

Abstract
A field, laboratory, and modeling study of As in groundwater discharging to Waquoit Bay, MA, shed light on coupled control of chemistry and hydrology on reactive transport of As in a coastal aquifer. Dissolved Fe(II) and As(III) in a reducing groundwater plume bracketed by an upper and a lower redox interface are oxidized as water flows towards the bay. This results in precipitation of Fe(III) oxides, along with oxidation and adsorption of As to sediment at the redox interfaces where concentrations of sedimentary HCl-leachable Fe (80~90% Fe(III)) are 734±232 mg kg-1, sedimentary phosphate extractable As (90~100% As(V)) are 316±111 μg kg-1, and are linearly correlated. Batch adsorption of As(III) onto orange, brown and gray sediments follows Langmuir isotherms, and can be fitted by a surface complexation model (SCM) assuming a diffuse layer for ferrihydrite. The sorption capacity and distribution coefficient for As increase with decreasing sediment Fe(II)/Fe. To allow accumulation of the amount of sediment As, similar hydrogeochemical conditions would have been operating for thousands of years at Waquoit Bay. The SCM simulated the observed dissolved As concentration better than a parametric approach based on Kd. Site specific isotherms should be established for Kd or SCM based models.
Introduction
To study contaminant transport, reactive transport models have been developed to couple solute transport in an aquifer with reactions in aqueous phases and solute-solid sorption equilibrium (1, 2). The majority of models developed for regulatory purposes use a single distribution coefficient (Kd) to describe sorption equilibrium due to ease of incorporation to transport codes (1). Because a single Kd value usually does not represent sorption behavior over a wide range of geochemical conditions, this leads to errors and uncertainties in the simulation (3) and is sometimes dealt with by adopting spatially variable Kd values that represent the characteristics of distinct geochemical zones (1). Langmuir isotherm with finite sorption capacity is more appropriate for many contaminant sorption reactions than the linear isotherm of the Kd approach because it accounts for the decrease in Kd value as the adsorbing surface is increasingly occupied by adsorbed species. Semi-mechanistic surface complexation models (SCM) that establish the reaction stoichiometry and apparent stability constants using site-specific material have been developed to simulate field observations for zinc (4), phosphate (5), and oxyanions of molybdenum (6) and uranium (7, 8) in aquifers. In addition to characterization of the solute-solid reactions using site-specific materials either for Kd (1) or SCM (7, 9), the aforementioned studies rely on an extensive collection of hydrologic parameters and geochemical data of both solute and solid phases. For example, the SCM approach has been shown to better capture the dynamic behavior of uranium than the Kd approach (8). These studies demonstrate that reactive-transport modeling is challenging but can be accomplished given an appropriate level of integration of field and laboratory investigations.
Reactive transport modeling has rarely been applied to the oxyanion of arsenic (As) despite the great interest in arsenic as a contaminant in sedimentary aquifers. The interests are two fold: 1) its wide spread natural occurrence in reducing groundwater used for drinking in many countries that threaten the health of hundreds of millions of people (10), 2) its ranking as the top contaminant in aquifers on the EPA National Priority List. In an effort to understand the potential for As impacts on subsurface water supplies from gold mining-related activities at Carlin, Nevada, a reactive transport model with a variably saturated reactive transport code (UNSATCHEM) was developed to simulate As transport using empirically determined pH-dependent isotherms to describe As sorption (11). A 1-dimensional reactive transport model using PHREEQC was constructed to determine the geochemical processes controlling As transport vertically in the Red River floodplain, Vietnam (12). To date, coupled chemical and hydrological processes regulating reactive transport of As in sedimentary aquifers remains largely unexplored.
To illustrate the coupled role of hydrology and chemistry in regulating groundwater As transport, parametric Kd and SCM based reactive transport models were constructed for a coastal aquifer at Waquoit Bay, Massachusetts. This approach allows us to take advantage of the carefully documented hydrological parameters (13, 14) and the wide range of chemical parameters established for the site (15) and built upon with this study. The objectives of the study are to determine what geochemical parameters are critical for reactive transport modeling of As, and to evaluate the performance of SCM vs. Kd in simulating the observed groundwater As distribution. The recent development of PHT3D (2), which couples transport simulator MODFLOW/MT3DMS with geochemical code PHREEQC makes this exercise feasible. The redox condition spans from anoxic to oxic in the aquifer. This redox gradient has been shown to regulate As mobility (16). Pore water and sediment core samples taken along a transect perpendicular to the shore were used for chemical characterization of solute and solid. Sorption experiments of As(III) were conducted with three sediment samples that captured the range of sedimentary Fe(II)/Fe. SCM modeling of the sorption experiments was performed with consideration given to the surface site densities of the sediment and oxidation of As(III). Finally, a multi-component reactive transport model (PHT3D) compared a parametric Kd approach with a SCM based approach in simulation of groundwater As distribution.
Methods
Study site
The Cape Cod aquifer is unconfined and is ~ 100-120 m thick with an upper permeable layer of 11 m thickness (17). The fresh groundwater in the upper permeable layer discharging to Waquoit Bay is the focus for investigation of As transport.
Sampling and Analysis
Between June 18th and 20th, 2007, three pore water (or groundwater) profiles up to 8 m deep along a 12 m transect perpendicular to the shore were collected at Waquoit Bay (Fig. S1). Sampling and analysis methods are described in the Supporting Information. The in-line filtered pore water samples were assayed for dissolved Fe(II) and As(III) immediately on site and for total dissolved As and Fe at Lamont-Doherty (Table S1). Three sediment cores spanning 2 m to 4 m depth were obtained along the same transect perpendicular to the shore at PZ7, PZ6, and PZ3 also in June of 2007, with samples stored within a nitrogen atmosphere to allow for the determination of HCl-leachable sediment Fe(II) and Fe within hours of sample collection. Another aliquot was extracted using an anaerobic phosphate solution to determine sorbed As(III) and As (Table S2). Sediment core penetrating to 6 m at PZ11 was collected in June 2006, and HCl-leachable Fe, and phosphate extractable As were obtained on samples stored wet and cold after 4 months of sample collection. All HCl-leachate were also analyzed for As.
Batch Adsorption Experiment
To establish sorption isotherms for As(III), three sediment samples at ~ 1 m depth from PZ7, PZ6, and PZ3 that captured the range of redox conditions indicated by HCl-leachable sedimentary Fe(II)/Fe ratios were selected for the batch adsorption experiment (Table 1). Arsenite was added to serum vials containing 10 g of sediment and topped off with N2-purged nano-pure water to result in an initial solute As(III) concentration ranging from 0 to 2.5 mg L-1 (Table S3). The vials were immediately crimp sealed and kept in ultra-pure N2 filled anaerobic chamber until the end of each time point when the contents were filtered (0.45 μm) after 1 week, and again after 2 weeks of equilibration time. The concentrations of As(III) and As in the supernatant and sorbed on the sediment were quantified.
Table 1.
Characteristics of Sediment and Langmuir Sorption Isotherm
| Sample ID | Depth | Characteristics of sediment |
Sorption propertis of sediment |
||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Color | 1.2N HCl leach FeII/Fe | 1.2N HCl leach FeIII | 1.2N HCl leach As | 1M P-ext As | 1M P-ext AsV | KLa | Kd at 10μg/L As | As sorption capacity | R2 | ||
| m | mg/kg | μg/kg | μg/kg | μg/kg | L/μg | L/kg | μg/kg | ||||
| PZ7 | 1.3 | Dark gray | 0.74 | 123 | 140 | 54 | - | 0.038 | 30 | 1515 | 0.94 |
| PZ6 | 0.6 | Brown | 0.25 | 562 | 387 | 197 | 197 | 0.067 | 42 | 1754 | 0.95 |
| PZ3 | 1.1 | Orange | 0.12 | 700 | 340 | 426 | - | 0.029 | 83 | 4762 | 1.00 |
Geochemical Modeling of Sorption Isotherms
Experimental data were first fitted to Langmuir isotherms to determine sorption capacity (Smax) and KLa, a constant representing the binding strength, and then the Kd values at equilibrium with 10 μg L-1 As were estimated from the isotherm fitted to batch sorption data.
A semi-mechanistic SCM of experimental data used the same acidity constants and equilibrium constants (K) as in a diffuse layer surface complexation model of ferrihydrite (19, 20). The surface site density of each sediment used in the sorption experiment was estimated first and then specified as 0.44, 2.0 and 4.9 μM g-1 in PHREEQC (version 2.15 with MINTEQA2 version 4.0 database) (20, 21). The solute consisted of Na+ and Cl- of 1 mmol L-1, As(III) or As(V) of 750-6000 μg L-1. The best fit of the data was achieved by adjusting the proportion of As(III) and As(V) when the solute is equilibrated with sediment at pH 7.
Reactive multi-component transport modeling
A 2D reactive transport of As in the discharging fresh groundwater of the upper aquifer was simulated coupling MODFLOW 2005 and PHT3D (version 2.0). The grid spacing for the x and z direction was 0.5 m and 0.2 m, respectively, corresponding to ~3% of the model length of 14 m and 6 m, respectively. The simulation time was 40,000 days (~110 years) with 6,400 reaction steps. The grid spacing and simulation time step were proven to be sufficient through sensitivity tests. Horizontal and vertical hydraulic conductivities were allocated to be 8.64 m d-1 and 0.864 m d-1, respectively (13). Hydraulic gradient was set to 0.009, corresponding to groundwater advection flow rate of 0.08 m d-1 or 29 m y-1 (14). Simulated Fe oxide and dissolved As at steady state were insensitive to variation of hydraulic gradient from 0.004 to 0.020. Longitudinal and vertical dispersivities were set to 1 m and 0.01 m, respectively (22). Constant head boundary for the upland side and river boundary for the Waquoit Bay side were selected, while all other sides were defined as no flow boundary. In our modeling, density-dependent flow is not taken into account because the nearshore circulation of seawater seems to be of minor importance at Waquoit Bay, and our simulated upward advection of fresh groundwater plume does not significantly differ from the simulation result incorporating a density-dependent flow code (23).
The geochemical gradient of the aquifer is represented by 3 vertical layers and 4 horizontal zones. The top (0-2 m) and bottom (4-6 m) layers are oxic with dissolved oxygen of 3 mg L-1 without dissolved Fe and As representing upland groundwater water entering the aquifer. They are assigned surface site density of 2 μM g-1 for the SCM and Kd of 60 L kg-1. The middle layer (2-4 m) has 4 horizontal redox zones downgradient to capture the redox transition from reducing to oxic toward the bay, with increasing surface site density from 1.1, 1.5, 3.0 to 4.0 μM g-1 for SCM, or with increasing Kd from 25, 60, 90 to 120 L kg-1. Upland groundwater contains dissolved Fe(II) of 5.6 mg L-1 of and As(III) of 15 μg L-1. A key feature of the simulation is that the recharge rate of oxygenated water increases downgradient from 0.001 m y-1 upland, typical of the annual average recharge rate, to 0.05 m y-1 near shore to reflect increasing nearshore circulation due to tides and waves (13, 14). Neither simulated Fe oxide nor dissolved As was sensitive to an increase or decrease of recharge rates by a factor of 4. The simulation is run for ~110 years to reach steady state for solute As or Fe, but the transient features are also reported. Arsenic adsorption along the flow path is governed only by spatially assigned surface site densities or Kd values, not by reaction with simulated Fe oxide. Detailed hydrologic and chemical settings are in Supporting Information.
Results and Discussion
Chemistry of Groundwater
The As containing fresh groundwater plume moves upward as the water flows towards the bay to discharge, with the center of the plume rising from ~ 3 m depth at PZ10 to ~ 2 m depth at PZ6 over a distance of 6 m (Fig. 1A). No pore water sample was taken from < 0.9 m depth at PZ3, but the more reduced sediment Fe(II)/Fe ratios between 0.1 m to 0.3 m depth at PZ3 (Fig. 1B) suggest that the plume may have risen to ~ 0.2 m at the shore. The peak concentration of dissolved As decreases from 14.3 μg L-1 at PZ10 to 2.4 μg L-1 at PZ6. The large concentration gradient corresponds to a shift of As speciation from As(III) to As(V) as the proportion of dissolved As(III) decreases from ~85% at PZ 10 to < 10% at PZ 6. Like As, the depth of the peak dissolved Fe concentration in the reducing plume of fresh groundwater also becomes shallower toward the bay, decreasing from 2.0 mg L-1 at PZ10 to 1.4 mg L-1 at PZ6 (Fig. 1A). Unlike As, dissolved Fe(II) is > 90% of total Fe at both PZ10 and PZ6 within the plume. The horizontal redox gradient indicated by Fe and Eh (Table S1) along the discharging flow path is subtle between PZ10 and PZ6 and is only evident in dissolved As speciation change.
Fig. 1.
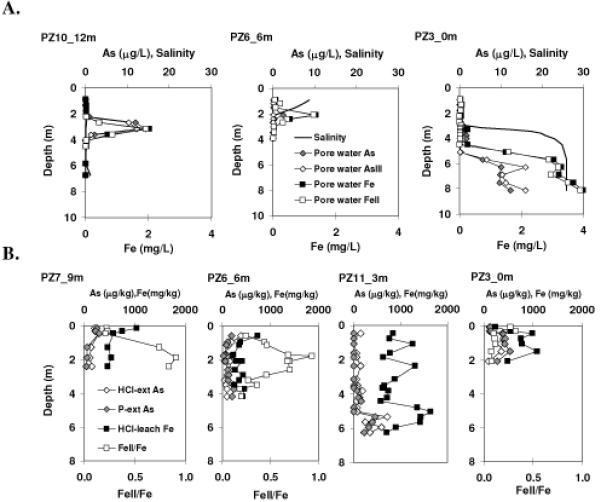
Field data from Waquoit Bay. A. Pore water profiles of salinity, dissolved Fe(II) and Fe, and dissolved As(III) and As. B. Sediment profiles of HCl-leachable Fe & Fe(II)/Fe, P-extractable As(III) & As.
The vertical redox zonation is well defined. The Eh and Fe data indicate three vertical redox zones in fresh groundwater (salinity < 1, Table S1). The proportion of dissolved Fe(II) decreases to < 50% above or below the reducing groundwater plume at all sites (Table S1). The pH values are higher (7.2 to 7.6) within the reducing plume, and are lower (6.0±0.4) above and below the plume (Table S1).
At PZ3 below the fresh, deeper redox interface between 1 m and 3 m, a fresh and saline water mixing zone between 3-4 m where the salinity increases from 1.5 to 23 corresponds to a third redox interface. Here, Fe(II) is ~ 50% of total Fe of ~0.03 mg L-1, and As(V) accounts for > 80% of total As of 1~2 μg L-1. The proportions of dissolved Fe(II) and As(III) increase with depth from 4 m to 8 m (Table S1). The saline and anoxic pore water at depth > 5 m displays high concentrations of Fe (> 90% Fe(II)), Mn and As (~ 100% As(III)), similar to those reported in Bone et al (16).
Chemistry of Aquifer Sediment
Sedimentary Fe(II)/Fe ratios (Fig. 1B) display consistent redox zones delineated by the dissolved constituents with depth and along the flow path to the bay. The sediment within the reducing groundwater plume is characterized by high Fe(II)/Fe ratios, with the maximum Fe(II)/Fe reaching 0.91 at 1.9 m at PZ7, and 0.94 at 1.8 m at PZ6. At PZ3, the sediment Fe(II)/Fe ratio is ~ 0.3 between 0.1 m and 0.3 m, and this may correspond to the reducing groundwater plume. In the upper and lower redox zones, sediment Fe(II)/Fe ratios are lower, with average values of 0.30±0.12 for the upper redox zones and 0.21±0.12 for the lower redox zone, respectively (Fig. 1B). The lower redox zone was not cored at PZ7 and the upper redox zone was not sampled at PZ3. The HCl leachable sedimentary Fe is < 500 mg kg-1 within the reducing plume, but is up to ~1000 mg kg-1 above and below.
Sedimentary As determined by 1 M P-extraction or 1.2 N HCl leaching displays low contents of 50~100 μg kg-1 within the reducing plume, but is up to ~ 500 μg kg-1 above or below. At PZ6, sediment P-extractable As(III) is < 11% of total As within the reducing plume, consistent with dissolved As(III) < 10% of total As found in pore water from the same depth intervals (Tables S1 and S2). In the upper and lower redox zones, > 95% of sediment P-extractable As is As(V).
Sediment data support a third redox interface at PZ11 between 5 m and 6 m (Fig. 1B), corresponding to the mixing zone between the deeper oxic fresh water and re-circulating reducing saline water (23). Concentrations of sedimentary As, Fe and Mn are elevated (Fig. 1B, Table S2), with ~ 450 μg kg-1 P-extractable As, ~ 720 μg kg-1 HCl leachable As, > 1000 mg kg-1 HCl leachable Fe. This saline water redox interface is also elevated with reductively leachable Mn ranging from 30 to 140 mg kg-1, higher than sedimentary Mn at the other redox interfaces in the freshwater regime (15).
Oxidation and Sorption of As by Fe(III) oxides at multiple redox interfaces
The iron curtain consisting of hydrous iron oxides (HFO) of ferrihydrite, goethite, and lepidocrocite at Waquoit Bay has been shown to sequester many elements in subterranean estuaries, resulting in a reduction of the chemical fluxes of these elements to the bay as the groundwater discharges through this natural reactive barrier (16, 24, 25). New insight from the pore water and sediment chemistry depth profiles presented here is that there are three distinct natural reactive barriers in the fresh and saline water regimes. The shallow oxic-anoxic interface between 0 and 1 m above the reducing groundwater plume is well recognized at PZ7, PZ6 and PZ11, where HCl leachable Fe has a peak, and the Fe(II)/Fe ratios are low (Fig. 1B). Gas-exchange between the upwelling reducing groundwater and the atmosphere and intrusion of seawater by nearshore circulation due to tides and waves can both supply oxygen (13). The deeper redox interface within the fresh groundwater regime is less intuitive, but is a feature that reflects the chemistry of the advecting anoxic groundwater plume and the oxic groundwater at depth (23). This is recognized at PZ6 at > 3 m, PZ11 at ~ 2.5m, and PZ3 between 0.3 m and 2 m. Lastly, a third and the deepest redox interface between 4 and 6 m at PZ11 results from mixing between the re-circulating anoxic saline groundwater and the advecting oxic fresh groundwater (26).
For sediment samples within the two redox interfaces in the freshwater regime where the sediment Fe(II)/Fe ratios are < 0.3, concentrations of sediment P-extractable As are > 100 μg kg-1 for all sites. The sedimentary P-extractable As reflects As adsorbed on the mineral surfaces (27). In comparison, in the reducing zones where the sedimentary Fe(II)/Fe ratios are > 0.7, the sediment P-extractable As concentrations are only ~50 μg kg-1, comparable to reductively extracted sedimentary As of ~75 μg kg-1 from reducing aquifer sediment in the Cape Cod aquifer (18).
That adsorption to amorphous Fe(III) oxyhydroxide is responsible for immobilization of As is further evidenced by an excellent correlation between P-extractable As and the HCl leachable sediment Fe(III) concentrations in all sediment samples (n = 26; R2 = 0.81, Fig. S2A). Although the HCl leachable As concentration increases with the HCl leachable sediment Fe(III) concentration (Fig. S2B), the correlation is not as good (R2 = 0.59) and the data form two clusters. This suggests that adsorption rather than coprecipitation is a dominant process for As immobilization. Because As(V) accounted for the majority of sedimentary P-extractable As whereas the reducing groundwater plume contained primarily As(III) (Fig. 1 and Table S1), oxidation of As(III) also occurred during or after adsorption.
Langmuir Sorption Isotherms and Kd
The equilibrium concentrations of As in the supernatant and sorbed on the sediment during batch sorption experiment fitted well to Langmuir sorption isotherms (Fig. 2) for the three sediment samples with distinct color of dark gray (PZ7), brown (PZ6) and orange (PZ3). The As sorption capacity (Smax) was estimated to be 1520, 1750, and 4760 μg kg-1 for the dark gray, brown and orange colored sediments from PZ7, PZ6, and PZ3, respectively (Table 1). Although As(III) was added and care was taken to conduct the experiment under anaerobic conditions, arsenic remaining in supernatant after equilibration of 2 weeks was 92~100% As(V) (Table S3). The concentrations of As sorbed on sediment were estimated first by mass balance from the supernatant concentration changes. The concentrations of As sorbed on sediment were also determined by extracting the equilibrated sediment sequentially with 1M phosphate under anaerobic condition and with 1.2 N HCl. The phosphate and HCl extract together recovered all As when compared to those estimated by mass balance (Table S3). P-extraction liberated 64±12%, 70±10%, and 89±10% of sorbed As from PZ7, PZ6, and PZ3, respectively, with > 90% as As(V).
Fig. 2.
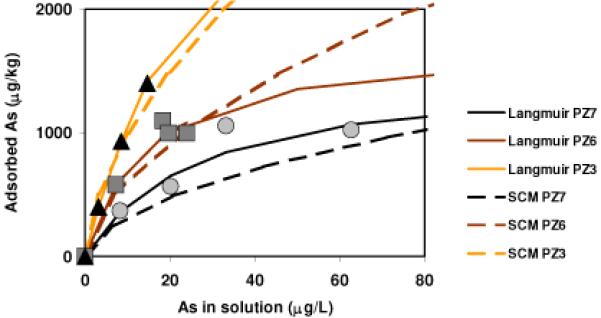
Batch As(III) sorption experiment results for dark gray (PZ7, circle), brown (PZ6, square), and orange (PZ3, triangle) sediment from Waquoit Bay. Solid lines are Langmuir isotherms. Dashed lines are surface complexation models.
Because the groundwater As concentration at the site is ~ 10 μg L-1 or less and the sediment As concentration is far less than the sorption capacity (Table 1), a calculation of Kd for sediment at equilibrium with a dissolved As of 10 μg L-1 is made so that one empirical but intuitive parameter instead of two parameters (e.g. KLa and Smax) can be used to compare sediment samples with distinct color and Fe(II)/Fe. The distribution coefficients (Kd) at equilibrium with 10 μg L-1 dissolved As for sediments from PZ7, PZ6, and PZ3 are 30, 42, and 83 L kg-1, respectively (Table 1). This increase of Kd corresponds to a change of sediment color from dark gray, to brown, and then to orange, and a decrease of sediment Fe(II)/Fe ratio (Table 1). Nevertheless, this Kd value should not be interpreted to imply infinite sorption sites or linear sorption isotherm.
Semi-Mechanistic Surface Complexation Modeling of Sorption Experiment
A semi-mechanistic SCM provided a reasonable fit to the sorption experimental data (Fig. 2). Surface complexation reactions, i.e., the equilibrium constants considered, are the same as those used in the ferrihydrite model system (Table S4) by Dzombak and Morel (20). The ratio of As(III) and As(V) in each sorption experimental system was adjusted until the fit to the experimental data was achieved. In this scenario, approximately 0, 30, and 40% of added As(III) is oxidized to As(V) during adsorption equilibrium with PZ7, PZ6, and PZ3 sediments, respectively. Whereas we do not believe the proportion of oxidation occurred during sorption is entirely quantitative, the increasing proportion of As(V) is consistent with the decreasing sediment Fe(II)/Fe ratio (Table 1). The SCM fit closely tracks the experimental data and the Langmuir isotherms when dissolved [As] are < 20 μg L-1 (Fig. 2), but diverges significantly when the dissolved [As] are < 40 μg L-1. Because the concentration of As in the Cape Cod aquifer is usually < 20 μg L-1 (18, 28), this experimentally derived semi-mechanistic SCM is therefore a reasonable representation of water-sediment sorption equilibrium for the Cape Cod aquifer.
The semi-mechanistic SCM of the sorption experiment used surface site density values of 0.44, 2.0, and 4.9 μM g-1 for the dark gray (PZ7), brown (PZ6) and orange (PZ3) colored sediment samples (Fig. 2). That the surface site density increases as sediment becomes more oxidized is consistent with the increases of As sorption capacity from 1520, 1750, to 4760 μg kg-1 (Table 1). The surface site density is estimated from the sediment Fe(III) concentrations. The dark gray, brown and orange colored sediments are characterized by 1.2N HCl leachable Fe(III) of 123, 562, and 700 mg kg-1 (Table 1). Because the 1.2N hot HCl extraction tends to leach amorphous and relatively labile crystalline Fe oxyhydroxides, the surface site density is calculated assuming all Fe(III) is in the form of ferrihydrite with 0.2 mol of surface site per mole of Fe (20). The results are 0.44 μM g-1, 2.0 μM g-1 and 2.5 μM g-1 for dark gray, brown and orange sediment, respectively. Because of the higher proportion of crystalline iron oxides in the orange sediment (25), the HCl leachable Fe(III) concentration did not leach other iron minerals that also contribute to sorption. Selective leaching aimed for both amorphous and crystalline Fe oxides for sediment collected from the same depth at PZ3 found an Fe concentration of ~2500 mg kg-1 (25), with approximately half as amorphous Fe oxides and half as crystalline Fe oxides. Assuming that the amorphous Fe oxide is ferrihydrite like with 0.2 mol of surface site per 1 mol Fe, and that the crystalline Fe oxides is goethite like with 0.02 mol of surface site per 1 mol Fe (20, 29), a weighted revised surface site density of 4.9 μM g-1 is the best estimate for the orange sediment.
That the HCl leachable Fe concentration under-represented secondary Fe minerals in the orange sediment is also consistent with the observation that P-extractable As concentration of 426 μg kg-1 is greater than the HCl leachable As concentration of 340 μg kg-1 (Table 1). The likely reason for this is that 1.2 N HCl leaching was not effective in attacking the crystalline Fe oxides that only part of adsorbed As was leached out from the amorphous Fe oxides, while phosphate extraction was effective in displacing As from sorption sites of both crystalline and amorphous Fe oxides. A previous study has indeed identified a variety of Fe mineral such as ferrihydrite, goethite, and lepidocrosite by XAS (24).
Reactive Transport Modeling of As
Both the Kd and SCM based reactive transport simulations show that dissolved As concentrations reach, or are very close to steady state in ~80 years (Fig. 3). The Kd-based steady state As concentrations at the center of the plume are 15, 12 and 6 μg L-1 at 12 m (PZ10), 6 m (PZ6) and 0 m (PZ3) from the shore, respectively, which are considerably higher than the observed groundwater concentrations. In contrast, the SCM-based As concentrations at the center of plume are 15, 5.5 and < 0.3 μg L-1 at 12 m (PZ10), 6 m (PZ6) and 0 m (PZ3) from the shore, and compare favorably with field results (Fig. 3). The reason why the SCM performed significantly better than parametric Kd-based model is that SCM can account for the effects of spatio-temporal chemical variation on the As adsorption and desorption reactions. The model also succeeded in generating the upper and lower iron oxides layers with 0.5~1 m thickness along the plume.
Fig. 3.
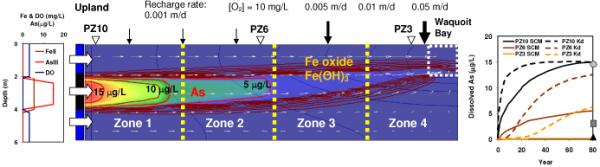
Composition of groundwater with three distinct vertical redox zonations flowinginto the model at the upland boundary (left), the simulation result of As plume migration and Fe oxide precipitation using SCM based PHT3D after 80 years of model run (middle), and the comparison of simulated groundwater As over 80 years between parametric Kd based model (dashed lines) and SCM based model (solid lines) (right). Arrows indicate variable recharge rates parameterized to simulate nearshore circulation due to tides and waves, and the white box (right corner, middle panel) on the bay side indicates a 2-m river boundary. In the middle reducing layer (2-4 m), the model has 4 zones to capture the geochemical gradient with increasing surface site density and Kd values towards the bay. Contour interval for As is 3 μg/L. Observed total dissolved As at PZ10 (circle), PZ6 (square), and PZ3 (triangle) compare well with SCM based model. Simulated Fe oxide ranging from 3 to 15 mg/kg has the contour intervalof 3 mg/kg.
History of Natural Reactive Barrier at Waquoit Bay
The time to accumulate 400 μg kg-1 As by a natural reactive barrier at PZ3 is estimated to be ~2300 years assuming that the reactive barrier is 1 m thick and 1.5 m long, and that groundwater containing 5 μg L-1 of As is transported at 0.26 m y-1, a slower rate due to retardation corresponding to the average Kd value of 60 L kg-1. This suggests that the natural reactive barrier at Waquoit Bay has operated for thousands of years. This is not entirely surprising given the geologic history of the area, namely, that the hydrologic conditions have been fairly close to present day conditions in the last few thousand years after the sea level had stabilized (30). What is surprising is how quickly the system has reached steady state. Even with a high retardation factor, groundwater As reaches steady state in ~100 years over a distance of 12 m (Fig. 3).
Implications for Arsenic Contaminated Aquifer
A critical issue in reactive transport of groundwater As is to determine whether Kd-based models can be used to study processes without invoking SCM. In the case of Cape Cod coastal aquifer, this is a difficult proposition because the Langmuir isotherms are characterized by low sorption capacity and high KLa (Table 1). This leads to a strong degree of dependence of Kd values on the equilibrated As concentrations (Fig. S4). But even in this case, if the geochemical conditions along the flow path of contaminant are similar, Kd based approach can still be useful. An example is shown by a simulation of an As(V) injection experiment into an anoxic zone of a sandy aquifer at the USGS research site on Cape Cod (28). We found that a Kd of 4 L kg-1 for an equilibrium As concentration of 75 μg L-1 (equivalent to a Kd of 30 L kg-1 if the equilibrium As concentration is 10 μg L-1) in PHT3D simulated the migration of As plume to a distance of 4.5 m down gradient over ~100 days (Fig. S3). Furthermore, the simulated maximum total As concentration in the center of the plume was 190 μg L-1, while the observed maximum total As value was ~150 μg L-1 in ~100 days. This implies that As sorption isotherms of the reducing aquifer sediment are similar at Waquoit Bay and at the USGS research site on Cape Cod.
If the site specific Langmuir sorption isotherms are characterized by high sorption capacity and low KLa values, then the non-linearity in the sorption is minimum, translating to a low degree of dependence of Kd on the equilibrium solute As concentration (Fig. S4). This may have been the case for the Bangladesh aquifer where a fairly consistent Kd value of 4 L kg-1 has been inferred by regional scale studies (31). Because of the wide range of Kd values observed for a variety of soils and sediment (Table S5), we recommend that investigators conduct site specific isotherm studies to determine whether a Kd-based approach is justified.
Supplementary Material
Acknowledgement
We thank Paul Henderson, Megan Gonneea and Katherine French for field assistance. We are grateful to Henning Prommer, Chunmiao Zheng and Rui Ma for helpful discussion of PHT3D modeling. HBJ received a University Fellowship and Mina Rees Dissertation Fellowship from the Graduate Center, CUNY. MAC received NSF awards (OCE-0425061 and OCE-0751525). YZ received awards from NIEHS SBRP 2 P42 ES10349 and NSF EAR-0738888.
Footnotes
Supporting Information Available Pore water (Table S1) and sediment Data (Table S2); sorption experiment results (Table S3) and SCM modeling reactions with equilibrium constants (Table S4); distribution coefficients (Kd) of As for soil and sediment (Table S5); location of pore water and sediment core transect (Fig. S1); sedimentary As vs. sedimentary Fe(III) and Fe(II)/Fe (Fig. S2); PHT3D simulation of an As(V) injection experiment (Fig. S3); Kd dependence on equilibrium As concentration (Fig. S4) are available free of charge via the Internet at http://pubs.acs.org.
Literature Cited
- (1).Zhu C, Anderson G. Environmental Applications of Geochemical Modeling. Cambridge University Press; London: 2002. [Google Scholar]
- (2).Prommer H, Barry DA, Zheng C. MODFLOW/MT3DMS-based reactive multicomponent transport modeling. Ground Water. 2003;41:247–257. doi: 10.1111/j.1745-6584.2003.tb02588.x. [DOI] [PubMed] [Google Scholar]
- (3).Bethke CM, Brady PV. How the K-d approach undermines ground water cleanup. Ground Water. 2000;38:435–443. [Google Scholar]
- (4).Kent DB, Abrams RH, Davis JA, Coston JA, Leblanc DR. Modeling the influence of variable pH on the transport of zinc in a contaminated aquifer using semiempirical surface complexation models. Water Resour. Res. 2000;36:3411–3425. [Google Scholar]
- (5).Parkhurst DL, Stollenwerk KG, Colman JA. Reactive-Transport Simulation of Phosphorus in the Sewage Plume at the Massachusetts Military Reservation, Cape Cod, Massachusetts. 2003. U.S. Geological Survey Water-Resources Investigations Report 03-4017. [Google Scholar]
- (6).Stollenwerk KG. Modeling the effects of variable groundwater chemistry on adsorption of molybdate. Water. Resour. Res. 1995;31:347–357. [Google Scholar]
- (7).Davis JA, Meece DE, Kohler M, Curtis GP. Approaches to surface complexation modeling of uranium(VI) adsorption on aquifer sediments. Geochim. Cosmochim. Acta. 2004;68:3621–3641. [Google Scholar]
- (8).Curtis GP, Davis JA, Naftz DL. Simulation of reactive transport of uranium(VI) in groundwater with variable chemical conditions. Water Resour. Res. 2006;42:15. [Google Scholar]
- (9).Stollenwerk KG. Molybdate transport in a chemically complex aquifer: Field measurements compared with solute-transport model predictions. Water Resour. Res. 1998;34:2727–2740. [Google Scholar]
- (10).Smedley PL, Kinniburgh DG. A review of the source, behaviour and distribution of arsenic in natural waters. Appl. Geochem. 2002;17:517–568. [Google Scholar]
- (11).Decker DL, Simunek J, Tyler SW, Papelis C, Logsdon MJ. Variably saturated reactive transport of arsenic in heap-leach facilities. Vadose Zone J. 2006;5:430–444. [Google Scholar]
- (12).Postma D, Larsen F, Hue NTM, Duc MT, Viet PH, Nhan PQ, Jessen S. Arsenic in groundwater of the Red River floodplain, Vietnam: Controlling geochemical processes and reactive transport modeling. Geochim. Cosmochim. Acta. 2007;71:5054–5071. [Google Scholar]
- (13).Michael HA, Mulligan AE, Harvey CF. Seasonal oscillations in water exchange between aquifers and the coastal ocean. Nature. 2005;436:1145–1148. doi: 10.1038/nature03935. [DOI] [PubMed] [Google Scholar]
- (14).Mulligan AE, Charette MA. Intercomparison of submarine groundwater discharge estimates from a sandy unconfined aquifer. J. Hydrol. 2006;327:411–425. [Google Scholar]
- (15).Gonneea ME, Morris PJ, Dulaiova H, Charette MA. New perspectives on radium behavior within a subterranean estuary. Mar. Chem. 2008;109:250–267. [Google Scholar]
- (16).Bone SE, Gonneea ME, Charette MA. Geochemical cycling of arsenic in a coastal aquifer. Environ. Sci. Technol. 2006;40:3273–3278. doi: 10.1021/es052352h. [DOI] [PubMed] [Google Scholar]
- (17).Cambareri TC, Eichner EM. Watershed delineation and ground water discharge to a coastal embayment. Ground Water. 1998;36:626–634. [Google Scholar]
- (18).Kent DB, Fox PM. The influence of groundwater chemistry on arsenic concentrations and speciation in a quartz sand and gravel aquifer. Geochem. Transact. 2004;5:1–12. doi: 10.1186/1467-4866-5-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Allison JD, Brown DS, Novo-Gradic KH. MINTEQA2/PRODEFA2, A Chemical Assessment Model for Environmental Systems: Version 4.0 User's Manual. U.S. Environmental Protection Agency; Athens, GA: 1991. EPA/600/3-91/021. [Google Scholar]
- (20).Dzombak DA, Morel FMM. Surface Complexation Modeling: Hydrous Ferric Oxide. Wiley-Interscience; New York: 1990. [Google Scholar]
- (21).Parkhurst DL, Appelo CAJ. User's guide to PHREEQC (version 2)-A computer program for speciation, batch-reaction, one-dimensional transport, and inverse geochemical calculations. 1999. U.S. Geological Survey Water-Resources Investigations Report 99-4259. [Google Scholar]
- (22).Garabedian SP, LeBlanc DR, Gelhar LW, Celia MA. Large-scale natural gradient tracer test in sand and gravel, Cape Cod, Massachusetts 2. Analysis of spatial moments for nonreactive tracer. Water Resour. Res. 1991;27:911–924. [Google Scholar]
- (23).Spiteri C, Slomp CP, Charette MA, Tuncay K, Meile C. Flow and nutrient dynamics in a subterranean estuary (Waquoit Bay, MA, USA): Field data and reactive transport modeling. Geochim. Cosmochim. Acta. 2008;72:3398–3412. [Google Scholar]
- (24).Charette MA, Sholkovitz ER. Oxidative precipitation of groundwater-derived ferrous iron in the subterranean estuary of a coastal bay. Geophys. Res. Lett. 2002;29 2001GL014512. [Google Scholar]
- (25).Charette MA, Sholkovitz ER, Hansel CM. Trace element cycling in a subterranean estuary: Part 1. Geochemistry of the permeable sediments. Geochim. Cosmochim. Acta. 2005;69:2095–2109. [Google Scholar]
- (26).Charette MA, Sholkovitz ER. Trace element cycling in a subterranean estuary: Part 2. Geochemistry of the pore water. Geochim. Cosmochim. Acta. 2006;70:811–826. [Google Scholar]
- (27).Jung HB, Zheng Y. Enhanced recovery of arsenite sorbed onto synthetic oxides by L-ascorbic acid addition to phosphate solution: calibrating a sequential leaching method for the speciation analysis of arsenic in natural samples. Water Res. 2006;40:2168–2180. doi: 10.1016/j.watres.2006.03.032. [DOI] [PubMed] [Google Scholar]
- (28).Hohn R, Isenbeck-Schroter A, Kent DB, Davis JA, Jakobsen R, Jann S, Niedan V, Scholz C, Stadler S, Tretner A. Tracer test with As(V) under variable redox conditions controlling arsenic transport in the presence of elevated ferrous iron concentrations. J. Contam. Hydrol. 2006;88:36–54. doi: 10.1016/j.jconhyd.2006.06.001. [DOI] [PubMed] [Google Scholar]
- (29).Mathur SS, Dzombak DA. Surface Complexation Modeling: Goethite. In: Lutzenkirchen J, editor. Surface Complexation Modeling. Elsevier; Amsterdam: 2006. [Google Scholar]
- (30).Redfield AC. Postglacial change in sea level in the Western North Atlantic Ocean. Science. 1967;157:687–692. doi: 10.1126/science.157.3789.687. [DOI] [PubMed] [Google Scholar]
- (31).van Geen A, Zheng Y, Goodbred S, Horneman A, Aziz Z, Cheng Z, Stute M, Mailloux B, Weinman B, Hoque MA, Seddique AA, Hossain MS, Chowdhury SH, Ahmed KM. Flushing history as a hydrogeological control on the regional distribution of arsenic in shallow groundwater of the Bengal Basin. Environ. Sci. Technol. 2008;42:2283–2288. doi: 10.1021/es702316k. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.