

Abstract
We previously reported that ischemic postconditioning with a series of mechanical interruptions of reperfusion reduced infarct volume 2 days after focal ischemia in rats. Here, we extend this data by examining long-term protection and exploring underlying mechanisms involving the Akt, mitogen-activated protein kinase (MAPK) and protein kinase C (PKC) signaling pathways. Post-conditioning reduced infarct and improved behavioral function assessed 30 days after stroke. Additionally, postconditioning increased levels of phosphorylated Akt (Ser473) as measured by western blot and Akt activity as measured by an in vitro kinase assay. Inhibiting Akt activity by a phosphoinositide 3-kinase inhibitor, LY294002, enlarged infarct in postconditioned rats. Postconditioning did not affect protein levels of phosphorylated-phosphatase and tensin homologue deleted on chromosome 10 or -phosphoinositide-dependent protein kinase-1 (molecules upstream of Akt) but did inhibit an increase in phosphorylated-glycogen synthase kinase 3β, an Akt effector. In addition, postconditioning blocked β-catenin phosphorylation subsequent to glycogen synthase kinase, but had no effect on total or non-phosphorylated active β-catenin protein levels. Furthermore, postconditioning inhibited increases in the amount of phosphorylated-c-Jun N-terminal kinase and extracellular signal-regulated kinase 1/2 in the MAPK pathway. Finally, postconditioning blocked death-promoting δPKC cleavage and attenuated reduction in phosphorylation of survival-promoting εPKC. In conclusion, our data suggest that postconditioning provides long-term protection against stroke in rats. Additionally, we found that Akt activity contributes to postconditioning’s protection; furthermore, increases in εPKC activity, a survival-promoting pathway, and reductions in MAPK and δPKC activity; two putative death-promoting pathways correlate with postconditioning’s protection.
Keywords: Akt, cerebral ischemia, mitogen-activated protein kinase, postconditioning, protein kinase C, β-catenin
Rapid ischemic postconditioning was first defined in the research field of myocardial ischemia as a series of mechanical interruptions of reperfusion performed immediately after reperfusion (Zhao et al. 2003b). We and others have demonstrated that rapid postconditioning also reduces infarction for a few days after focal cerebral ischemia (Zhao et al. 2006b; Pignataro et al. 2007; Zhao 2007). In addition, it was reported that delayed postconditioning performed 2 days after reperfusion also blocked hippocampal neuronal death after transient global cerebral ischemia (Burda et al. 2006). Whether rapid postconditioning protects against brain ischemia long-term is not known. In addition, spared brain tissue may not translate into preservation of brain function (Dumas and Sapolsky 2001); whether rapid postconditioning improves neurological function after focal ischemia needs further study. Moreover, its mechanism(s) of protection largely remain elusive.
The pathological mechanisms after brain ischemia involve the protein kinase C (PKC) (Bright et al. 2004; Raval et al. 2005), the phosphoinositide 3-kinase (PI3K)/Akt (Noshita et al. 2001; Yano et al. 2001; Zhao et al. 2005; Kitano et al. 2007), and the mitogen-activated protein kinase (MAPK) pathways, including c-jun N-terminal kinase (JNK) and extracellular signal-regulated kinase (ERK) 1/2 (Gonzalez-Zulueta et al. 2000; Lee and Lo 2003). The PKC pathways include δPKC and εPKC (Bright et al. 2004; Raval et al. 2005). δPKC activity reflected by increases in δPKC cleavage or membrane translocation promotes neuronal death (Raval et al. 2005; Shimohata et al. 2007b), while an increase in εPKC levels improves neuronal survival (Shimohata et al. 2007a). The involvement of PKC pathways in the protective effect of postconditioning is not known.
Akt is presumed to be activated by phosphorylation via activated PI3K and phosphoinositide-dependent protein kinase-1 (PDK1) (Franke et al. 2003), but is inactivated by phosphatase and tensin homologue deleted on chromosome 10 (PTEN), while PTEN is activated by dephosphorylation (Vazquez et al. 2000). Activated Akt blocks apoptosis by phosphorylating glycogen synthase kinase 3β (GSK3β) (Franke et al. 2003; Hanada et al. 2004). Like PTEN, GSK3β is activated by dephosphorylation (Cross et al. 1995). In turn, GSK3β phosphorylates β-catenin, leading to proteasomal degradation of β-catenin and ultimately apoptosis (Nusse 2003). Conversely, active β-catenin which is not phosphorylated remains stable (van Noort et al. 2002). As we recently reviewed, most previous studies examining the Akt pathway in postconditioning assessed phosphorylation of Akt alone without characterizing phosphorylation of other molecules in the Akt pathway, and an assay for true Akt activity was usually neglected (Zhao et al. 2006a). Importantly, Akt’s phosphorylation level does not represent its true activity (Zhao et al. 2005).
c-Jun N-terminal kinase activity has been definitively shown to worsen ischemic brain injury (Gao et al. 2005); by contrast, ERK1/2 may either protect (Shamloo et al. 1999) or exacerbate ischemic injuries (Noshita et al. 2002; Wang et al. 2004). Pignataro et al. (2007) showed that postconditioning enhanced phosphorylation of ERK1/2 and had no effect on JNK. Given the controversial data for ERK1/2 in past studies, we assume that the role of ERK1/2 is determined by ischemic models, activating factors, and cell types. Thus, the roles of ERK1/2 as well as JNK in postconditioning warrant further studies in different ischemic models.
In summary, we studied the long-term protection of postconditioning in rats subjected to focal ischemia generated by bilateral common carotid artery occlusion plus permanent distal middle cerebral artery (MCA) occlusion, and tested whether the PKC, Akt, and MAPK pathways are involved in postconditioning.
Materials and methods
Focal cerebral ischemia and postconditioning
Focal ischemia was generated as described previously in male Sprague—Dawley rats (350–390 g) (Chen et al. 1986; Zhao et al. 2003a, 2004). Experimental protocols were approved by the Stanford University Administrative Panel on Laboratory Animal Care. Core body temperatures were monitored with a rectal probe and were maintained at 37°C throughout the experiment under anesthesia with isoflurane. The distal MCA was exposed and cauterized permanently above the rhinal fissure. The bilateral common carotid arteries (CCA) were occluded for 30 min with suture tightening. Post-conditioning was performed as described (Zhao et al. 2006b): three cycles of 30 s of CCA suture release followed by 10 s of occlusion.
General histology and infarct size measurement
Infarction was measured by 2,3,5-triphenyl-2H-tetrazolium chloride (TTC) or cresyl violet staining (Zhao et al. 2005). Rats killed 2 days after stroke were perfused transcardially with phosphate-buffered saline. Brains were sectioned into five coronal blocks rostral (level 1) to caudal (level 5) and stained with 1% TTC solution. The percentage of infarct in the cortex was calculated according to the formula: [contralateral cortex — (ipsilateral cortex — infarct cortex)/contralateral cortex] × 100%.
Rats killed 30 days after ischemia were perfused transcardially with phosphate-buffered saline followed by 4% paraformaldehyde. Brains were sliced and stained with cresyl violet. The injury size was normalized to the contralateral non-ischemic cortex and expressed as percentages according to the formula: [(contralateral cortex — normal ipsilateral cortex)/contralateral cortex] × 100% (Zhao et al. 2005).
Behavioral testing
The vibrissae test was used to quantify motor asymmetry (Schallert et al. 2000; Zhao et al. 2005) caused by a unilateral cortical stroke and was performed by a person who was blinded to the experimental conditions. Rats were divided into three groups: (i) sham surgery without ischemia; (ii) ischemia for 30 min only (control ischemia), (iii) ischemia plus postconditioning. Rats were handled for 3 days before stroke, and baseline was tested on the day before surgery. Two to three persons who were blinded to the experimental conditions performed all behavior tests for 2, 3, 7, 14, and 30 days post-stroke. The method of Vibrissae-elicited forelimb placement test was detailed by Zhao et al. (2005).
Drug delivery
To study whether Akt inhibition abolishes the protection of postconditioning, 10 μL of the PI3K inhibitor LY294002 (10 mM) or vehicle was infused into the ventricular space ipsilateral to the ischemia, as described (Zhao et al. 2005). Infarct size was measured 2 days after stroke by TTC staining.
Western blots
Brain tissue from three groups was prepared: (i) sham surgery without ischemia; (ii) ischemia for 30 min only (control ischemia), (iii) ischemia plus postconditioning. In group 1, rats were subjected to sham surgery without ischemia. In groups 2 and 3, brains were harvested at 1, 5, and 24 h after stroke onset; tissues corresponding to the ischemic penumbra and core were dissected for western blot (Fig. 1). The ischemic penumbra was defined as the tissue saved by postconditioning 2 days post-stroke, and the corresponding region from the control ischemic brain was dissected for comparison. Whole cell protein was extracted from the fresh brain tissue, and western blot was performed as described with modification (Zhao et al. 2003a, 2004, 2005). In each lane, 12 μg protein was subjected to sodium dodecyl sulfate—polyacrylamide gel electrophoresis using 4–15% Ready Gel (Bio-Rad Laboratories, Hercules, CA, USA) for 1.5 h. Protein bands were transferred from the gel to polyvinylidinene fluoride (Millipore, Bedford, MA, USA) membranes for 1 h.
Fig. 1.
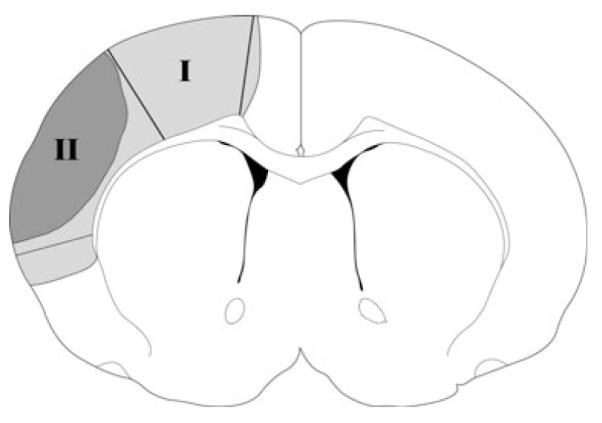
Tissue corresponding to the ischemic penumbra and core. The gray region (I), including the black region (II) represents ischemic injury in controls with ischemia alone; the black region (II) represents infarction in ischemia plus postconditioning. The region spared by postconditioning is defined as the penumbra (region I) and the black area is defined as the ischemic core (region II). These regions were dissected for western blotting. The corresponding non-ischemic cortex from sham animal without ischemia was dissected for comparison.
To determine phosphorylation or total protein levels of Akt, PTEN, PDK1, GSK3β, JNK, ERK1/2, δPKC, εPKC, and phosphorylated β-catenin, primary antibodies (see Table 1) were incubated with membranes overnight at 4°C with gentle agitation. Next, membranes were incubated for 1 h with horseradish peroxidase-conjugated secondary anti-rabbit antibody (1 : 1000, Cell Signaling Technology, Beverly, MA, USA), and finally for 5 min with enhanced chemiluminescence + substrate (GE Healthcare, Sunnyvale, CA, USA) for 5 min. Membranes were scanned using Typhoon trio (GE Healthcare).
Table 1.
Antibodies and their concentrations, manufacturers, and application for western blot
| Primary antibodies | S | Dilutions | Manufacturer | Cat# |
|---|---|---|---|---|
| P-Akt (Ser473) | Rab | 1 : 1000 | Cell signaling, MA, USA |
9271 |
| Akt | Rab | 1 : 1000 | Cell signaling | 9272 |
| P-PTEN (Ser380) | Rab | 1 : 1000 | Cell signaling | 9551 |
| PTEN | Rab | 1 : 1000 | Cell signaling | 9552 |
| P-PDK1 (Ser241) | Rab | 1 : 1000 | Cell signaling | 3061 |
| PDK1 | Rab | 1 : 1000 | Cell signaling | 3062 |
| P-GSK3b (Ser 9) | Rab | 1 : 1000 | Cell signaling | 9336 |
| GSK3b | Rab | 1 : 1000 | Cell signaling | 9332 |
| P-β-catenin | Rab | 1 : 1000 | Cell signaling | 9561 |
| β-catenin | Ms | 1 : 1000 | Upstate Biotechnology, NY, USA |
05-482 |
| Active β-catenin | Ms | 1 : 1000 | Calbiochem, CA, USA |
219357 (7D11) |
| P-JNK | Rab | 1 : 1000 | Cell signaling | 9251 |
| JNK | Rab | 1 : 1000 | Cell signaling | 9252 |
| P-ERK1/2 | Rab | 1 : 1000 | Cell signaling | 9101 |
| ERK1/2 | Rab | 1 : 1000 | Cell signaling | 9102 |
| P-δPKC (Thr505) | Rab | 1 : 1000 | Cell signaling | 9374 |
| δ-PKC | Rab | 1 : 5000 | Santa Cruz Biotechnology, CA, USA |
sc-213 |
| P-εPKC | Rab | 1 : 1000 | Upstate Biotechnology |
06-821 |
| ε-PKC | Rab | 1 : 10 000 | Santa Cruz Biotechnology |
sc-214 |
| β-actin | Ms | 1 : 20 000 | Sigma, MO, USA | A-3854 |
| β-actin | Rab | 1 : 20 000 | Bethyl, TX, USA | A300-491A |
PKC, protein kinase C; P-εPKC, phosphorylated P-εPKC; P-δPKC, phosphorylated P-δPKC; ERK extracellular signal-regulated kinase; P-ERK, phosphorylated ERK; JNK, c-jun N-terminal kinase; P-JNK, phosphorylated-JNK; GSK3β, glycogen synthase kinase; P-GSK3β, phosphorylated GSK3β; PDK1, phosphoinositide-dependent protein kinase-1; P-PDK1, phosphorylated PDK1; PTEN, phosphatase and tensin homologue deleted on chromosome 10; P-PTEN, phosphorylated PTEN; P-β-catenin, phosphorylated β-catenin S, species; Rab, rabbit; Ms, Mouse.
After scanning, the same membranes without stripping were incubated with β-actin (Sigma, St. Louis, MO, USA) (Table 1) for 30 min at 23°C, and then with Alexa Fluor®647 donkey anti-mouse IgG (H + L) secondary antibody (1 : 5000, Invitrogen, Eugene, OR, USA) for 1 h at 23°C. Membranes were scanned using the same Typhoon trio.
To detect total or active β-catenin with β-actin, membranes were incubated overnight at 4°C with gentle agitation in a mixture of primary antibodies consisting of anti-β-catenin or anti-active β-catenin and β-actin (1 : 20 000, rabbit, Cat no. A300–491A; Bethyl, Texas, USA) (Table 1). Secondary antibodies Alexa Fluor®647 donkey anti-mouse (1 : 1500) for detecting β-catenin and Alexa Fluor®488 donkey anti-rabbit (1 : 5000) for detecting β-actin were then incubated with membranes for 1 h at 23°C. Protein bands of β-catenin and β-actin were scanned simultaneously by the Typhoon trio.
The optical densities of all protein bands were analyzed using IMAGEQUANT 5.2 software (GE Healthcare). The same samples from rats with sham surgery were used to compare with control and postconditioning, and all samples were run on the same gel. The protein bands were rearranged solely to ease comparison in figures. Thus, only one value for sham is shown.
In vitro Akt kinase assay
To determine whether postconditioning affects true Akt activity, we performed an exact in vitro Akt kinase assay as described (Cat# 9840, Cell Signaling Technology; Zhao et al. 2005). Rat brains subjected to ischemia with or without postconditioning or sham surgery were harvested at 5 and 24 h after stroke. To determine whether the PI3K inhibitor, LY294002, blocks Akt activity after postconditioning, ischemic brains treated with the PI3K inhibitor and postconditioning were harvested 5 h after stroke. Whole cell extracts were prepared from the ischemic penumbra. Akt was immunoprecipitated with immobilized Akt primary antibody bead slurry and incubated with GSK3 fusion protein. The protein level of phosphorylated GSK3 fusion protein, which was detected by western blots reflects Akt activity.
Statistics
For behavioral tests, one-way repeated measures ANOVA was used to compare a test at different time points in the same group, and two-way ANOVA was used to compare tests between postconditioning and control ischemia, followed by Student—Newman—Keuls test. For infarction, two-way ANOVA was used, followed by Student—Newman—Keuls test. For western blot, two-way ANOVA was used to analyze various protein bands between postconditioning and control ischemia; one-way ANOVA was used to compare protein bands after ischemia with sham; tests were followed by Fisher’s least square difference post hoc test. All were considered statistically significant for p-values ≤ 0.05. Data are presented as mean ± SEM.
Results
Postconditioning provided long-term protection
Postconditioning reduced lesion size approximately 40% in rats subjected to ischemia when measured 30 days after ischemia, suggesting long-term protection (Fig. 2a). Neurological functions were assayed by the vibrissae test. The percentage of contralateral forelimb placements dropped from 100% before ischemia to approximately 25–60% after ischemia, indicating an asymmetrical deficit in forelimb use. Postconditioning attenuated the overall deficit from 2 to 30 days after ischemia (Fig. 2b).
Fig. 2.

Long-term protective effect of postconditioning. (a). Post-conditioning reduced ischemic damage up to 1 month after focal ischemia. Representative whole brains (top) and the cresyl violet staining of four coronal sections (second panel) from rats that received sham surgery, postconditioning, or control ischemia are presented. Arrows indicate injury sites. A bar graph (third panel) presents average lesion sizes measured 1 month after ischemia. Postconditioning reduced infarction from 36 ± 4% (n = 6) to 22 ± 3% (n = 7) in control ischemic animals (*p = 0.006, postconditioning vs. control). Post, ischemia plus postconditioning; Con, control ischemia without postconditioning. (b). Postconditioning attenuates the asymmetrical behavioral abnormalities measured up to 1 month after stroke. In the vibrissae test, the number of contralateral forelimb placements was compared with that of the ipsilateral forelimb, n = 5–7/group. Postconditioning attenuates the overall deficit from 2 to 30 days after stroke (two-way ANOVA, p < 0.001). *p < 0.05 and **p < 0.01 versus sham (one-way repeated measures ANOVA); #p < 0.05 versus postconditioning at 21 days (two-way ANOVA, Student—Newman—Keuls).
Increased Akt activity contributed to the protective effect postconditioning
Western blot analysis was used to evaluate the effect of postconditioning on Akt phosphorylation (P-Akt). Levels of P-Akt increased at 1 and 5 h and returned to normal levels at 24 h postischemia in the penumbra of control rats; they did not change significantly in the ischemic core (Fig. 3a). Postconditioning significantly increased P-Akt levels compared with control ischemia both in the ischemic penumbra and in the core at 5 and 24 h after stroke (Fig. 3a). However, levels of total Akt decreased after stroke in the ischemic penumbra and the core, and this was not prevented by postconditioning (densitometry quantitation not shown). Thus, postconditioning increased P-Akt without affecting total (decreased) Akt in the ischemic penumbra and core.
Fig. 3.

Akt activity contributed to the protective effect of postconditioning. (a) Changes in phosphorylated-Akt (P-Akt) and total Akt after stroke in rats with and without postconditioning. Representative protein bands for P-Akt (Ser473), total Akt and β-actin in both the ischemic penumbra and the core are shown. Relative optical densities of protein bands in ischemic rats were normalized to those in sham rats and calibrated with β-actin. Postconditioning up-regulated the overall protein level of P-Akt across all time points compared with control ischemia (two-way ANOVA: p = 0.005 in the penumbra, p = 0.027 in the ischemic core). *p < 0.05 and **p < 0.01 versus sham, respectively; #p < 0.01 and ##p < 0.05 versus control at a corresponding time point, respectively. Total Akt decreased after ischemia in both the ischemic core and penumbra; postconditioning had no statistically significant effect. *p < 0.01 and *p < 0.05 versus sham, n = 5–7/group. c, control; p, postconditioning. (b) Postconditioning increased in vitro Akt activity at 5 h. Akt activity as assessed by protein levels of phosphophorylated Akt substrate, a fusion protein of glycogen synthase kinase (GSK3β) is expressed as percentages of phosphorylated GSK3β (P-GSK3) from sham animals. *p < 0.05 versus sham; #p < 0.05 versus 5-h control, n = 5/group. (c) Phosphoinositide 3-kinase (PI3K) inhibitor, LY294002 inhibited Akt activity at 5 h in postconditioned animals. Antibody directed against P-GSK3β was used for detection. *p < 0.05 versus sham; #p < 0.05 versus PI3K inhibitor. (d) The PI3K inhibitor LY294002, partially blocked the protective effect of postconditioning. Infarct size was detected by 2,3,5-triphenyl-2H-tetrazolium chloride (TTC) staining 2 days after stroke. The left columns show representative TTC staining from rat brains that received postconditioning or control ischemia treated with vehicle [dimethylsulfoxide (DMSO)] or with the PI3K inhibitor LY294002. Bar graph (right panel) shows quantitation of the data. *p < 0.001 versus other groups and #p < 0.05 versus DMSO/con and inhib/con. Post, postconditioning; con, control ischemia; inhib, PI3K inhibitor.
An in vitro Akt kinase assay was used to assay Akt activity. Despite the robust increase in P-Akt, 5 or 24 h after control ischemia (Fig. 3a), Akt activity decreased to approximately 70% of sham (Fig. 3b). Postconditioning significantly increased Akt kinase activity to approximately 110% at 5 h post-stroke (p < 0.05 vs. control ischemia), but had no effect at 24 h (Fig 3b). Injection of the PI3K inhibitor, LY294002, reduced Akt activity in rat brains that received postconditioning (Fig. 3c). Injection of LY294002 enlarged infarct by 42% in rats with postconditioning (Fig. 3d). However, it did not completely reverse the protective effect of postconditioning, as the infarct in rats that received postconditioning was still smaller than that in rats receiving vehicle. Notably, LY294002 did not change the infarction volume in rats treated with control ischemia. These data indicate that postconditioning increases Akt activity, and that inhibition of upstream Akt-activating PI3K activity does not completely abolish the protective effects of postconditioning on infarct size.
The levels of phosphorylated PDK1, which activates Akt and of phosphorylated PTEN (P-PTEN), which cannot inhibit Akt as well as their total protein levels were examined next (Fig. 4). Levels of phosphorylated PDK1and P-PTEN decreased in both the ischemic penumbra and the core after ischemia (Fig. 4a and b). Postconditoning did not significantly affect their levels compared with control ischemia, though postconditioning slightly enhanced levels of P-PTEN at 1, 5, and 24 h in the penumbra. Total protein levels of PDK1 and PTEN decreased after ischemia; postconditioning did not affect their levels (densitometry quantitation not shown). Thus, postconditioning had no effect on the decrease of phosphorylated or total PDK1 or total PTEN, though it slightly increased P-PTEN (inactive PTEN).
Fig. 4.

The effect of postconditioning on phosphorylated and total protein levels of phosphoinositide-dependent protein kinase-1 (PDK1), phosphatase and tensin homologue deleted on chromosome 10 (PTEN), and glycogen synthase kinase 3β (GSK 3β) after stroke. Representative protein bands of phosphorylated and total PDK1, PTEN, and GSK 3β are shown along with their corresponding mean optical densities (bar graphs). For brevity, β-actin bands were omitted. Levels of phosphorylated proteins are shown in a, b, and c for PDK1, PTEN, and GSK 3β, respectively. Protein levels of phosphorylated-PDK1 (P-PDK1), phosphorylated-PTEN (P-PTEN), and phosphorylated glycogen synthase kinase 3β (P-GSK3β) decreased after ischemia. (a) The P-PDK1 levels were reduced at 24 h in postconditioned control ischemic brains in the penumbra; they were reduced at 24 h postischemia with or without postconditioning in the ischemic core.*p < 0.05, **p < 0.01 versus sham. (b) Although postconditioning slightly attenuated the reduction in P-PTEN levels at 1, 5, and 24 h in the penumbra, no significant difference was detected between postconditioning and control ischemia. *p < 0.05, **p < 0.01, and ***p < 0.001 versus sham. (c) P-GSK3β levels were transiently increased at 1 h in only the penumbra, and then reduced at 5 and 24 h in both the penumbra and core in rats with control ischemia; they were reduced in rats with postconditioning in both the penumbra and core postischemia. *p < 0.05, **p < 0.01,***p < 0.001 versus sham; #p < 0.05 versus 1 h/con in the penumbra; ##p < 0.01 versus 1 h/con in the core, n = 4–5/group.
The levels of Akt substrate GSK3β and its inactive phosphorylated form, P-GSK3β were examined in the context of postconditioning. The P-GSK3β level was transiently increased at 1 h and then decreased at 5 and 24 h in the penumbra after stroke (Fig. 4c); postconditioning blocked the increase at 1 h, but had no effect on its levels at 5 and 24 h. In the ischemic core, P-GSK3β levels were decreased at 5 h in brains subjected to control ischemia; with postconditioning, it was decreased as early as 1 h. Total protein levels of GSK3β did not change significantly in ischemic brains with or without postconditioning (densitometry quantitation not shown). Postconditioning therefore had no effect on total GSK3β levels; its effect of lowering P-GSK3β levels was limited to the 1 h time point.
The last Akt signaling cascade molecule we examined was β-catenin. Levels of phosphorylated β-catenin were increased as early as 1 h, peaked at 5 h and then returned to normal levels in the penumbra of control ischemic brains; postconditioning blocked the increase at 1 h, but had no effect on phosphorylated β-catenin levels at other time points (Fig. 5a). In the ischemic core, the level of phosphorylated β-catenin was slightly increased at 5 h, but did not reach a significant difference compared with sham; postconditioning reduced phosphorylated β-catenin at 1 h, whereas it increased phosphorylated β-catenin at 5 h. Protein levels of total β-catenin were decreased from 1 through 24 h postischemia; postconditioning had no effect on them (Fig. 5b). We also measured the level of active β-catenin, i.e., non-phosphorylated (Fig. 5c) and obtained similar results with total β-catenin. Postconditioning thus decreased phosphorylated β-catenin levels at 1 h in the ischemic penumbra and core, while increasing phosphorylated β-catenin at 5 h in the core, without affecting total phosphorylated β-catenin levels.
Fig. 5.

Effects of postconditioning on levels of phosphorylated, total, and active β-catenin after stroke. Western blot protein bands and corresponding mean optical densities (bar graphs) are shown. (a) Phosphorylated-β-catenin levels postischemia with and without postconditoning. *p < 0.05, **p < 0.01, ***p < 0.001 versus sham; and #p < 0.05, versus 1 h/con. (b) Decreased levels of β-catenin postischemia. An antibody that recognizes total β-catenin was used for detection.†p < 0.05 versus other time points, n = 3–5/group. (c) Decreased levels of active β-catenin postischemia. An antibody recognizing the active, non-phosphorylated form of β-catenin was used for detection. †p < 0.05 versus other time points, n = 3–5/group.
Inhibition of MAPK signaling molecules JNK and ERK1/2 correlates with postconditioning’s protection
We determined the effect of postconditioning on levels of phosphorylated-JNK (P-JNK) and total JNK protein in both the ischemic penumbra and the core (Fig. 6a). P-JNK transiently increased at 1 h in the ischemic penumbra, but decreased to approximately 50% of sham from 1 through 24 h in the ischemic core. Postconditioning blocked the increase of P-JNK at 1 h in the penumbra, but did not affect P-JNK levels in the ischemic core. Total protein levels of JNK did not decrease until 24 h after ischemia and they were unaffected by postconditioning (densitometry quantitation not shown). Thus, postconditioning’s effect on P-JNK levels were limited to blocking the increase of P-JNK at 1 h in the ischemic penumbra.
Fig. 6.

The effects of postconditoning on the mitogen-activated protein kinase pathway. Representative western blot protein bands (top panels) and corresponding mean optical densities (bar graphs) are shown. (a) Postconditioning reduced overall increases in phosphorylated-c-jun N terminal kinase from 1 to 24 h in the penumbra (two-way ANOVA, p = 0.023) but not in the core. *p < 0.05 versus sham; ##p < 0.01 versus control/1 h; ††p < 0.01 versus other time points. (b) Postconditioning reduced overall levels of phosphorylated-ERK (P-ERK) from 1 to 24 h in the penumbra but not in the core (p = 0.015, two-way ANOVA). *p < 0.05 versus sham; #p < 0.05 versus control at a corresponding time point, n = 5–9/group.
We then measured protein levels of phosphorylated-ERK1/2 (P-ERK1/2) and ERK1/2 (Fig. 6b). P-ERK1/2 transiently increased after reperfusion in both the ischemic penumbra and core of control ischemic rats. Postconditioning reduced overall levels of P-ERK1/2 from 1 to 24 h in the penumbra, and reduced P-ERK1/2 levels at 5 h but not at 1 and 24 h in the ischemic core. Total protein levels of ERK1/2 did not change significantly after ischemia with or without postconditioning (densitometry quantitation not shown).
The effect of postconditioning on the PKC pathway
We assayed for changes in total and cleaved δPKC, phosphorylated δPKC (Thr 505; P-δPKC), and phosphorylated εPKC (P-εPKC). Total δPKC decreased in the ischemic core of both control and postconditioning groups at 5 and 24 h (p < 0.05) and in the penumbra of control at 5 h (p < 0.05); postconditioning had no significant effect on protein levels of total δPKC (bar graphs for densitometry quantitation of total δPKC are not shown). However, levels of the cleaved form of δPKC, indicative of δPKC activity were transiently increased 1 h after stroke in the penumbra; postconditoning attenuated this increase (Fig. 7a). P-δPKC levels were decreased by 24 h to less than 70% of sham in the penumbra and to less than 40% in the core in ischemic brains (Fig. 7b); postconditioning did not significantly change P-δPKC levels compared with control. P-εPKC decreased to approximately 60–20% of sham at 1 and 24 h after stroke, respectively; postconditioning attenuated such decreases in the penumbra but not in the core (Fig. 7c). Taken together, these data suggest postconditioning blocked the increase of cleaved δPKC at 1 h and attenuated the decrease in P-εPKC at 1 h; these effects were limited to the ischemic penumbra.
Fig. 7.

The effect of postconditioning on the protein kinase C (PKC) pathway. Representative western blot protein bands (top panels) and corresponding mean optical densities, normalized to same protein in sham rats (bar graphs) are shown. (a) Postconditioning reduced δPKC cleavage in the ischemic penumbra. The levels of cleaved δPKC were increased in the ischemic penumbra after stroke (see bar graph). The increase at 1 h was significantly inhibited by postconditioning. *p < 0.05, ***p < 0.001 versus sham; ###p < 0.001 versus 1 h/control, n = 5–10/group. (b) Phosphorylated-δPKC level decreased postischemia. No significant difference was found between control ischemia and postconditioning. *p < 0.05 versus sham. †††p < 0.001 versus the rest of the groups in the core, n = 3–5/group. (c) Postconditioning increased P-εPKC levels at 1 h compared with control ischemia. *p < 0.05, **p < 0.01 versus sham; #p < 0.05 versus 1 h/con; ††p < 0.01 versus the rest of the groups in the core, n = 3– 10/group.
Discussion
We demonstrated that postconditioning not only reduced infarction, but also improved neurological function up to 1 month after focal ischemia in rats, suggesting long-term protection. We also showed that postconditioning differentially regulated Akt activity in the Akt pathway, as postconditioning increased phosphorylation of Akt, but not of PDK1, PTEN or GSK3β. Additionally, postconditioning enhanced Akt activity and inhibition of Akt activity partly abolished the protective effect of postconditioning, suggesting a critical protective role of Akt in postconditioning. Furthermore, postconditioning reduced β-catenin phosphorylation at 1 h but not at 5 h or 24 h after stroke; it had no effect on the amount of total β-catenin or active β-catenin which were decreased after stoke. Postconditioning also inhibited the levels of phosphorylated JNK and Erk1/2, blocked δPKC cleavage and increased levels of phosphorylated εPKC.
Postconditioning provided long-term protection and improved behavioral recovery
Some neuroprotectants, such as postischemic hypothermia (Dietrich et al. 1993) and rapid ischemic preconditioning (Perez-Pinzon 2004, review) provide protection for only a few days after ischemia. In contrast, we show that rapid postconditioning reduced lesion size up to 1 month after stroke.
Nevertheless, reducing injured brain tissue may not translate into preservation of neurological function (Dumas and Sapolsky 2001). Although preconditioning is generally believed to spare ischemic tissues and improve neurological function (Kirino 2002), there are a few exceptions. For instance, brief ischemic preconditioning alone does not cause infarction, but does elicit behavioral deficits (Hua et al. 2005). In another study, preconditioning attenuated delayed neuronal loss in the hippocampal region CA1 after transient global ischemia, but did not preserve neurological function (Corbett and Crooks 1997). Nevertheless, we demonstrated that postconditioning attenuated asymmetry of forelimb use in the vibrissae test, suggesting that it preserves neurological function.
Implication of the Akt pathways in the protective effect of postconditioning
Dysfunction of the Akt pathway is involved in ischemic damage (Noshita et al. 2001; Osuka et al. 2004; Zhao et al. 2005). Most recently, Pignataro et al. (2007) have demonstrated that postconditioning increases P-Akt, and that the PI3K inhibitor, LY 294002 blocks the protective effect of postconditioning (Pignataro et al. 2007). However, actual Akt activity was not measured and inhibition of Akt activity by the PI3K inhibitor was not confirmed. We have previously shown that increasing P-Akt levels does not equate with increasing Akt activity (Zhao et al. 2005). Thus, we performed in vitro Akt kinase assays. Here, we showed that postconditioning increased not only P-Akt levels, but also Akt activity. We further demonstrated that LY294002 partly abolished the protective effect of postconditioning, and confirmed that LY294002 indeed inhibited Akt activity in rat brains treated with postconditioning.
Akt activity is regulated by several molecules in Akt pathways. Thus, we further studied how postconditioning mediates PTEN, PDK1, and GSK3β. We found that postconditioning enhanced P-Akt and its activity, but had no significant effect on phosphorylation of PTEN and PDK1. To our surprise, levels of P-GSK3β unexpectedly increased at 1 h postischemia but decreased thereafter. The increase in P-GSK3β levels is at odds with the decreased Akt activity, as decreased Akt activity should lead to decreased P-GSK3β levels. This mismatch suggests that Akt may not be the upstream signal regulating GSK3β phosphorylation, or that GSK3β phosphorylation is regulated by other signaling pathways. In addition, this P-GSK3β result contradicts that of our previous study which showed that GSK3β was dephosphorylated as early as 30 min after reperfusion. However, in that study, focal ischemia was induced by permanent MCA occlusion with a longer period (1 h) of CCA occlusion. The milder ischemia in this current study might allow hyperphosphorylation of GSK3β. To our surprise, postconditioning blocked increases in P-GSK3β, suggesting that hyperphosphorylation of GSK3β does not necessarily lead to neuroprotection.
β-catenin is a molecule downstream of GSK3β. Dephosphorylated, activated GSK3β phosphorylates β-catenin, leading to its degradation (Nusse 2003). Indeed, our data showing reduced phosphorylation of GSK3β, at least at 5 and 24 h are associated with increased levels of phosphorylated β-catenin, and this increased phosphorylation of β-catenin might be responsible for the reduced total protein levels of β-catenin (Nusse 2003). In addition, we showed that the level of active (i.e., non-phosphorylated) β-catenin was decreased after stroke. β-catenin has been shown to decrease in brains of patients with Alzheimer’s disease (Fuentealba et al. 2004; Li et al. 2007). In addition, β-catenin knockdown results in apoptosis, whereas β-catenin over-expression prevents neuronal death in vitro (Fuentealba et al. 2004; Li et al. 2007). Although total and active β-catenin decreased after stroke, postconditioning did not attenuate their reductions, suggesting that these reductions do not play critical roles in the protective effect of postconditioning.
The effect of postconditioning on the MAPK pathway
The current study suggests that inhibition of ERK1/2 activity may contribute to the protective effect of postconditioning, which conflicts the findings from Pignataro et al. (2007), that postconditioning enhanced ERK1/2 phosphorylation. Stroke-induced injury involves increases in P-ERK1/2 (Xu et al. 2006), one of the major signaling molecules of the MAPK pathway. However, the role of increased P-ERK1/2 after ischemia is a matter of debate, as increasing P-ERK1/2 correlates with both beneficial and detrimental effects. For instance, the beneficial effects are associated with growth factors, which further promote the increase in P-ERK1/2 after reperfusion, and inhibition of P-ERK1/2 abolishes the protection of growth factors (Zhang et al. 2006). Conversely, detrimental effects are correlated with oxidative stress and inflammation which increase P-ERK1/2 (Noshita et al. 2002; Levinthal and Defranco 2005). Such detrimental effects are further confirmed by the fact that inhibition of P-ERK1/2 reduces infarction (Noshita et al. 2002). We confirmed that P-ERK1/2 was increased from 1 to 24 h after stroke, and found that postconditioning reduced its level in the penumbra. Our results favor the detrimental role of P-ERK1/2 after ischemia. The discrepancy between our result and that of Pignataro et al. (2007)is probably because of differing ischemic models and postconditioning parameters employed in these two studies (Pignataro et al. 2007). First, Pignataro et al. (2007) used MCA suture occlusion model while we used a model with permanent distal MCA occlusion plus bilateral CCA occlusion. Secondly, in their study, postconditioning was induced by a single 10 min re-occlusion after 10 min of reperfusion, but postconditioning in our study was executed by a few cycles of brief CCA occlusion plus reperfusion. Such differences in ischemic models and postconditioning may result in disparate pathological responses after stroke in each model.
Unlike the controversial role of ERK1/2, JNK activities aggravate ischemic damage (Kamada et al. 2007), which is supported by our current study. Again, our result is inconsistent with that of Pignataro et al. (2007), who showed that postconditoning had no effect on JNK phosphorylation while we found that it reduced JNK phosphorylation. P-JNK transiently increases after reperfusion (Gao et al. 2005). JNK exacerbates ischemic apoptotic pathways by promoting activities of Bim, Bax and Fas; critical molecules that trigger apoptosis (Gao et al. 2005). Conversely, JNK inhibition prevents ischemia-induced translocation of Bax and Bim to the mitochondria, which causes the executive stage of apoptosis via release of cytochrome c and caspase-3 and -9 activation (Gao et al. 2005). Here, we showed that P-JNK transiently increased in the penumbra after stroke, which postconditioning blocked, suggesting that JNK inhibition may contribute to the protection of postconditioning. However, we also showed that reduction in P-JNK levels does not always result in neuroprotection, as P-JNK also decreased in the ischemic core in control rats, where tissues were destined to die. Such reduction in P-JNK is perhaps the consequence of ischemic tissue degeneration, which is distinct from the reduction of P-JNK caused by postconditioning in the penumbra.
Postconditioning blocked δPKC cleavage and promoted εPKC phosphorylation
Inhibition of δPKC cleavage and εPKC dephosphorylation may also contribute to the protective effect of postconditioning. δPKC promotes whereas εPKC inhibits apoptosis (Anantharam et al. 2002; Brodie and Blumberg 2003). The activities of both δPKC and εPKC are regulated by subcellular translocation, proteolytic cleavage, and phosphorylation (Shimohata et al. 2007b). Although we have previously shown that both δPKC and εPKC translocate into the particulate membrane after stroke in a more severe focal ischemia model with a longer 1 h CCA occlusion (Shimohata et al. 2007a,b), pilot data in the current study (not shown) did not suggest significant PKC translocation in the model with shorter CCA occlusion (30 min). Thus, we concentrated on δPKC cleavage and εPKC phosphorylation and found that postconditioning blocked δPKC cleavage and enhanced protein levels of P-εPKC, suggesting that inhibition of δPKC activity and enhancement of εPKC activity may contribute to the protective effect of postconditioning.
The current study has some limitations. We had several lines of strong evidence to support Akt’s protective role, using an Akt inhibitor, P-Akt levels, and Akt activity, but we have not performed similar experiments to support the roles of the MAPK and PKC pathways. In addition, there may be cross-talk between pathways during injury development. It is clear that postconditioning regulates all of them, but we have not studied the interactive effects between them.
In summary, postconditioning provided long-term protection against cerebral ischemia by reducing lesion size and improving behavioral tests in a rat focal ischemia model. Postconditioning’s ability to increase Akt activity contributed to this protection; in addition, postconditioning’s protection is correlated with inhibition of ERK1/2 and JNK activities, promotion of εPKC phosphorylation, and reduction of δPKC cleavage.
Acknowledgments
The authors thank Elizabeth Hoyte for preparing the figures and Dr. Angela Lee Riepel for editorial assistance. This study was supported by AHA National Scientist Development Grant 0730113N (HZ), NINDS grants R01 NS27292 (GKS) and P01 NS37520 (GKS).
Abbreviations used
- CCA
common carotid arteries
- ERK
extracellular signal-regulated kinase
- GSK3β
glycogen synthase kinase 3β
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- MCA
middle cerebral artery
- P- δPKC
phosphorylated δPKC
- PDK
phosphoinositide-dependent protein kinase-1
- P-ERK
phosphorylated ERK
- P-GSK3β
phosphorylated GSK3β
- PI3K
phosphoinositide 3-kinase
- P-JNK
phosphorylated-JNK
- PKC
protein kinase C
- P-PTEN
phosphorylated PTEN
- PTEN
phosphatase and tensin homologue deleted on chromosome 10
- TTC
2,3,5-triphenyl-2H-tetrazolium chloride
References
- Anantharam V, Kitazawa M, Wagner J, Kaul S, Kanthasamy AG. Caspase-3-dependent proteolytic cleavage of protein kinase C delta is essential for oxidative stress-mediated dopaminergic cell death after exposure to methylcyclopentadienyl manganese tricarbonyl. J. Neurosci. 2002;22:1738–1751. doi: 10.1523/JNEUROSCI.22-05-01738.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bright R, Raval AP, Dembner JM, Perez-Pinzon MA, Steinberg GK, Yenari MA, Mochly-Rosen D. Protein kinase C delta mediates cerebral reperfusion injury in vivo. J. Neurosci. 2004;24:6880–6888. doi: 10.1523/JNEUROSCI.4474-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodie C, Blumberg PM. Regulation of cell apoptosis by protein kinase c delta. Apoptosis. 2003;8:19–27. doi: 10.1023/a:1021640817208. [DOI] [PubMed] [Google Scholar]
- Burda J, Danielisova V, Nemethova M, Gottlieb M, Matiasova M, Domorakova I, Mechirova E, Ferikova M, Salinas M, Burda R. Delayed postconditioning initiates additive mechanism necessary for survival of selectively vulnerable neurons after transient ischemia in rat brain. Cell. Mol. Neurobiol. 2006;26:1141–1151. doi: 10.1007/s10571-006-9036-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen ST, Hsu CY, Hogan EL, Maricq H, Balentine JD. A model of focal ischemic stroke in the rat: reproducible extensive cortical infarction. Stroke. 1986;17:738–743. doi: 10.1161/01.str.17.4.738. [DOI] [PubMed] [Google Scholar]
- Corbett D, Crooks P. Ischemic preconditioning: a long term survival study using behavioural and histological endpoints. Brain Res. 1997;760:129–136. doi: 10.1016/s0006-8993(97)00294-1. [DOI] [PubMed] [Google Scholar]
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature. 1995;378:785–789. doi: 10.1038/378785a0. [DOI] [PubMed] [Google Scholar]
- Dietrich WD, Busto R, Alonso O, Globus MY, Ginsberg MD. Intraischemic but not postischemic brain hypothermia protects chronically following global forebrain ischemia in rats. J. Cereb. Blood Flow Metab. 1993;13:541–549. doi: 10.1038/jcbfm.1993.71. [DOI] [PubMed] [Google Scholar]
- Dumas TC, Sapolsky RM. Gene therapy against neurological insults: sparing neurons versus sparing function. Trends Neurosci. 2001;24:695–700. doi: 10.1016/s0166-2236(00)01956-1. [DOI] [PubMed] [Google Scholar]
- Franke TF, Hornik CP, Segev L, Shostak GA, Sugimoto C. PI3K/Akt and apoptosis: size matters. Oncogene. 2003;22:8983–8998. doi: 10.1038/sj.onc.1207115. [DOI] [PubMed] [Google Scholar]
- Fuentealba RA, Farias G, Scheu J, Bronfman M, Marzolo MP, Inestrosa NC. Signal transduction during amyloid-beta-peptide neurotoxicity: role in Alzheimer disease. Brain Res. Brain Res. Rev. 2004;47:275–289. doi: 10.1016/j.brainresrev.2004.07.018. [DOI] [PubMed] [Google Scholar]
- Gao Y, Signore AP, Yin W, Cao G, Yin XM, Sun F, Luo Y, Graham SH, Chen J. Neuroprotection against focal ischemic brain injury by inhibition of c-Jun N-terminal kinase and attenuation of the mitochondrial apoptosis-signaling pathway. J. Cereb. Blood Flow Metab. 2005;25:694–712. doi: 10.1038/sj.jcbfm.9600062. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Zulueta M, Feldman AB, Klesse LJ, Kalb RG, Dillman JF, Parada LF, Dawson TM, Dawson VL. Requirement for nitric oxide activation of p21(ras)/extracellular regulated kinase in neuronal ischemic preconditioning. Proc. Natl Acad. Sci. US A. 2000;97:436–441. doi: 10.1073/pnas.97.1.436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanada M, Feng J, Hemmings BA. Structure, regulation and function of PKB/AKT — a major therapeutic target. Biochim. Biophys. Acta. 2004;1697:3–16. doi: 10.1016/j.bbapap.2003.11.009. [DOI] [PubMed] [Google Scholar]
- Hua Y, Wu J, Pecina S, Yang S, Schallert T, Keep RF, Xi G. Ischemic preconditioning procedure induces behavioral deficits in the absence of brain injury? Neurol. Res. 2005;27:261–267. doi: 10.1179/016164105X25270. [DOI] [PubMed] [Google Scholar]
- Kamada H, Nito C, Endo H, Chan PH. Bad as a converging signaling molecule between survival PI3-K/Akt and death JNK in neurons after transient focal cerebral ischemia in rats. J. Cereb. Blood Flow Metab. 2007;27:521–533. doi: 10.1038/sj.jcbfm.9600367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirino T. Ischemic tolerance. J. Cereb. Blood Flow Metab. 2002;22:1283–1296. doi: 10.1097/01.WCB.0000040942.89393.88. [DOI] [PubMed] [Google Scholar]
- Kitano H, Young JM, Cheng J, Wang L, Hurn PD, Murphy SJ. Gender-specific response to isoflurane preconditioning in focal cerebral ischemia. J. Cereb. Blood Flow Metab. 2007;27:1377–1386. doi: 10.1038/sj.jcbfm.9600444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SR, Lo EH. Interactions between p38 mitogen-activated protein kinase and caspase-3 in cerebral endothelial cell death after hypoxia-reoxygenation. Stroke. 2003;34:2704–2709. doi: 10.1161/01.STR.0000096540.40826.BA. [DOI] [PubMed] [Google Scholar]
- Levinthal DJ, Defranco DB. Reversible oxidation of ERK-directed protein phosphatases drives oxidative toxicity in neurons. J. Biol. Chem. 2005;280:5875–5883. doi: 10.1074/jbc.M410771200. [DOI] [PubMed] [Google Scholar]
- Li HL, Wang HH, Liu SJ, et al. Phosphorylation of tau antagonizes apoptosis by stabilizing beta-catenin, a mechanism involved in Alzheimer’s neurodegeneration. Proc. Natl Acad. Sci. USA. 2007;104:3591–3596. doi: 10.1073/pnas.0609303104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Noort M, Meeldijk J, van der Zee R, Destree O, Clevers H. Wnt signaling controls the phosphorylation status of beta-catenin. J. Biol. Chem. 2002;277:17901–17905. doi: 10.1074/jbc.M111635200. [DOI] [PubMed] [Google Scholar]
- Noshita N, Lewen A, Sugawara T, Chan PH. Evidence of phosphorylation of Akt and neuronal survival after transient focal cerebral ischemia in mice. J. Cereb. Blood Flow Metab. 2001;21:1442–1450. doi: 10.1097/00004647-200112000-00009. [DOI] [PubMed] [Google Scholar]
- Noshita N, Sugawara T, Hayashi T, Lewen A, Omar G, Chan PH. Copper/zinc superoxide dismutase attenuates neuronal cell death by preventing extracellular signal-regulated kinase activation after transient focal cerebral ischemia in mice. J. Neurosci. 2002;22:7923–7930. doi: 10.1523/JNEUROSCI.22-18-07923.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nusse R. Wnts and Hedgehogs: lipid-modified proteins and similarities in signaling mechanisms at the cell surface. Development. 2003;130:5297–5305. doi: 10.1242/dev.00821. [DOI] [PubMed] [Google Scholar]
- Osuka K, Watanabe Y, Usuda N, Nakazawa A, Tokuda M, Yoshida J. Modification of endothelial NO synthase through protein phosphorylation after forebrain cerebral ischemia/reperfusion. Stroke. 2004;35:2582–2586. doi: 10.1161/01.STR.0000143454.14159.28. [DOI] [PubMed] [Google Scholar]
- Perez-Pinzon MA. Neuroprotective effects of ischemic preconditioning in brain mitochondria following cerebral ischemia. J. Bioenerg. Biomembr. 2004;36:323–327. doi: 10.1023/B:JOBB.0000041762.47544.ff. [DOI] [PubMed] [Google Scholar]
- Pignataro G, Meller R, Inoue K, Ordonez AN, Ashley MD, Xiong Z, Simon RP. In vivo and in vitro characterization of a novel neuroprotective strategy for stroke: ischemic postconditioning. J. Cereb. Blood Flow Metab. 2007 doi: 10.1038/sj.jcbfm.9600559. Online; http://www.nature.com/jcbfm/journal/vaop/ncurrent/abs/9600559a.html. [DOI] [PubMed]
- Raval AP, Dave KR, Prado R, Katz LM, Busto R, Sick TJ, Ginsberg MD, Mochly-Rosen D, Perez-Pinzon MA. Protein kinase C delta cleavage initiates an aberrant signal transduction pathway after cardiac arrest and oxygen glucose deprivation. J. Cereb. Blood Flow Metab. 2005;25:730–741. doi: 10.1038/sj.jcbfm.9600071. [DOI] [PubMed] [Google Scholar]
- Schallert T, Fleming SM, Leasure JL, Tillerson JL, Bland ST. CNS plasticity and assessment of forelimb sensorimotor outcome in unilateral rat models of stroke, cortical ablation, parkinsonism and spinal cord injury. Neuropharmacology. 2000;39:777–787. doi: 10.1016/s0028-3908(00)00005-8. [DOI] [PubMed] [Google Scholar]
- Shamloo M, Rytter A, Wieloch T. Activation of the extracellular signal-regulated protein kinase cascade in the hippocampal CA1 region in a rat model of global cerebral ischemic preconditioning. Neuroscience. 1999;93:81–88. doi: 10.1016/s0306-4522(99)00137-2. [DOI] [PubMed] [Google Scholar]
- Shimohata T, Zhao H, Steinberg GK. Epsilon PKC may contribute to the protective effect of hypothermia in a rat focal cerebral ischemia model. Stroke. 2007a;38:375–380. doi: 10.1161/01.STR.0000254616.78387.ee. [DOI] [PubMed] [Google Scholar]
- Shimohata T, Zhao H, Sung JH, Sun G, Mochly-Rosen D, Steinberg GK. Suppression of delta PKC activation after focal cerebral ischemia contributes to the protective effect of hypothermia. J. Cereb. Blood Flow Metab. 2007b;27:636–641. doi: 10.1038/sj.jcbfm.9600450. [DOI] [PubMed] [Google Scholar]
- Vazquez F, Ramaswamy S, Nakamura N, Sellers WR. Phosphorylation of the PTEN tail regulates protein stability and function. Mol. Cell. Biol. 2000;20:5010–5018. doi: 10.1128/mcb.20.14.5010-5018.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang ZQ, Wu DC, Huang FP, Yang GY. Inhibition of MEK/ERK 1/2 pathway reduces pro-inflammatory cytokine interleukin-1 expression in focal cerebral ischemia. Brain Res. 2004;996:55–66. doi: 10.1016/j.brainres.2003.09.074. [DOI] [PubMed] [Google Scholar]
- Xu Y, Zhang W, Klaus J, Young J, Koerner I, Sheldahl LC, Hurn PD, Martinez-Murillo F, Alkayed NJ. Role of cocaine- and amphetamine-regulated transcript in estradiol-mediated neuroprotection. Proc. Natl Acad. Sci. USA. 2006;103:14489–14494. doi: 10.1073/pnas.0602932103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yano S, Morioka M, Fukunaga K, Kawano T, Hara T, Kai Y, Hamada J, Miyamoto E, Ushio Y. Activation of Akt/protein kinase B contributes to induction of ischemic tolerance in the CA1 subfield of gerbil hippocampus. J. Cereb. Blood Flow Metab. 2001;21:351–360. doi: 10.1097/00004647-200104000-00004. [DOI] [PubMed] [Google Scholar]
- Zhang F, Signore AP, Zhou Z, Wang S, Cao G, Chen J. Erythropoietin protects CA1 neurons against global cerebral ischemia in rat: potential signaling mechanisms. J. Neurosci. Res. 2006;83:1241–1251. doi: 10.1002/jnr.20816. [DOI] [PubMed] [Google Scholar]
- Zhao H. The protective effect of ischemic postconditioning against ischemic injury: from the heart to the brain. J. Neuroimmune Pharmacol. 2007;2:313–318. doi: 10.1007/s11481-007-9089-8. [DOI] [PubMed] [Google Scholar]
- Zhao H, Yenari MA, Cheng D, Sapolsky RM, Steinberg GK. Bcl-2 overexpression protects against neuron loss within the ischemic margin following experimental stroke and inhibits cytochrome c translocation and caspase-3 activity. J. Neurochem. 2003a;85:1026–1036. doi: 10.1046/j.1471-4159.2003.01756.x. [DOI] [PubMed] [Google Scholar]
- Zhao ZQ, Corvera JS, Halkos ME, Kerendi F, Wang NP, Guyton RA, Vinten-Johansen J. Inhibition of myocardial injury by ischemic postconditioning during reperfusion: comparison with ischemic preconditioning. Am. J. Physiol. Heart Circ. Physiol. 2003b;285:H579–H588. doi: 10.1152/ajpheart.01064.2002. [DOI] [PubMed] [Google Scholar]
- Zhao H, Yenari MA, Cheng D, Barreto-Chang OL, Sapolsky RM, Steinberg GK. Bcl-2 transfection via herpes simplex virus blocks apoptosis-inducing factor translocation after focal ischemia in the rat. J. Cereb. Blood Flow Metab. 2004;24:681–692. doi: 10.1097/01.WCB.0000127161.89708.A5. [DOI] [PubMed] [Google Scholar]
- Zhao H, Shimohata T, Wang JQ, Sun G, Schaal DW, Sapolsky RM, Steinberg GK. Akt contributes to neuroprotection by hypothermia against cerebral ischemia in rats. J. Neurosci. 2005;25:9794–9806. doi: 10.1523/JNEUROSCI.3163-05.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Sapolsky RM, Steinberg GK. Phosphoinositide-3-kinase/akt survival signal pathways are implicated in neuronal survival after stroke. Mol. Neurobiol. 2006a;34:249–270. doi: 10.1385/MN:34:3:249. [DOI] [PubMed] [Google Scholar]
- Zhao H, Sapolsky RM, Steinberg GK. Interrupting reperfusion as a stroke therapy: ischemic postconditioning reduces infarct size after focal ischemia in rats. J. Cereb. Blood Flow Metab. 2006b;26:1114–1121. doi: 10.1038/sj.jcbfm.9600348. [DOI] [PubMed] [Google Scholar]