanning mutagenesis. J Exp Med. 2000;191:835–846. doi: 10.1084/jem.191.5.835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conte LL, Chothia C, Janin J. The atomic structure of protein-protein recognition sites. J Mol Biol. 1999;285:2177–2198. doi: 10.1006/jmbi.1998.2439. [DOI] [PubMed] [Google Scholar]
- De Genst E, Handelberg F, Van Meirhaeghe A, Vynck S, Loris R, Wyns L, Muyldermans S. Chemical basis for the affinity maturation of a camel single domain antibody. J Biol Chem. 2004;279:53593–53601. doi: 10.1074/jbc.M407843200. [DOI] [PubMed] [Google Scholar]
- DeLano WL. The PyMOL User’s Manual. San Carlos, CA: DeLano Scientific; 2002a. [Google Scholar]
- DeLano WL. Unraveling hot spots in binding interfaces: progress and challenges. Curr Opin Struct Biol. 2002b;12:14–20. doi: 10.1016/s0959-440x(02)00283-x. [DOI] [PubMed] [Google Scholar]
- Ding YH, Baker BM, Garboczi DN, Biddison WE, Wiley DC. Four A6-TCR/peptide/HLA-A2 structures that generate very different T cell signals are nearly identical. Immunity. 1999;11:45–56. doi: 10.1016/s1074-7613(00)80080-1. [DOI] [PubMed] [Google Scholar]
- Fields BA, Malchiodi EL, Li H, Ysern X, Stauffacher CV, Schlievert PM, Karjalainen K, Mariuzza RA. Crystal structure of a T-cell receptor β-chain complexed with a superantigen. Nature. 1996;384:188–192. doi: 10.1038/384188a0. [DOI] [PubMed] [Google Scholar]
- Garcia KC, Degano M, Pease LR, Huang M, Peterson PA, Teyton L, Wilson IA. Structural basis of plasticity in T cell receptor recognition of a self peptide-MHC antigen. Science. 1998;279:1166–1172. doi: 10.1126/science.279.5354.1166. [DOI] [PubMed] [Google Scholar]
- Gascoigne NR, Zal T. Molecular interactions at the T cell-antigen-presenting cell interface. Curr Opin Immunol. 2004;16:114–119. doi: 10.1016/j.coi.2003.11.008. [DOI] [PubMed] [Google Scholar]
- Guerois R, Nielsen JE, Serrano L. Predicting changes in the stability of proteins and protein complexes: a study of more than 1000 mutations. J Mol Biol. 2002;320:369–387. doi: 10.1016/S0022-2836(02)00442-4. [DOI] [PubMed] [Google Scholar]
- Huo S, Massova I, Kollman PA. Computational alanine scanning of the 1:1 human growth hormone-receptor complex. J Comput Chem. 2002;23:15–27. doi: 10.1002/jcc.1153. [DOI] [PubMed] [Google Scholar]
- Keskin O, Ma B, Nussinov R. Hot regions in protein-protein interactions: the organization and contribution of structurally conserved hot spot residues. J Mol Biol. 2005;345:1281–1294. doi: 10.1016/j.jmb.2004.10.077. [DOI] [PubMed] [Google Scholar]
- Kieke MC, Sundberg E, Shusta EV, Mariuzza RA, Wittrup KD, Kranz DM. High affinity T cell receptors from yeast display libraries block T cell activation by superantigens. J Mol Biol. 2001;307:1305–1315. doi: 10.1006/jmbi.2001.4560. [DOI] [PubMed] [Google Scholar]
- Kortemme T, Baker D. A simple physical model for binding energy hot spots in protein-protein complexes. Proc Natl Acad Sci USA. 2002;99:14116–14121. doi: 10.1073/pnas.202485799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraulis PJ. MOLSCRIPT: a program to produce both detailed and schematic plots of protein structures. J Appl Crystallogr. 1991;24:946–950. [Google Scholar]
- Li Y, Li H, Yang F, Smith-Gill SJ, Mariuzza RA. X-ray snapshots of the maturation of an antibody response to a protein antigen. Nat Struct Biol. 2003;10:482–488. doi: 10.1038/nsb930. [DOI] [PubMed] [Google Scholar]
- Li Y, Huang Y, Swaminathan CP, Smith-Gill SJ, Mariuzza RA. Magnitude of the hydrophobic effect at central versus peripheral sites in protein-protein interfaces. Structure (Camb) 2005;13:297–307. doi: 10.1016/j.str.2004.12.012. [DOI] [PubMed] [Google Scholar]
- Lowman HB. Bacteriophage display and discovery of peptide leads for drug development. Annu Rev Biophys Biomol Struct. 1997;26:401–424. doi: 10.1146/annurev.biophys.26.1.401. [DOI] [PubMed] [Google Scholar]
- Ma B, Wolfson HJ, Nussinov R. Protein functional epitopes: hot spots, dynamics and combinatorial libraries. Curr Opin Struct Biol. 2001;11:364–369. doi: 10.1016/s0959-440x(00)00216-5. [DOI] [PubMed] [Google Scholar]
- Massova I, Kollman PA. Computational alanine scanning to probe protein-protein interactions: a novel approach to evaluate binding free energies. J Am Chem Soc. 1999;121:8133–8143. [Google Scholar]
- McRee DE. XtalView/Xfit–a versatile program for manipulating atomic coordinates and electron density. J Struct Biol. 1999;125:156–165. doi: 10.1006/jsbi.1999.4094. [DOI] [PubMed] [Google Scholar]
- Merritt EA, Bacon DJ. Raster3D: photorealistic molecular graphics. Methods Enzymol. 1997;277:505–524. doi: 10.1016/s0076-6879(97)77028-9. [DOI] [PubMed] [Google Scholar]
- Midelfort KS, Hernandez HH, Lippow SM, Tidor B, Drennan CL, Wittrup KD. Substantial energetic improvement with minimal structural perturbation in a high affinity mutant antibody. J Mol Biol. 2004;343:685–701. doi: 10.1016/j.jmb.2004.08.019. [DOI] [PubMed] [Google Scholar]
- Mizutani R, Miura K, Nakayama T, Shimada I, Arata Y, Satow Y. Three-dimensional structures of the Fab fragment of murine N1G9 antibody from the primary immune response and of its complex with (4-hydroxy-3-nitrophenyl)acetate. J Mol Biol. 1995;254:208–222. doi: 10.1006/jmbi.1995.0612. [DOI] [PubMed] [Google Scholar]
- Murshudov GN, Vagin AA, Dodson EJ. Refinement of macromolecular structures by the maximum-likelihood method. Acta Crystallogr D Biol Crystallogr. 1997;53:240–255. doi: 10.1107/S0907444996012255. [DOI] [PubMed] [Google Scholar]
- Nooren IM, Thornton JM. Diversity of protein-protein interactions. EMBO J. 2003;22:3486–3492. doi: 10.1093/emboj/cdg359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otwinowski Z, Minor W. Processing X-ray diffraction data collected in oscillation mode. Methods Enzymol. 1997;276:307–326. doi: 10.1016/S0076-6879(97)76066-X. [DOI] [PubMed] [Google Scholar]
- Pal G, Ultsch MH, Clark KP, Currell B, Kossiakoff AA, Sidhu SS. Intramolecular cooperativity in a protein binding site assessed by combinatorial shotgun scanning mutagenesis. J Mol Biol. 2005;347:489–494. doi: 10.1016/j.jmb.2005.01.040. [DOI] [PubMed] [Google Scholar]
- Pawson T, Nash P. Protein-protein interactions define specificity in signal transduction. Genes Dev. 2000;14:1027–1047. [PubMed] [Google Scholar]
- Reichmann D, Rahat O, Albeck S, Meged R, Dym O, Schreiber G. The modular architecture of protein-protein binding interfaces. Proc Natl Acad Sci USA. 2005;102:57–62. doi: 10.1073/pnas.0407280102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reiser JB, Gregoire C, Darnault C, Mosser T, Guimezanes A, Schmitt-Verhulst AM, Fontecilla-Camps JC, Mazza G, Malissen B, Housset D. A T cell receptor CDR3β loop undergoes conformational changes of unprecedented magnitude upon binding to a peptide/MHC class I complex. Immunity. 2002;16:345–354. doi: 10.1016/s1074-7613(02)00288-1. [DOI] [PubMed] [Google Scholar]
- Reiser JB, Darnault C, Gregoire C, Mosser T, Mazza G, Kearney A, van der Merwe PA, Fontecilla-Camps JC, Housset D, Malissen B. CDR3 loop flexibility contributes to the degeneracy of TCR recognition. Nat Immunol. 2003;4:241–247. doi: 10.1038/ni891. [DOI] [PubMed] [Google Scholar]
- Sharp KA. Calculation of HyHel10-lysozyme binding free energy changes: effect of ten point mutations. Proteins. 1998;33:39–48. [PubMed] [Google Scholar]
- Sheinerman FB, Norel R, Honig B. Electrostatic aspects of protein-protein interactions. Curr Opin Struct Biol. 2000;10:153–159. doi: 10.1016/s0959-440x(00)00065-8. [DOI] [PubMed] [Google Scholar]
- Sundberg EJ, Urrutia M, Braden BC, Isern J, Tsuchiya D, Fields BA, Malchiodi EL, Tormo J, Schwarz FP, Mariuzza RA. Estimation of the hydrophobic effect in an antigen-antibody protein-protein interface. Biochemistry. 2000;39:15375–15387. doi: 10.1021/bi000704l. [DOI] [PubMed] [Google Scholar]
- Sundberg EJ, Andersen PS, Schlievert PM, Karjalainen K, Mariuzza RA. Structural, energetic and functional analysis of a protein-protein interface at distinct stages of affinity maturation. Structure. 2003;11:1151–1161. doi: 10.1016/s0969-2126(03)00187-4. [DOI] [PubMed] [Google Scholar]
- Teufel DP, Kao RY, Acharya KR, Shapiro R. Mutational analysis of the complex of human RNase inhibitor and human eosinophil-derived neurotoxin (RNase 2) Biochemistry. 2003;42:1451–1459. doi: 10.1021/bi026852o. [DOI] [PubMed] [Google Scholar]
- Warren AJ. Eukaryotic transcription factors. Curr Opin Struct Biol. 2002;12:107–114. doi: 10.1016/s0959-440x(02)00296-8. [DOI] [PubMed] [Google Scholar]
- Wedemayer GJ, Patten PA, Wang LH, Schultz PG, Stevens RC. Structural insights into the evolution of an antibody combining site. Science. 1997;276:1665–1669. doi: 10.1126/science.276.5319.1665. [DOI] [PubMed] [Google Scholar]
- Willcox BE, Gao GF, Wyer JR, Ladbury JE, Bell JI, Jakobsen BK, van der Merwe PA. TCR binding to peptide-MHC stabilizes a flexible recognition interface. Immunity. 1999;10:357–365. doi: 10.1016/s1074-7613(00)80035-7. [DOI] [PubMed] [Google Scholar]
- Winter G, Griffiths AD, Hawkins RE, Hoogenboom HR. Making antibodies by phage display technology. Annu Rev Immunol. 1994;12:433–455. doi: 10.1146/annurev.iy.12.040194.002245. [DOI] [PubMed] [Google Scholar]
- Wodak SJ, Janin J. Structural basis of macromolecular recognition. Adv Protein Chem. 2002;61:9–73. doi: 10.1016/s0065-3233(02)61001-0. [DOI] [PubMed] [Google Scholar]
- Yang PL, Schultz PG. Mutational analysis of the affinity maturation of antibody 48G7. J Mol Biol. 1999;294:1191–1201. doi: 10.1006/jmbi.1999.3197. [DOI] [PubMed] [Google Scholar]
- Yang J, Swaminathan CP, Huang Y, Guan R, Cho S, Kieke MC, Kranz DM, Mariuzza RA, Sundberg EJ. Dissecting cooperative and additive binding energetics in the affinity maturation pathway of a protein-protein interface. J Biol Chem. 2003;278:50412–50421. doi: 10.1074/jbc.M306848200. [DOI] [PubMed] [Google Scholar]
- Yuhasz SC, Parry C, Strand M, Amzel LM. Structural analysis of affinity maturation: the three-dimensional structures of complexes of an anti-nitrophenol antibody. Mol Immunol. 1995;32:1143–1155. doi: 10.1016/0161-5890(95)00063-1. [DOI] [PubMed] [Google Scholar]