

Abstract
Quinones such as ubiquinone are the lipid soluble electron and proton carriers in the membranes of mitochondria, chloroplasts and oxygenic bacteria. Quinones undergo controlled redox reactions bound to specific sites in integral membrane proteins such as the cytochrome bc1 oxidoreductase. The quinone reactions in bacterial photosynthesis are amongst the best characterized, presenting a model to understand how proteins modulate cofactor chemistry. The free energy of ubiquinone redox reactions in aqueous solution and in the QA and QB sites of the bacterial photosynthetic reaction centers (RCs) are compared. In the primary QA site ubiquinone is reduced only to the anionic semiquinone (Q•−) while in the secondary QB site the product is the doubly reduced, doubly protonated quinol (QH2). The ways in which the protein modifies the relative energy of each reduced and protonated intermediate are described. For example, the protein stabilizes Q•− while destabilizing Q= relative to aqueous solution through electrostatic interactions. In addition, kinetic and thermodynamic mechanisms for stabilizing the intermediate semiquinones are compared. Evidence for the protein sequestering anionic compounds by slowing both on and off rates as well as by binding the anion more tightly is reviewed.
Keywords: Quinone, Photosynthesis, Em, Continuum electrostatics, Ligand, Ubiquinone, Electrochemistry
Quinones are the primary intramembrane, mobile, electron carriers in the energy-coupling electron transfer chains in mitochondria, chloroplasts and bacterial cell membranes (Cape et al. 2006). The relative concentration of the oxidized quinones, Q, and doubly reduced, quinol, QH2, help define the redox poise in these membranes. Quinone oxidation/reduction reactions occur in proteins such as the NADH:quinone reductase (Complex I), the cytochrome bc1 oxidoreductase (Complex II) and succinate–quinone reductase (Complex III) in mitochondria and oxygenic bacteria (Saraste 1999). In photosynthetic electron transfer, reactions involving quinones are found in the type II reaction centers (PSII or bacterial reaction centers (RCs)), b6f (or in bacteria the bc1) complexes and in the type I reaction centers (PSI). Quinone function is the characteristic that distinguishes between type I reaction centers where bound quinone is only transiently reduced to semiquinone and type II reaction centers where quinone is doubly reduced to QH2 and released to the membrane Qpool (Heathcote 2002). All proteins that use quinones must also guard against side reactions involving the free radical intermediates of quinone oxidation/reduction reactions, which can catalyze the formation of dangerous reactive oxygen species (Forquer et al. 2006).
Quinones, with their two electron, two proton electrochemistry have nine redox states (Fig. 1). All but Q and QH2 are unstable in water or membrane under physiological conditions. However, the quinone reactions in biology occur in specific binding sites that can stabilize free radical, semiquinone intermediates. This permits a general mechanism using two reaction cycles to go between Q and QH2. Thus, the bound quinones couple the obligate single electron carriers such as (bacterio)chlorophylls, hemes and iron sulfur complexes to the quinone pool where only Q and QH2 are found. For example, in photosynthetic reaction centers, considered in this review, a primary quinone (QA) is first reduced to the anionic semiquinone state (QA•−). This then reduces the oxidized secondary quinone (QB) to the semiquinone QB•(H) (Q•(H) here indicates either the anionic (Q•−) or protonated (Q•H) semiquinone). A second turnover of the protein re-reduces QA, which reduces QB•(H). Thus, QB•(H) must live long enough for the protein to undergo a second turnover to re-reduce QA. In addition, QA must be a good enough reductant to reduce both QB and Q•(H). This is the same type of mechanism used for the QI site of the bc1 complex. The QO site of QH2 oxidation in the bc1 complex uses a fundamentally different strategy (Osyczka et al. 2005; Cape et al. 2006). Here two different, simultaneously accessible reduction partners (cyt BH and the Rieske iron sulfur complex) are found. Thus, oxidation of quinol at this site does not rely on a stable semiquinone waiting for a second turnover. Until recently no semiquinone had been seen in the QO site (Cape et al. 2007; Zhang et al. 2007).
Fig. 1.
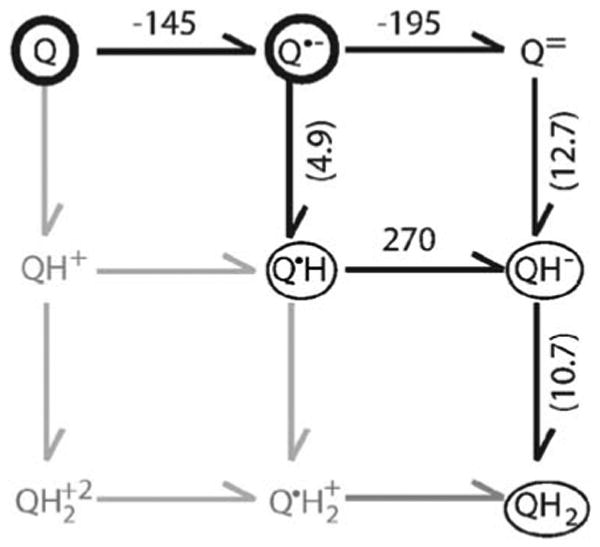
The Ems on the horizontal arrows are in millivolt. The pKas, are in parenthesis on the vertical arrows. The quinone species in the dark circles are found in both the QA and QB sites. Thin circles indicate species found only in the QB site. The species in grey are not seen in RCs or in aqueous solution as they are expected to have pKas below pH 0. All values are taken from (Zhu and Gunner 2005, see references therein)
To move between Q and QH2 the protein binding sites can change the order of electron and proton transfers from that found in solution (Fig. 1) by modifying the relative energy of the intermediates. They can also kinetically trap the intermediates so they are not lost to the surroundings. This review discusses the electrochemistry of ubiquinone in aqueous solution, the general strategies that proteins use to modify the energies by changing electrostatic interactions and the evidence that the proteins modulate the on and off rates of reactants and products. The specific reaction sequence, thermodynamics and rates for reduction of quinones in the QA and QB sites in the photosynthetic reaction centers of the purple, non-sulfur bacteria Rhodobacter sphaeroides are described. This review focuses on the reactions of ubiquinone, however the chemistry of menaquinone (Rich and Bendall 1979) and plastoquinone are quite similar (Wraight 2004; Swallow 1982).
Electrostatic modification of the thermodynamics of quinone redox reactions
Proteins can greatly modify the in situ electrochemistry of bound cofactors. For example, the Ems of protein bound hemes range from −400 to +450 mV, representing a change in the free energy of the oxidation reaction of over 18.4 kcal/mol (Churg and Warshell 1986; Gunner and Honig 1991; Voigt and Knapp 2003; Reedy and Gibney 2004; Zheng and Gunner 2008). It has been suggested that electrostatic interactions with the protein are the primary means by which proteins control electrochemistry (Warshel and Russell 1984; Honig and Nicholls 1995; Shurki et al. 2004; Gunner et al. 2006).
To understand how proteins change quinone electrochemistry, it is helpful to start with a description of their chemistry in solution (Rich and Bendall 1979; Swallow 1982; Prince et al. 1983, 1986; Wraight 2004; Zhu and Gunner 2005). The role of the protein then becomes clear when these Ems and pKas of different quinone redox states are compared to those in a particular binding site. While it is not easy to measure the high energy free, radical intermediates between ubiquinone and ubiquinol in solution, a consensus view of the free energy changes linking each species can be found in Fig. 1 (see Zhu and Gunner (2005) and references therein).
There are many methods for analyzing reaction chemistry in proteins, including classical, free energy perturbation techniques embedded in Molecular Dynamics Simulations (Marchi et al. 1993); semi-microscopic PDLD (PDLD/S) analysis (Churg et al. 1983; Parson et al. 1990; Sham et al. 1997, 1999); Density Function Theory (Mallik and Datta 2004; Cape et al. 2006) and QM/MM treatments (Hasegawa et al. 2003). For reactions such as electron or proton transfers where the primary difference between reactant and product is a change in charge, classical continuum electrostatics (CE) analysis has provided important insights into reaction mechanism (Beroza et al. 1995; Lancaster et al. 1996; Rabenstein et al. 1998; Alexov and Gunner 1999; Ishikita et al. 2003; Ishikita and Knapp 2004; Zhu and Gunner 2005; Klingen et al. 2007).
In a continuum electrostatics analysis the influence of the protein on the free energy of a reaction can be divided into three terms: the desolvation penalty where the stabilization of a charge by water is lessened when a charged group is moved into protein; the pairwise interactions between the reactants and the protein charges and dipoles; and dielectric relaxation, where the protein and surroundings change position to stabilize either the reactant or the product (Honig and Nicholls 1995; Gunner and Alexov 2000; Baker 2005; Gunner et al. 2006).
1. The desolvation penalty
Charged and polar solutes have favorable interactions with water that are diminished when they are bound to protein. This loss forms the basis of the substantial destabilization of charged groups in proteins (Kassner 1972; Warshel and Russell 1984). The desolvation energy for moving a sphere between solvents with different dielectric constants can be estimated with the Born equation (Bockris and Reddy 1973; Rashin and Honig 1985), which gives the energy of transfer from water to protein as:
| (1) |
With the constant 331, the distance in Å and the charge in multiples of the charge on an electron the outcome is provided in Kcal/mol. The desolvation energy increases with the charge (q) squared and becomes less important as the charge is delocalized over a molecule with a larger radius (r). With water as the reference solvent, the external dielectric constant is 80. The appropriate value of the interior dielectric constant is subject of much debate, with values from 2 to 80 being used (Gunner and Alexov 2000; Schutz Warshel 2001). This review will discuss calculations carried out with an εprot of 4 (Zhu and Gunner 2005). Roughly, the loss of solvation energy for a cofactor in a protein comes from the larger distance of the charge to water, i.e. the protein adds to the effective cofactor radius. Since proteins are of finite size, even deeply buried groups retain significant solvation energy (Kim et al. 2005).
Charged species always loose more solvation energy then neutral, polar species when moved into the protein (Kassner 1972). Thus the desolvation energy destabilizes the anionic Q•− relative to Q or Q•H, lowering the Em. Likewise the neutral intermediate, Q•H has less solvation energy to loose then Q•−, raising the pKa relative to the reaction in water.
To avoid formation of a buried, anionic semiquinone the reaction sequence could ensure tightly coupled electron and proton transfer (Q + H+ + e− → Q•H). However the electron and proton transfers in proteins occur over very different distance scales. The electron-transfer reactions occur by electron tunneling between reaction partners that are generally more then 7 Å apart (Moser et al. 2003, 2006). In contrast, the proton must be transferred directly between two residues that are close enough that they could make a good hydrogen bond (Sham et al. 1999). Thus, mechanisms that require strictly coupled electron and proton transfer need a proton donor near the electron acceptor (and a separate proton acceptor near the distant electron donor). Instead, the anionic quinone state is often found as a stable intermediate in the various proteins in the bioenergetic electron transfer chains. Thus each protein is designed to provide favorable interactions to stabilize the bound anion despite the loss of solvation energy.
2. Pairwise interactions with protein side chains and backbone
The second contribution the protein makes to the change in the free energy of a redox reaction is via the interactions of the protein charges and dipoles with each redox and protonation state. While, the desolvation penalty always destabilizes both buried anions and cations, the pairwise interactions that stabilize an anion in a particular location will destabilize a cation. The shift in reaction free energy (ΔΔG°) is proportional to the charges on the quinone and the interacting group, the distance between them and the effective screening by the protein and surrounding water (Gunner et al. 1997, 2006).
3. The effects of dielectric relaxation
The third contribution of the protein to the reaction comes from changes in electronic polarization, atomic position and nearby residue protonation in response to the changes in charge distribution caused by the redox reaction. In standard Continuum Electrostatics simulation methods the atomic positions are kept fixed. The energy of electronic polarization and atomic reorganization are accounted for by the assigned protein dielectric constant. These methods do incorporate explicit changes in the protonation state of the residues in the protein (Bashford and Karplus 1990; Beroza et al. 1991; Yang et al. 1993). This relatively simple style of analysis has been able to provide detailed, molecular insight into how specific protein residues play a role in proton coupled electron transfer (Beroza et al. 1995; Lancaster et al. 1996; Rabenstein et al. 1998; Ullmann and Knapp 1999; Ishikita et al. 2003; Haas and Lancaster 2004). In hybrid methods the protein side chains, but not backbone, can change position (You and Bashford 1995; Beroza and Case 1996; Alexov and Gunner 1997). These methods identify more of the ‘dielectric response’ with specific conformational changes rather then with an averaged dielectric constant (Gunner and Alexov 2000; Simonson 2001; Baker 2005; Gunner et al. 2006). The work described below uses the hybrid MultiConformation Continuum Electrostatics (MCCE) method (Georgescu et al. 2002; Alexov and Gunner 1999).
Redox reactions can be stabilized by coupling electron and proton transfers. If one proton is bound per electron transferred then the reaction will not change the charge of the system. This will reduce the contributions of electrostatic interactions to the reaction free energy. However, one of key features of the chemiosmotic mechanism, elucidated by Michel, for storing energy in a transmembrane proton gradient is the alternation of cofactors that bind protons and those which do not (Mitchell 1975a, b). Thus, in principle, the electron transfer sequence has both reactions where the protein changes charge and those that are electroneutral.
The free energy of redox reactions where the reactants and products have different numbers of bound protons will be pH dependent. The proton can be bound to the cofactor itself (Rich and Bendall 1979; Fig. 1) or to the acidic and basic residues in the surrounding protein (Alexov and Gunner 1999; Haas and Lancaster 2004). The presence of nearby ionized groups and the cost of coupled changes in protonation affects the Em (Fig. 2a). Changes in conformation coupled to the redox reaction can be analyzed in a similar manner, by considering the free energy of making the conformational change in the absence of the charge change (Mao et al. 2003). In standard continuum electrostatics methods the costs of these conformational changes are averaged using the protein dielectric constant. In MCCE a mixed response is used, with an averaged protein response of four, while side chain motions are included explicitly.
Fig. 2.
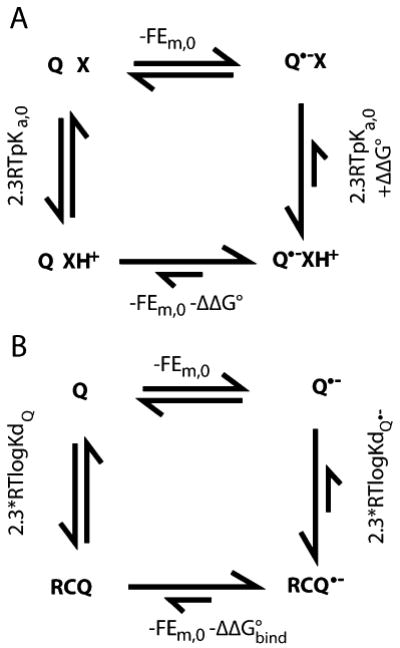
Modification of quinone Ems by proton or protein binding. a Energetics of coupled electron and proton transfer. X is a base associated with a quinone binding site. When X is neutral the Em for Q is Em,0; when Q is neutral the pKa for X is pKa,0. Favorable interactions between X+ and Q•− raise the Em for Q by −ΔΔG°/nF. The same interaction stabilizes XH+, raising the pKa by ΔΔG°/2.3RT. A ΔΔG° of −1.36 kcal/mol shifts the Em up by 58 mV and the pKa up by 1 pH unit. If the pKa of X remains below the pH in the presence of Q•− then X will never be protonated and Em=Em,0. If the pH is below the pKa of X with Q oxidized, XH+ will be present throughout the reaction and the observed Em will be Em=Em,0−ΔΔG°/F. In this case the reduction of Q feels the full stabilization by the adjacent base. In either case the reaction is pH independent. However, if the pH is at least ≈2 pH units below the pKa with Q and ≈2 pH units above the pKa in the presence of Q•− (i.e. ΔΔG° >≈5.4 kcal/mol) then binding ≈1 proton will be coupled to electron transfer (when the pH is 2 pH units below the pKa X will remain 1% protonated; when the pH is 2 pH units above the pKa X will remain 1% XH+. Thus, on average 0.98 more protons will be bound). The resultant Em will be Em,0− [ΔΔG°+(pH−pKa,0)/2.3RT]/F. The free energy needed to protonate X at this pH is (pH−pKa,0)/2.3RT. It is this term which leads to the classic 60 meV/pH unit Em shift with pH indicative of 1 proton bound/electron. The cost of rearranging the surroundings diminishes the Em shift from that found if XH+ were present at the start of the reaction. If pKa,0 for X is near the pH even a small ΔΔG° of interaction with a more distant Q•− will lead to substoichiometric changes in protonation of X, leading to a pH dependence smaller then 60 mV/pH unit. b The relationship between the thermodynamics of quinone binding and quinone electrochemistry. If the Em is more positive in the quinone binding site then the semiquinone is bound more tightly then the quinone. ΔΔG°bind is 2.3RT(logKdQ•−− logKdQ)
If the protein shifts the reaction free energy then its affinity for the reactant and product states must be different (Fig. 2b). A positive Em shift indicates that the reduced cofactor is more tightly bound. A similar connection can be made between the pKa and Kd, where a higher pKa indicates that the protonated species binds more favorably then the unprotonated one (Zhu and Gunner 2005).
The reaction sequence in bacterial photosynthetic reaction centers
The photosynthetic reaction centers from purple bacteria (RCs) were the first membrane protein with a structure known to atomic resolution (Feher et al. 1989; Gunner 1991; Woodbury and Allen 1995). Since a flash of light initiates the reactions, it is possible to measure single turnovers, so the sequence and kinetics of the individual electron transfer reactions are known in detail. The electrochemistry of metastable intermediates have been determined by equilibrium redox titrations. The free energy differences between many reaction intermediates have also been established by measuring reaction rates that rely on a stable species remaining in equilibrium with a high energy intermediate (Woodbury et al. 1986; Xu and Gunner 2000). The overall reaction in RCs uses the energy of two photons to take electrons off two cytochromes c, reducing ubiquinone to the dihydroubiquinone. No protons are pumped across the protein. However, the protons bound to the reduced quinone are taken up from the cell interior, adding to the transmembrane ΔpH (Fig. 3).
Fig. 3.
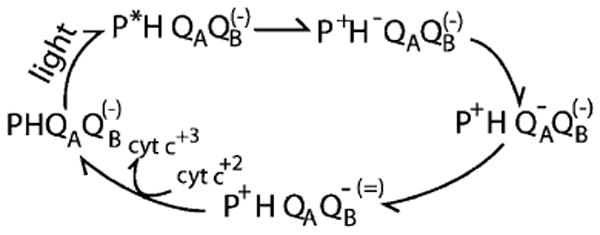
The overview of the sequence of electron transfer reactions in the bacterial type II reaction centers. The reaction starts with a dimer of bacteriochlorophylls (P) excited by light. Electron transfer from the excited state (P*) leads to reduction of a nearby bacteriopheophytin (H). The reduced H•− is used to reduce the primary quinone, QA, which in turn reduces the secondary quinone, QB, to the anionic semiquinone (QB•−). Absorption of a second photon again leads to formation of QA•− which now reduces QB•− to the quinol, QH2, which dissociates from the protein and quinone is rebound. The QB charge in parenthesis is found on the second turnover. The order in which electrons and protons are added to QB is described in Fig. 4 B. In the membrane of the purple non-sulfur photosynthetic bacteria the cytochrome c is rereduced and the QH2 reoxidized by the cytochrome bc1 oxidoreductase with the concomitant increase in the transmembrane proton gradient
The RCs bind UQ in two distinct quinone binding sites, which are then designated QA and QB. Only the oxidized QA and anionic semiquinone QA•− are found. QA does not dissociate from the protein during the reaction cycle. In contrast, QB serves as the two-electron gate, where two single electron transfers from QA•− form the doubly reduced QBH2 for release into the membrane (Wraight 1979; Okamura et al. 2000). RCs are found with QB in three relatively stable redox states: unreduced quinone (Q), anionic semiquinone (Q−) and fully reduced and protonated dihydroquinone (QH2). The anionic semiquinone is tightly bound to the protein, while the Q and QH2 freely exchange with the quinone pool in the membrane (Diner et al. 1984; Madeo and Gunner 2005). The pathway for the second reduction indicates that of the two possible intermediates, QBH is easier to form than QB−2, so proton binding occurs prior to electron transfer (Graige et al. 1996; Zhu and Gunner 2005). Thus, of the nine possible redox states for QB five are found on the reaction pathway (Figs. 1, 4 B). Computational analysis using variations of classical continuum electrostatics (either with a rigid protein (SCCE) or with conformational flexibility (MCCE)) have found fairly good agreement between experiment and calculation, providing insight into the reaction mechanism (Beroza et al. 1995; Lancaster et al. 1996; Rabenstein et al. 1998; Alexov and Gunner 1999; Rabenstein et al. 2000; Ishikita et al. 2003; Alexov et al. 2000). There is a second distal binding site for QB seen in the crystal structures (Lancaster 1998; Stowell et al. 1997). Kinetic measurements find no evidence for quinone reduction in this outer site (Remy and Gerwert 2003; Breton et al. 2002; Xu et al. 2002; Breton 2004; Pokkuluri et al. 2004) and simulation suggests the Em of this quinone too low for it to be reduced by QA•− (Zhu and Gunner 2005).
Fig. 4.
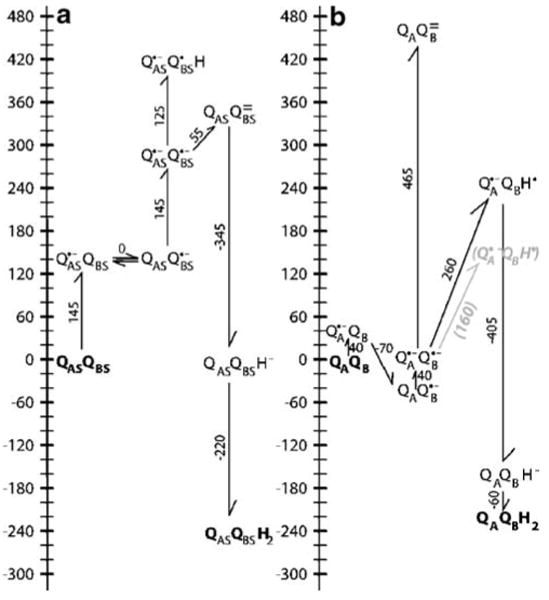
Energy levels for sequential electron transfer from a primary semiquinone to a secondary quinone. A QAS:QBS indicate a complex of two quinones which have the same electrochemistry as two isolated ubiquinones in solution at pH 7 (Fig. 1). To start the cycle one quinone is reduced to the semiquinone forming QAS•−:QBS. Given a solution Eh of 0 mV or a chemical electron donor with an Em of 0 mV, the single reduction of quinone with an Em of −145 mV to Q•− is uphill by 145 meV. Since these quinones are identical, electron transfer from QAS•− to QBS will be isoenergetic. Given the semiquinone pKa of 4.9, protonating either semiquinone would be uphill by ≈120 meV. Thus, the lowest energy, singly reduced state will be a 50:50 mixture of QAS•−:QBS and QAS:QBS•−. The second turnover starts with the second reduction of QAS forming QAS•−:QBS•−. The thermodynamically preferred pathway has the electron transfer occurring before the first proton is bound. The formation of QAS:QBS= requires only 55 meV because the second reduction of QBS is coupled to the favorable oxidation of QAS. The reaction path where QBS•− is protonated to form QAS•−:QBSH before it is reduced is 120 meV uphill. QAS:QBSH− where one quinone has two electrons and one proton is 290 meV lower in energy then QAS•−:QBS•− and is essentially isoenergetic with the initial QAS:QBS state at pH 7. Once QAS:QBSH− is formed the second protonation to form QAS:QBSH2 is downhill by −220 meV. B The ubiquinone energy levels in R. sphaeroides RCs at pH 7 and Eh 0. In the initial reaction QA is reduced to the semiquinone forming QA•−:QB. Calculations put this state near 0 mV, close to the measured values between −45 and −70 mV (all calculated values are from Zhu and Gunner (2005)). QB•− is stabilized by 30 (calculation) to ≈70 meV more then QA•−. The second turnover starts with formation of QA•−:QB•−. Calculations and the experiments of Graige and Okamura show that in the protein QA•−:QB•H is lower in energy then QA:QB= (Graige et al. 1998). The calculations place the energy of QA•−:QB•H 260 meV above QA•−:QB•−, while the kinetics of forward electron transfer support a value of 160 meV (grey text; Graige et al. 1999). The second reduction of QBH− is favorable. The anionic QAQBH− is 110 meV more stable in the protein then in solution, while QAQBH2 is 50 meV less stable favoring quinol dissociation
Quinone electrochemistry in solution
In many photosynthetic type II reaction centers the same chemical species of quinone is used for both the primary (QA) and secondary (QB) quinone acceptors. This is ubiquinone in R. Sphaeroides reaction centers (RCs) and the electrochemistry of this quinone will form the basis of the discussion. The protein shifts the quinone Ems and pKas in the two sites differently so each is changed from what would be found in solution. To understand how the protein perturbs the quinone electrochemistry it is useful to start with describing the intrinsic chemistry of the cofactor in water (Fig. 1). The uncertainty in quinone electrochemistry in aqueous solution is discussed in (Zhu and Gunner 2005).
The free energy of the different pathways for reduction of ubiquinone (Q) to ubiquinol (QH2) in water using a semiquinone electron donor is shown in Fig. 4 A. The reaction starts with QAS:QBS. The final product QAS:QBSH2 is formed via intermediates with reduced primary quinone, first with the secondary quinone oxidized (QAS•−:QBS) and then with it reduced to the semiquinone (QAS•−:QBS•(H)). QAS and QBS have pKas and Ems unperturbed from their solution values (identified by the subscript s). The reaction sequence is described in detail in the legend to Fig. 4 A. While reduction of quinone to the dihydroquinone (quinol) is favorable at pH 7 and Eh 0, all protonated or reduced intermediates are at higher energy. The lowest energy pathway has the quinone donor, QAS•−, reducing the secondary quinone twice forming QAS:QBS= before any protons are bound.
Quinone electrochemistry in bacterial photosynthetic reaction centers
The energy levels of the quinone redox intermediates (Fig. 4 A) can be compared in solution and in RCs (Fig. 4 B). Both experimental and calculated Ems and pKas for reactions in the protein will be considered. In RCs there are only limits for the energies for most intermediates. In contrast, it is possible to calculate energy levels for all states, predicting the energies of unobserved states. The calculated shifts in state energies from solution can then be quantitatively analyzed in light of the protein structure to understand the physical forces that yield the changes in electrochemistry. The overall picture, ranking the energy levels is the same in both simulation and experiment (Rabenstein et al. 1998; Alexov Gunner 1999; Ishikita et al. 2003; Zhu and Gunner 2005). All calculated values quoted here are from (Zhu and Gunner 2005). In general calculated and experimental values differ by less then 60 mV or 1 pH unit, which will be viewed here as representing good agreement. Reactions where experiment and simulation do not match this well highlight interesting changes in energy levels accomplished by the protein.
The protein uses the low potential bacteriopheophytin with an Em (≈−500 mV) as the electron donor (Gunner 1991), making QA reduction an essentially irreversible step in photosynthetic charge separation. Thus, reduction of QA by BPh•− would be favorable even if the Q/Q•− Em was unchanged from the −145 mV found in solution. However, the Em of both QA and QB are raised in the protein. One reason for this could be that it ensures the semiquinone is bound more tightly to the protein then the quinone (Fig. 2b). The calculated Em for QA is near 0 mV, close to the measured values of −45 mV (Dutton et al. 1973) and −70 mV (Rutherford and Evans 1980).
QA•−, in its deeply buried site, is calculated have almost 400 meV less favorable interaction with water then the free semiquinone (Zhu and Gunner 2005). If the quinone Em was lowered by this amount (to −545 mV) even the very low potential BPh•− would have difficulty reducing it. However, interactions of QA•− with the backbone dipoles and with the protein side chains are each calculated to stabilize the anion by ≈−250 meV. QA can be reduced in samples frozen to 4 K (Kleinfeld et al. 1984b; Gunner and Dutton 1989; Xu Gunner 2000), and this is consistent with the simulation showing small conformational changes when QA is reduced (Alexov and Gunner 1999). The reduction of QA provides a clear example of a charged cofactor being stabilized in a deeply buried site within the protein (Gunner et al. 1996). In essence the protein replaces the stabilization of the charge by water's mobile dipoles with charge:charge interactions with permanent dipoles and charged residues within the protein.
The semiquinone is stabilized in the QB site by 30 meV (calculation; Zhu and Gunner 2005) to ≈70 meV (experiment; Mancino et al. 1984; Kleinfeld et al. 1984a) more then in the QA site. The simulation shows that the two quinones loose the same amount of solvation energy, the backbone dipoles stabilize QB•− more and the side chains less then QA•−. The various simulations all suggest that there is significant rearrangement of the charges and dipoles in the QB site required to stabilize the anion (Rabenstein et al. 1998; Alexov and Gunner 1999; Rabenstein et al. 2000; Ishikita et al. 2003; Ishikita and Knapp 2004; Alexov et al. 2000). Without these rearrangements electron transfer would be unfavorable. This is constant with the electron transfer from QA•− to QB freezing out at 200 K (Kleinfeld et al. 1984b; Xu and Gunner 2001). Interestingly, the protein frozen when it is equilibrated around QB•− will allow electron transfer to reform QB•− at temperatures <20 K, indicating that the protein can be trapped in the higher energy, active conformation (Xu and Gunner 2001). The electron transfer rate is independent of the driving force (Graige et al. 1998; Li et al. 2000) which implies that conformational changes, rather then the electron transfer itself, represent the rate determining step in the reaction.
Once the state PHQAQB•− is formed, the second turnover starts with BPh•− reducing QA in the presence of QB•− (Fig. 3). The intermediates QA:QB= and QA•−:QB•H are unstable and are never seen. In solution, electron transfer to form QA:QB= would precede proton uptake (Fig. 4 A). However, the calculations show that the protein switches the relative stability of the two intermediates (Fig. 4 B). This is in agreement with the elegant experiments of Graige and Okamura (Graige et al. 1996). Two changes contribute to the reordering of the relative energy of the intermediates. One is that QA•− is more stable in the protein then in solution so that oxidation of the primary quinone donor contributes less driving force to the reaction. More significant is that the reduction to the unprotonated Q=, with its −2 charge, is very unfavorable in the protein.
The destabilization of Q= can be explained using a continuum electrostatics analysis (Zhu and Gunner 2005). The anionic semiquinone, QB•−, is calculated to be stabilized by the fixed backbone dipoles, the proton side chains, and an adjacent non-heme iron by ≈500 meV. The calculated, favorable interactions are more then doubled for QB= to ≈1,300 mV. The interactions of a rigid protein with QB= would be twice that with QB•−. However, there are significant rearrangements made to the protein side chains to stabilize the first reduction (Alexov and Gunner 1999; Ishikita and Knapp 2004). The reaction free energy reflects the energy needed to move from the protein equilibrated around QB to that equilibrated around QB•− (or QB=; Fig. 2) (Mao et al. 2003). Thus, pairwise interactions with the protein favor the double reduction because the first reduction pre-organizes the protein into conformation and protonation states that favor an anion in the QB site. However, the solvation energy varies, to first order, with q2 (Eq. 1). The stabilization of the dianion by water is seen in the comparison of the reaction in water where Em for reduction of Q is −145 and for reduction of Q•− to Q= is −195 and in the aprotic solvent dimethyl formamide where the Q•−/Q= Em is more then 600 mV more negative then that that of Q/Q•− (Prince et al. 1983). The loss of solvation energy is ≈4 times larger for QB= then for QB•−, resulting in a calculated Em for QB•−/QB= ≈500 mV lower then for QB/QB•−. Thus, the separation of the two energy levels moving from water to the QB site reflects the desolvation penalty increasing with ≈q2 while the favorable pairwise interactions only increases with ≈q.
In water Q•− has a pKa of 4.9, while QB•− is calculated to have a pKa of 2.7. At pH 7 the neutral radical would therefore be 260 meV higher in energy then the anion. The ΔG° between the two states can be estimated from the rate of electron transfer from QA•− to QB•−, which has been shown to proceed by rapid, uphill preprotonation of QB•− followed by the electron transfer reaction (Graige et al. 1998; Graige et al. 1999). The rate is determined by the fraction of the RCs in the state QA•−:QB•H. The rate determining step is then the quantum tunneling of the electron from QA•− to QB•H. The energy of the protonated intermediate thus factors into the rate of electron transfer since the concentration of QB•H is diminished by a factor of 10 for every 60 mV increase in the ΔG°. These kinetic measurements suggest QB•H is between 80 and 240 meV higher in energy then QB•−, with a best estimate of 160 meV (Graige et al. 1998). The higher calculated energy for QB•H would slow the observed rate of the second electron transfer by ≈40 fold.
The apparent disagreement between experimental and calculated pKa for QB•− raises an interesting question about how the protein stabilizes the individual quinone redox states. The work of Okamura and Graige places the in situ pKa for QB•− at 4.5, a negligible shift from solution, indicating the proton stabilizes the charged QB•− and neutral QB•H by essentially the same amount. In contrast, in the simulations QB•− is stabilized relative to both neutral Q and QB•H by ≈140 meV, lowering the calculated semiquinone pKa from 4.9 to 2.7. If the experimental assignment of the quinone pKa is correct then the protein must stabilize QB•H relative to Q by ≈140 meV more then found in the current simulations.
The final steps in formation of the quinol in the QB site involve the second reduction of QB followed by binding the second proton to form QBH2. The doubly reduced QBH− is calculated to be more stable in the protein then in solution (Fig. 4 B). However, the calculated pKa for proton binding is ≈8 while the pKa is 10.7 in solution indicating the doubly protonated QH2 is destabilized in the binding site, with the quinol bound ≈50 fold less tightly then the quinone. This favors quinol dissociation, allowing a new quinone to be bound to restart the reaction cycle (Fig. 2 B).
Thermodynamics and kinetics of quinone binding
The reduction of ubiquinone in the QB site uses two turnovers of the single electron donor QA•− to form the doubly reduced, product QH2, which is released into the Qpool (Fig. 3). The semiquinone is a potentially dangerous high-energy free radical, which is considered to be a primary source of damaging reactive oxygen species (Kramer et al. 2004). In addition, the efficiency of the conversion of light energy to chemical energy relies on QA•− and QB•− remaining bound to the protein. Thus, the semiquinone at the QA•− site must be sufficiently stable that QB has time to bind and be reduced. Then QB•− must live long enough that a second photon can rereduce QA even under low light conditions where photons are scarce.
Given the importance of trapping the bound semiquinones at the QA and QB site it might seem as though the quinone binding sites should be designed to bind the semiquinone extremely tightly. However, stabilization of any individual redox intermediate will have consequences for the overall electron and proton transfers (Fig. 4 B). An over-stabilized QA•− would not be able to transfer electrons to QB. If QB•− were too stable, the succeeding intermediates would not be formed. Alternatively the protein could bind all quinone redox states very tightly to ensure the semiquinone were not lost while not changing the reaction free energy landscape. However, this would slow down turnover since, the product QH2 must be released from the QB site at the end of the reaction sequence.
A study of the binding of neutral and anionic quinones to the QA site of RCs may provide insight into how the protein can sequester an anionic semiquinone while not making it too stable (Madeo and Gunner 2005). Quinone on and off rates were measured for a series of tailless neutral, methyl and methoxy substituted benzoquinones and naphthoquinones. The semiquinone is not stable in aqueous solution so its affinity and binding rates cannot be determined directly. Rather hydroxyl quinones were used and measurements carried out above the hydroxyl pKa. While these anionic quinones are not reduced in the QA site they are found to be good, reversible competitive inhibitors for the binding site.
If the reactant and product are bound with different affinities the electrochemistry of the reaction will be shifted in the protein (Fig. 2 B). The Em for the reduction to the anionic semiquinone is 70–100 mV more positive in the QA site then in water. This indicates the semiquinone binds 15–45 times more tightly then the quinone. The koff for the tailless ubiquinone0 (2,3-dimethoxy, 5 methyl-benzoquinone), is 6/s (Madeo and Gunner 2005). If the change in affinity were translated into a change in the off rate (Keq = koff / kon) the t1/2 for semiquinone release would be 2.5–7.5 s. However, the series of neutral, alkyl quinones tested show a negligible change in koff with affinity. Rather for the neutral quinones the changes in affinity strongly correlate with changes in kon. If the semiquinone behaved similarly it would dissociate from the protein in 100 s of milliseconds and so could be easily lost.
The neutral, alkyl substituted and anionic, hydroxy substituted quinones studied have similar structures and similar Kds for the binding site, ranging from 0.1 to 10 μM (Madeo and Gunner 2005). However, the anionic, hydroxyl quinones have a binding mechanism that is different and remarkably slower then the neutral quinones. Thus, the rate-determining step for binding the neutral quinones is bimolecular while it is first order for the anionic quinone, indicating the latter process is gated in some way. The binding rates kon cannot be directly compared for the two types of compounds since the bimolecular reaction has units of per molar per second while it is per second for the unimolecular reaction. However the off rates both have units of per second. The anionic compounds are found to dissociate a remarkable 104 times more slowly then do the neutral compounds. The anionic, hydroxyl quinones bind and dissociate in tens of minutes while the neutral compounds do this in milliseconds. Thus, the protein is seen to decouple the kinetic and thermodynamic stability to firmly bind anionic quinones. If the anionic, hydroxyl quinones are a good analog for the anionic semiquinone, the protein could shift the Kd for QA•− relative to QA by less then 100 fold, constant with the modest Em shift, while slowing the rate of QA•− dissociation by 104 fold.
Conclusion
The photosynthetic reaction centers are a well-studied system where we have remarkable knowledge of the protein structures, reaction pathways, and cofactor electrochemistry and binding constants. This information makes it possible to understand how quinone properties are altered when bound to specific binding sites in the transmembrane electron transfer proteins. A continuum electrostatics analysis shows how the protein can stabilize an anionic semiquinone (QB•−) while destabilizing the dianion (QB=). Comparison of experimental and calculated results show that the protonated semiquinone is bound especially tightly, enabling the rapid preprotonation of QB•−. Further analysis of anionic inhibitor binding suggests that the protein uses kinetic barriers to sequester the valuable, high-energy semiquinone intermediates.
The lessons learned here may provide insight into how the other transmembrane electron transfer proteins such as the cytochrome bc1 oxidoreductase control their electron transfer reactions. As in RCs the bc1 complex binds two ubiquinones whose roles are defined by their binding sites. The Qi site carries out a clear two-step reaction sequence that is similar to that found in the QB site, with two turnovers leading to quinone reduction. At the QO site there is only transient semiquinone production as the bound QH2 reduces two different reaction partners (Cape et al. 2007; Zhang et al. 2007). The protein controls the relative energy levels of the different redox intermediates for function and stability. For example, increasing the thermodynamic stability of the semiquinone in the QO site leads to the production of harmful reactive oxygen species (Cape et al. 2005; Forquer et al. 2006). It has been shown here that proteins can decouple thermodynamics and kinetics of reactions, slowing the binding and dissociation of anionic quinones without greatly increasing their affinity for the proteins. Mechanisms of this kind might play a role in inhibiting thermodynamically favorable undesirable reactions such as the reduction of the high potential cytochrome c1 by the lower potential b-type cytochromes in the bc1 complex (Rich 2004; Osyczka et al. 2005)
Acknowledgments
I would like to thank Colin Wraight and David Kramer to their shared, long-standing interest in quinone electrochemistry. The financial support of NSF MCB-0517589 and of NIH 5G12 RR03060 for infrastructure support are gratefully acknowledged.
References
- Alexov EG, Gunner MR. Incorporating protein conformational flexibility into the calculation of pH-dependent protein properties. Biophys J. 1997;72:2075–2093. doi: 10.1016/S0006-3495(97)78851-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexov EG, Gunner MR. Calculated protein and proton motions coupled to electron transfer: electron transfer from QA− to QB in bacterial photosynthetic reaction centers. Biochemistry. 1999;38:8253–8270. doi: 10.1021/bi982700a. [DOI] [PubMed] [Google Scholar]
- Alexov E, Miksovska J, Baciou L, Schiffer M, Hanson DK, Sebban P, Gunner MR. Modeling the effects of mutations on the free energy of the first electron transfer from QA− to QB in photosynthetic reaction centers. Biochemistry. 2000;39:5940–5952. doi: 10.1021/bi9929498. [DOI] [PubMed] [Google Scholar]
- Baker NA. Improving implicit solvent simulations: a Poisson-centric view. Cur Opin Struct Biol. 2005;15:137–143. doi: 10.1016/j.sbi.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Bashford D, Karplus M. The pKas of ionizable groups in proteins: atomic detail from a continuum electrostatic model. Biochemistry. 1990;29:10219–10225. doi: 10.1021/bi00496a010. [DOI] [PubMed] [Google Scholar]
- Beroza P, Case D. Including side chain flexibility in continuum electrostatic calculations of protein titration. J Phys Chem. 1996;100:20156–20163. [Google Scholar]
- Beroza P, Fredkin DR, Okamura MY, Feher G. Protonation of interacting residues in a protein by a Monte Carlo method: application to Lysozyme and the photosynthetic reaction center of Rhodobacter sphaeroides. Proc Natl Acad Sci USA. 1991;88:5804–5808. doi: 10.1073/pnas.88.13.5804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beroza P, Fredkin DR, Okamura MY, Feher R. Electrostatic calculations of amino acid titration electron transfer, Q−AQB→ QAQB−, in the reaction center. Biophys J. 1995;68:2233–2250. doi: 10.1016/S0006-3495(95)80406-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bockris JOM, Reddy AKN. Modern electrochemistry. Vol. 1. Plenum; New York: 1973. [Google Scholar]
- Breton J. Absence of large-scale displacement of quinone QB in bacterial photosynthetic reaction centers. Biochemistry. 2004;43:3318–3326. doi: 10.1021/bi049811w. [DOI] [PubMed] [Google Scholar]
- Breton J, Boullais C, Mioskowski C, Sebban P, Bacious L, Nabedryk E. Vibrational spectroscopy favors a unique QB binding site at the proximal position in wild-type reaction centers and in the Pro-L209 → Tyr mutant from Rhodobacter sphaeroides. Biochemistry. 2002;41:12921–12927. doi: 10.1021/bi026565k. [DOI] [PubMed] [Google Scholar]
- Cape JL, Strahan JR, Lenaeus MJ, Yuknis BA, Le TT, Shepherd JN, Bowman MK, Kramer DM. The respiratory substrate rhodoquinol induces Q-cycle bypass reactions in the yeast cytochrome bc1 complex: mechanistic and physiological implications. J Biol Chem. 2005;280:34654–34660. doi: 10.1074/jbc.M507616200. [DOI] [PubMed] [Google Scholar]
- Cape JL, Bowman MK, Kramer DM. Computation of the redox and protonation properties of quinones: towards the prediction of redox cycling natural products. Phytochemistry. 2006;67:1781–1788. doi: 10.1016/j.phytochem.2006.06.015. [DOI] [PubMed] [Google Scholar]
- Cape JL, Bowman MK, Kramer DM. Understanding the cytochrome bc complexes by what they don't do. The Q-cycle at 30. Trends Plant Sci. 2006;11:46–55. doi: 10.1016/j.tplants.2005.11.007. [DOI] [PubMed] [Google Scholar]
- Cape JL, Bowman MK, Kramer DM. A semiquinone intermediate generated at the Qo site of the cytochrome bc1 complex: importance for the Q-cycle and superoxide production. Proc Natl Acad Sci USA. 2007;104:7887–7892. doi: 10.1073/pnas.0702621104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Churg AK, Warshel A. Control of the redox potential of cytochrome c and microscopic dielectric effects in proteins. Biochemistry. 1986;25:1675–1681. doi: 10.1021/bi00355a035. [DOI] [PubMed] [Google Scholar]
- Churg AK, Weiss RM, Warshel A, Takano T. On the action of cytochrome c: correlating geometry changes upon oxidation with activation energies of electron transfer. J Phys Chem. 1983;87:1683–1694. [Google Scholar]
- Diner BA, Schenck CC, DeVitry C. Effect of inhibitors, redox state and isoprenoid chain length on the affinity of ubiquinone for the secondary acceptor binding site in the reaction centers of photosynthetic bacteria. Biochim Biophys Acta. 1984;766:9–20. [Google Scholar]
- Dutton PL, Leigh JS, Wraight CA. Direct measurement of the midpoint potential of the primary electron acceptor in Rhodopseudomonas spheroides in situ and in the isolated state: some relationships with pH and o-phenanthroline. FEBS Lett. 1973;36:169–173. doi: 10.1016/0014-5793(73)80361-8. [DOI] [PubMed] [Google Scholar]
- Feher G, Allen JP, Okamura MY, Rees DC. Primary processes in bacterial photosynthesis: structure and function of bacterial photosynthetic reaction centres. Nature. 1989;339:111–116. [Google Scholar]
- Forquer I, Covian R, Bowman MK, Trumpower B, Kramer DM. Similar transition states mediate the Q-cycle and superoxide production by the cytochrome bc1 complex. J Biol Chem. 2006;281:38459–38465. doi: 10.1074/jbc.M605119200. [DOI] [PubMed] [Google Scholar]
- Georgescu RE, Alexov EG, Gunner MR. Combining conformational flexibility and continuum electrostatics for calculating pKas in proteins. Biophys J. 2002;83:1731–1748. doi: 10.1016/S0006-3495(02)73940-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graige MS, Paddock ML, Bruce JM, Feher G, Okamura MY. Mechanism of proton-coupled electron transfer for quinone (QB) reduction in reaction centers of R. sphaeroides. J Am Chem Soc. 1996;118:9005–9016. [Google Scholar]
- Graige MS, Feher G, Okamura MY. Conformational gating of the electron transfer reaction QA−QB → QAQB− in bacterial reaction centers of Rhodobacter sphaeroides determined by a driving force assay. Proc Natl Acad Sci USA. 1998;95:11679–11684. doi: 10.1073/pnas.95.20.11679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graige MS, Paddock ML, Feher G, Okamura MY. Observation of the protonated semiquinone intermediate in isolated reaction centers from Rhodobacter sphaeroides: implications for the mechanism of electron and proton transfer in proteins. Biochemistry. 1999;38:11465–11473. doi: 10.1021/bi990708u. [DOI] [PubMed] [Google Scholar]
- Gunner MR. The reaction center protein from purple bacteria: structure and function. Curr Top Bioenerg. 1991;16:319–367. [Google Scholar]
- Gunner MR, Alexov E. A pragmatic approach to structure based calculation of coupled proton and electron transfer in proteins. Biochim Biophys Acta. 2000;1458:63–87. doi: 10.1016/s0005-2728(00)00060-8. [DOI] [PubMed] [Google Scholar]
- Gunner MR, Dutton PL. Temperature and −ΔG° dependence of the electron transfer from BPh− to QA in reaction center protein from Rhodobacter sphaeroides with different quinones as QA. J Am Chem Soc. 1989;111:3400–3412. [Google Scholar]
- Gunner MR, Honig B. Electrostatic control of midpoint potentials in the cytochrome subunit of the Rhodopseudomonas viridis reaction center. Proc Natl Acad Sci USA. 1991;88:9151–9155. doi: 10.1073/pnas.88.20.9151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunner MR, Nicholls A, Honig B. Electrostatic potentials in Rhodopseudomonas viridis reaction center: implications for the driving force and directionality of electron transfer. J Phys Chem. 1996;100:4277–4291. [Google Scholar]
- Gunner MR, Alexov E, Torres E, Lipovaca S. The importance of the protein in controlling the electrochemistry of heme metalloproteins: methods of calculation and analysis. J Biol Inorg Chem. 1997;2:126–134. [Google Scholar]
- Gunner MR, Mao J, Song Y, Kim J. Factors influencing energetics of electron and proton transfers in proteins. What can be learned from calculations. Biochim Biophys Acta. 2006;1757:942–968. doi: 10.1016/j.bbabio.2006.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas AH, Lancaster CR. Calculated coupling of transmembrane electron and proton transfer in dihemic quinol:fumarate reductase. Biophys J. 2004;87:4298–4315. doi: 10.1529/biophysj.104.042945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa Jy, Ishida M, Nakatsuji H, Lu Z, Liu H, Yang W. Energetics of the electron transfer from bacteriopheophytin to ubiquinone in the photosynthetic reaction center of Rhodopseudomonas viridis: theoretical study. J Phys Chem B. 2003;107:838–847. [Google Scholar]
- Heathcote P. Reaction centers: the structure and evolution of biological solar power. Trends Biochem Sci. 2002;27:79–86. doi: 10.1016/s0968-0004(01)02034-5. [DOI] [PubMed] [Google Scholar]
- Honig B, Nicholls A. Classical electrostatics in biology and chemistry. Science. 1995;268:1144–1149. doi: 10.1126/science.7761829. [DOI] [PubMed] [Google Scholar]
- Ishikita H, Knapp EW. Variation of Ser-L223 hydrogen bonding with the QB redox state in reaction centers from Rhodobacter sphaeroides. J Am Chem Soc. 2004;126:8059–8064. doi: 10.1021/ja038092q. [DOI] [PubMed] [Google Scholar]
- Ishikita H, Morra G, Knapp EW. Redox potential of quinones in photosynthetic reaction centers from Rhodobacter sphaeroides: dependence on protonation of Glu-L212 and Asp-L213. Biochemistry. 2003;42:3882–3892. doi: 10.1021/bi026781t. [DOI] [PubMed] [Google Scholar]
- Kassner RJ. Effects of nonpolar environments on the redox potentials of heme complexes. Proc Natl Acad Sci USA. 1972;69:2263–2267. doi: 10.1073/pnas.69.8.2263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Mao J, Gunner MR. Are acidic and basic groups in buried proteins predicted to be ionized? J Mol Biol. 2005;348:1283–1298. doi: 10.1016/j.jmb.2005.03.051. [DOI] [PubMed] [Google Scholar]
- Kleinfeld D, Okamura MY, Feher G. Electron transfer in reaction centers of Rhodopseudomonas sphaeroides: I. Determination of the charge recombination pathway of D+ QAQB− and free energy and kinetic relations between QA−QB and QAQB−. Biochim Biophys Acta. 1984a;766:126–140. doi: 10.1016/0005-2728(84)90224-x. [DOI] [PubMed] [Google Scholar]
- Kleinfeld D, Okamura MY, Feher G. Electron-transfer kinetics in photosynthetic reaction centers cooled to cryogenic temperatures in the charge separated state: evidence for light-induced structural changes. Biochemistry. 1984b;23:5780–5786. doi: 10.1021/bi00319a017. [DOI] [PubMed] [Google Scholar]
- Klingen AR, Palsdottir H, Hunte C, Ullmann GM. Redox-linked protonation state changes in cytochrome bc1 identified by Poisson–Boltzmann electrostatics calculations. Biochim Biophys Acta. 2007;1767:204–221. doi: 10.1016/j.bbabio.2007.01.016. [DOI] [PubMed] [Google Scholar]
- Kramer DM, Roberts AG, Muller F, Cape J, Bowman MK. Q-cycle bypass reactions at the Qo site of the cytochrome bc1 (and related) complexes. Methods Enzymol. 2004;382:21–45. doi: 10.1016/S0076-6879(04)82002-0. [DOI] [PubMed] [Google Scholar]
- Lancaster CRD. Ubiquinone reduction and protonation in photosynthetic reaction centres from Rhodopseudomonas viridis: X-ray structures and their functional implications. Biochim Biophys Acta. 1998;1365:143–150. [Google Scholar]
- Lancaster CRD, Michel H, Honig B, Gunner MR. Calculated coupling of electron and proton transfer in the photosynthetic reaction center of Rhodopseudomonas viridis. Biophys J. 1996;70:2469–2492. doi: 10.1016/S0006-3495(96)79820-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Takahashi E, Gunner MR. −ΔG°AB and pH dependence on the electron transfer from P+ QA−QB to P+ QAQB− in Rhodobacter sphaeroides reaction centers. Biochemistry. 2000;39:7445–7454. doi: 10.1021/bi992591f. [DOI] [PubMed] [Google Scholar]
- Madeo J, Gunner MR. Modeling binding kinetics at the QA site in bacterial reaction centers. Biochemistry. 2005;44:10994–11004. doi: 10.1021/bi050544j. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallik B, Datta SN. Semiemprical quantum chemical treatment of the standard reduction potentials of quinone and plastoquinone in water. Int J Quant Chem. 2004;52:629–649. [Google Scholar]
- Mancino LJ, Dean DP, Blankenship RE. Kinetics and thermodynamics of the P870+ QA−>P870+ QB− reaction in isolated reaction centers from the photosynthetic bacterium Rhodopseudomonas sphaeroides. Biochim Biophys Acta. 1984;764:46–54. [Google Scholar]
- Mao J, Hauser K, Gunner MR. How cytochromes with different folds control heme redox potentials. Biochemistry. 2003;42:9829–9840. doi: 10.1021/bi027288k. [DOI] [PubMed] [Google Scholar]
- Marchi M, Gehlen JN, Chandler D, Newton M. Diabatic surfaces and the pathway for primary electron transfer in a photosynthetic reaction center. J Am Chem Soc. 1993;115:4178–4190. [Google Scholar]
- Mitchell P. Proton motive redox mechanism of the cytochrome bc1 complex in the respiratory chain: proton motive ubiquinone cycle. FEBS Lett. 1975a;56:1–6. doi: 10.1016/0014-5793(75)80098-6. [DOI] [PubMed] [Google Scholar]
- Mitchell P. The proton motive Q cycle: a general formulation. FEBS Lett. 1975b;59:137–139. doi: 10.1016/0014-5793(75)80359-0. [DOI] [PubMed] [Google Scholar]
- Moser CC, Page CC, Cogdell RJ, Barber J, Wraight CA, Dutton PL. Length, time, and energy scales of photosystems. Adv Protein Chem. 2003;63:71–109. doi: 10.1016/s0065-3233(03)63004-4. [DOI] [PubMed] [Google Scholar]
- Moser CC, Farid TA, Chobot SE, Dutton PL. Electron tunneling chains of mitochondria. Biochim Biophys Acta. 2006;1757:1096–1109. doi: 10.1016/j.bbabio.2006.04.015. [DOI] [PubMed] [Google Scholar]
- Okamura MY, Paddock ML, Graige MS, Feher G. Proton and electron transfer in bacterial reaction centers. Biochim Biophys Acta. 2000;1458:148–163. doi: 10.1016/s0005-2728(00)00065-7. [DOI] [PubMed] [Google Scholar]
- Osyczka A, Moser CC, Dutton PL. Fixing the Q cycle. Trends Biochem Sci. 2005;30:176–182. doi: 10.1016/j.tibs.2005.02.001. [DOI] [PubMed] [Google Scholar]
- Parson WW, Chu ZT, Warshel A. Electrostatic control of charge separation in bacterial photosynthesis. Biochim Biophys Acta. 1990;1017:251–272. doi: 10.1016/0005-2728(90)90192-7. [DOI] [PubMed] [Google Scholar]
- Pokkuluri PR, Laible PD, Crawford AE, Mayfield JF, Yousef MA, Ginell SL, Hanson DK, Schiffer M. Temperature and cryoprotectant influence secondary quinone binding position in bacterial reaction centers. FEBS Let. 2004;43:9909–9917. doi: 10.1016/j.febslet.2004.06.042. [DOI] [PubMed] [Google Scholar]
- Prince RC, Dutton PL, Bruce JM. Electrochemistry of ubiquinones. FEBS Lett. 1983;160:273–276. [Google Scholar]
- Prince RC, Lloyd-Williams P, Bruce JM, Dutton PL. Voltammetric measurements of quinines. Methods Enzymol. 1986;125:109–119. doi: 10.1016/s0076-6879(86)25010-7. [DOI] [PubMed] [Google Scholar]
- Rabenstein B, Ullmann GM, Knapp EW. Energetics of electron-transfer and protonation reactions of the quinones in the photosynthetic reaction center of Rhodopseudomonas viridis. Biochemistry. 1998;37:2488–2495. doi: 10.1021/bi971921y. [DOI] [PubMed] [Google Scholar]
- Rabenstein B, Ullmann GM, Knapp EW. Electron transfer between the quinones in the photosynthetic reaction center and its coupling to conformational changes. Biochemistry. 2000;39:10487–10496. doi: 10.1021/bi000413c. [DOI] [PubMed] [Google Scholar]
- Rashin AA, Honig B. Reevaluation of the born model of ion hydration. J Phys Chem. 1985;89:5588–5593. [Google Scholar]
- Reedy CJ, Gibney BR. Heme protein assemblies. Chem Rev. 2004;104:617–649. doi: 10.1021/cr0206115. [DOI] [PubMed] [Google Scholar]
- Remy A, Gerwert K. Coupling of light-induced electron transfer to proton uptake in photosynthesis. Nat Struct Biol. 2003;10:637–644. doi: 10.1038/nsb954. [DOI] [PubMed] [Google Scholar]
- Rich PR. The quinone chemistry of bc complexes. Biochem Biophys Acta. 2004;1658:165–171. doi: 10.1016/j.bbabio.2004.04.021. [DOI] [PubMed] [Google Scholar]
- Rich PR, Bendall DS. A mechanism for the reduction of cytochromes by quinols in solution and its relevance to biological electron transfer reactions. FEBS Lett. 1979;105:189–194. doi: 10.1016/0014-5793(79)80608-0. [DOI] [PubMed] [Google Scholar]
- Rutherford AW, Evans MCW. Direct measurement of the redox potential of the primary and secondary quinone electron acceptors in Rhodopseudomonas sphaeroides (wild-type) by EPR spectrometry. FEBS Lett. 1980;110:257–261. doi: 10.1016/0014-5793(80)80086-x. [DOI] [PubMed] [Google Scholar]
- Saraste M. Oxidative phosphorylation at the fin de siecle. Science. 1999;283:1488–1493. doi: 10.1126/science.283.5407.1488. [DOI] [PubMed] [Google Scholar]
- Schutz CN, Warshel A. What are the “dielectric constants” of proteins and how to validate electrostatic models? Proteins Struct Funct Genet. 2001;44:400–417. doi: 10.1002/prot.1106. [DOI] [PubMed] [Google Scholar]
- Sham YY, Chu ZT, Warshel A. Consistent calculations of pKa's of ionizable residues in proteins: semi-microscopic and microscopic approaches. J Phys Chem. 1997;101:4458–4472. [Google Scholar]
- Sham Y, Muegge I, Warshel A. Simulating proton translocations in proteins: probing proton transfer pathways in the Rhodobacter sphaeroides reaction center. Proteins Struct Funct Genet. 1999;36:484–500. [PubMed] [Google Scholar]
- Shurki A, Strajbl M, Schutz CN, Warshel A. Electrostatic basis for bioenergetics. Methods Enzymol. 2004;380:52–84. doi: 10.1016/S0076-6879(04)80003-X. [DOI] [PubMed] [Google Scholar]
- Simonson T. Macromolecular electrostatics: continuum models and their growing pains. Curr Opin Struct Biol. 2001;11:243–252. doi: 10.1016/s0959-440x(00)00197-4. [DOI] [PubMed] [Google Scholar]
- Stowell MHB, McPhillips TM, Rees DC, Soltis SM, Abresch E, Feher G. Light-induced structural changes in photosynthetic reaction center: implications for mechanism of electron–proton transfer. Science. 1997;276:812–816. doi: 10.1126/science.276.5313.812. [DOI] [PubMed] [Google Scholar]
- Swallow AJ. Physical chemistry of semiquinones. In: Trumpower BL, editor. Function of quinones in energy conserving systems. Academic; New York: 1982. pp. 59–72. [Google Scholar]
- Ullmann GM, Knapp EW. Electrostatic models for computing protonation and redox equilibria in proteins. Eur Biophys J. 1999;28:533–551. doi: 10.1007/s002490050236. [DOI] [PubMed] [Google Scholar]
- Voigt P, Knapp EW. Tuning heme redox potentials in the cytochrome c subunit of photosynthetic reaction centers. J Biol Chem. 2003;278:51993–52001. doi: 10.1074/jbc.M307560200. [DOI] [PubMed] [Google Scholar]
- Warshel A, Russell ST. Calculations of electrostatic interactions in biological systems and in solutions. Q Rev Biophys. 1984;17:283–422. doi: 10.1017/s0033583500005333. [DOI] [PubMed] [Google Scholar]
- Woodbury NW, Allen JP. The pathway, kinetics and thermodynamics of electron transfer in wild type and mutant reaction centers of purple nonsulfur bacteria. In: Blankenship RE, Madigan MT, Bauer CE, editors. Anoxygenic photosynthetic bacteria. Kluwer; Dordrecht: 1995. [Google Scholar]
- Woodbury NW, Parson WW, Gunner MR, Prince RC, Dutton PL. Radical-pair energetics and decay mechanisms in reaction center containing anthraquinones or benzoquinones in place of ubiquinone. Biochim Biophys Acta. 1986;851:6–22. doi: 10.1016/0005-2728(86)90243-4. [DOI] [PubMed] [Google Scholar]
- Wraight CA. Electron acceptors of bacterial photosynthetic reaction centers II. H+ binding coupled to secondary electron transfer in the quinone acceptor complex. Biochim Biophys Acta. 1979;548:309–327. doi: 10.1016/0005-2728(79)90138-5. [DOI] [PubMed] [Google Scholar]
- Wraight CA. Proton and electron transfer in the acceptor quinone complex of photosynthetic reaction centers from Rhodobacter sphaeroides. Front Biosci. 2004;9:309–337. doi: 10.2741/1236. [DOI] [PubMed] [Google Scholar]
- Xu Q, Gunner MR. Temperature dependence of the free energy, enthalpy and entropy of P+ QA− charge recombination in photosynthetic reaction centers. J Phys Chem B. 2000;104:8035–8043. [Google Scholar]
- Xu Q, Gunner MR. Trapping conformational intermediate states in the reaction center protein from photosynthetic bacteria. Biochemistry. 2001;40:3232–3241. doi: 10.1021/bi002326q. [DOI] [PubMed] [Google Scholar]
- Xu Q, Baciou L, Sebban P, Gunner MR. Exploring the energy landscape for QA− to QB electron transfer in bacterial photosynthetic reaction centers: effect of substrate position and tail length on the conformational gating step. Biochemistry. 2002;41:10021–10025. doi: 10.1021/bi025573y. [DOI] [PubMed] [Google Scholar]
- Yang AS, Gunner MR, Sampogna R, Sharp K, Honig B. On the calculation of pKa's in proteins. Proteins Struct Funct Genet. 1993;15:252–265. doi: 10.1002/prot.340150304. [DOI] [PubMed] [Google Scholar]
- You TJ, Bashford D. Conformation and hydrogen ion titration of proteins: a continuum electrostatic model with conformational flexibility. Biophys J. 1995;69:1721–1733. doi: 10.1016/S0006-3495(95)80042-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Osyczka A, Dutton PL, Moser CC. Exposing the complex III Qo semiquinone radical. Biochim Biophys Acta. 2007;1767:883–887. doi: 10.1016/j.bbabio.2007.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Gunner MR. Energetics of quinone-dependent electron and proton transfers in Rhodobacter sphaeroides photosynthetic reaction centers. Biochemistry. 2005;44:82–96. doi: 10.1021/bi048348k. [DOI] [PubMed] [Google Scholar]
- Zheng Z, Gunner MR. Analysis of the electrochemistry of hemes with Ems spanning 800 mV. Proteins. 2008 doi: 10.1002/prot.22282. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]