

Abstract
We screened 116 patients with a strong family history of pancreatic cancer using a combination of endoscopic ultrasound and computed tomography. Ten of these patients underwent surgical resection at our institution, providing an opportunity to define the morphology of pancreatic precursor lesions in patients with a strong family history of pancreatic cancer. Eight of the 10 pancreata were available and these were entirely submitted for histologic examination. The number of pancreatic intraepithelial neoplasia (PanIN) lesions and intraductal papillary mucinous neoplasms (IPMNs) were compared with age-matched controls. Parenchymal changes were defined. Selected precursor neoplasms from 6 pancreata were microdissected and analyzed for KRAS gene mutations. PanINs were significantly more common in the 8 cases (mean of 10.7% of the duct profiles, range 1.0% to 27.3%) than in the controls (mean 1.9%, range 0% to 9.2%, P<0.01). Different KRAS gene mutations were identified in separately microdissected precursor lesions in 2 of 6 cases. IPMNs were identified in 4 of the 8 cases, including 2 pancreata each having 2 distinct IPMNs. Both the IPMNs and the PanINs, even the low-grade PanIN-1 lesions, were associated with lobular parenchymal atrophy. Some individuals with a strong family history of pancreatic cancer develop multifocal, noninvasive epithelial precursor lesions of the pancreas. PanINs and IPMNs produce obstructive lobular atrophy, and this atrophy is likely the source of the chronic pancreatitis-like changes seen in these patients. The multifocal nature of familial pancreatic neoplasia suggests that surveillance of these patients is warranted after partial pancreatectomy.
Keywords: familial cancer, pancreatic cancer, pancreatic intraepithelial neoplasia, intraductal papillary mucinous neoplasm
It has been estimated that 10% of pancreatic cancers have a familial basis.31,39 Having a first-degree relative with pancreatic cancer doubles the risk of developing pancreatic cancer,1 and the risk increases with increasing numbers of affected relatives.33 Segregation analyses have suggested that a major gene is responsible for this increased risk,32 but the gene responsible for the familial aggregation of pancreatic cancer has not yet been identified and effective screening tests for asymptomatic noninvasive disease have not been fully developed.
Careful examination of the precursor lesions associated with the familial clustering of a cancer can provide insight into the biologic properties of a familial cancer gene. For example, innumerable colonic adenomas are present in patients with familial adenomatous polyposis (FAP), and APC, the gene responsible for FAP, has been therefore classified as a “gatekeeper” gene.27–29 By contrast, hereditary nonpolyposis colorectal cancer syndrome, as the name suggests, is not associated with increased numbers of noninvasive precursor lesions, and the genes responsible for hereditary nonpolyposis colorectal cancer syndrome function as “genome-maintenance” genes.27,36 By extension, careful examination of the histopathology of the precursor lesions associated with the familial aggregation of pancreatic cancer will help to define the biologic properties of the familial pancreatic cancer gene(s).
In addition, characterization of the pathology of noninvasive precursor lesions in patients with familial pancreatic cancer may also form a basis for the development of screening tests for early pancreatic neoplasia. For example, the demonstration that many intraductal carcinomas of the breast are associated with microcalcifications has helped guide mammography for the early detection of breast cancer,12 which has reduced breast cancer mortality.15,50
We recently screened a series of 116 asymptomatic patients with a strong family history of pancreatic cancer for evidence of pancreatic neoplasia using a combination of physical examination, endoscopic ultrasound (EUS), multidetector computed tomography, and, in some cases, endoscopic retrograde pancreatography.7,8 Ten of these patients underwent pancreatic resection at The Johns Hopkins Hospital, providing a unique opportunity to define the morphology of pancreatic precursor lesions in patients with a strong family history of pancreatic cancer. As the pancreata were removed early, before an invasive neoplasm developed, they also provide a unique opportunity to study early relatively unmodified precursor lesions in the pancreas.
MATERIALS AND METHODS
Patients
The National Familial Pancreas Tumor Registry (NFPTR) was established at The Johns Hopkins Medical Institutions in 1994.31 As of January 1, 2006, 1545 kindreds had enrolled in this registry, including 632 kindreds in which at least a pair of first-degree relatives had been diagnosed with pancreatic cancer (“familial” pancreatic cancer kindreds) and 913 kindreds in which a family member had been diagnosed with pancreatic cancer, but there were no pairs of affected first-degree relatives (“sporadic” pancreatic cancer kindreds).33,51 Two screening studies of asymptomatic members of the familial pancreatic cancer kindreds have recently been completed. 7,8 Participants in these studies had to have 3 or more blood relatives with pancreatic cancer, they had to have at least 1 first-degree relative with pancreatic cancer, and they had to be 40 years of age or older, or 10 years younger than the youngest family member with pancreatic cancer. Patients with the Peutz-Jeghers syndrome could also enroll in the screening studies as the Peutz-Jeghers syndrome has been associated with a significantly increased risk of pancreatic cancer.16,17 One hundred ten individuals were screened in these studies, called “Cancer of the Pancreas Screening Study 1 and 2” (CAPS 1 and CAPS 2). Thirty-eight patients were screened as part of CAPS 1 and 78 individuals were screened as part of CAPS 2.
Surgery was recommended for patients with a suspected pancreatic neoplasm, on the basis of the presence of a focal lesion such as a mass or cyst in one or more abnormal imaging tests (EUS, computed tomography or endoscopic retrograde pancreatography) and/or significantly atypical cells in EUS-guided fine needle aspirates from the pancreas. Serum tumor marker studies were universally normal at baseline and did not influence the decision to perform surgery.
Twelve of the individuals in CAPS 1 and CAPS 2 underwent surgical resection for radiologic evidence of early asymptomatic pancreatic neoplasia, 10 of whom had their surgery at The Johns Hopkins Hospital. One pancreas resected at Johns Hopkins was extensively frozen for molecular studies. The freezing and thawing of this case introduced significant histologic artifacts that made it unsuitable for detailed microscopic examination. This case was therefore not included in further analyses. Another patient who had surgery at Johns Hopkins had Peutz-Jeghers syndrome and a large intraductal papillary mucinous neoplasm (IPMN) that caused dramatic secondary changes in the adjacent pancreatic parenchyma. As our intent was to study unaltered early changes in the pancreas, this case also was not included in the analyses. Three of the 8 remaining patients included in this analysis had pancreatoduodenectomies and 5 had distal pancreatectomies. The clinical details of these patients have been reported elsewhere and are summarized in Table 1.7,8
TABLE 1.
Clinical Features
| Case | Age | Sex | Family History+ |
Preoperative Diabetes |
No. EUS Findings of Chronic Pancreatitis (Total of 9)++ |
Specific EUS Pancreatic Findings |
|---|---|---|---|---|---|---|
| 1 | 47 | M | 3 Affected | No | 2 | Dilated main PD, echogenic PD (no echogenic foci and strands) |
| 2 | 38 | F | 3 Affected | No | 2 | Echogenic foci and strands |
| 3 | 54 | M | 3 Affected | Yes | 2 | Echogenic foci and strands |
| 4 | 73 | M | 5 Affected | Yes | 3 | Echogenic foci and strands, lobularity |
| 5 | 39 | M | 4 Affected | Yes | 5 | Echogenic foci and strands, lobularity, echogenic PD, duct irregularity, single hypoechoic nodule |
| 6 | 72 | F | 3 Affected | No | 6 | Echogenic foci and strands, lobularity, dilated main PD, duct irregularity, side branch ectasia |
| 7 | 56 | F | 3 Affected | No | 5 | Echogenic foci and strands, lobularity, echogenic PD, duct irregularity |
| 8 | 57 | M | 3 Affected | No | 5 | Multiple echogenic foci and strands, lobularity, dilated main PD, echogenic PD |
It should be noted that, to date, only 1 of the patients screened in CAPS 1 and CAPS 2 has developed pancreatic cancer. This high-risk patient had a deleterious germline BRCA2 mutation, EUS changes of chronic pancreatitis, and a cystic lesion in the head of the pancreas that increased in size during follow-up. Surgery was recommended to this patient but the patient delayed surgery and presented 3 months later with biopsy-proven adenocarcinoma of the pancreas metastatic to the liver.
Microscopic Examination
The 8 pancreata were serially sectioned and submitted in their entirety for histologic examination.
Pancreatic Intraepithelial Neoplasia (PanIN)
PanIN was defined according to the currently accepted international nomenclature.21 Briefly, a PanIN is a microscopic papillary or flat, noninvasive epithelial neoplasm arising in a pancreatic duct. It is composed of columnar to cuboidal cells with varying amounts of mucin and degrees of cytologic and architectural atypia. PanINs usually involve ducts <5mm in diameter. PanINs were further subclassified into PanIN-1, PanIN-2 and PanIN-3 lesions as has been described.21
IPMN
IPMN was defined according to the current internationally accepted nomenclature for IPMNs.22,38 An IPMN is a grossly visible, noninvasive mucin-producing, predominantly papillary or rarely flat, epithelial neoplasm arising from the main pancreatic duct or branch ducts, with varying degrees of ductal dilatation. IPMNs were further classified as adenoma (mild dysplaia), moderate dysplasia, and marked dysplasia as has been described.22,38
The duct profiles and the number of duct profiles containing a PanIN lesion in each histologic section were counted. Only duct profiles likely to contain PanIN lesions were counted. The ducts had to be large enough to have a well-defined connective tissue layer by light microscopy. At the other extreme, larger duct profiles representing interlobular ducts were not counted, as proliferative lesions in these larger ducts are much more likely to be IPMNs than PanINs.21,22,38 The ducts counted were therefore larger than intercalated ducts but smaller than interlobular ducts.
In addition to the changes within the pancreatic ducts, the parenchymal lesions associated with the duct lesions were documented. Particular attention was paid to the pancreatic lobules associated with PanIN lesions and any associations between PanINs and parenchymal pathology were carefully documented.
Controls
Two age-matched controls were examined for each case. Controls were selected from patients who underwent surgical resection of a portion of their pancreas for serous cystadenoma (n=12), lymphoepithelial cyst (n=3), or a solid-pseudopapillary neoplasm (n=1) at our institution between February 1, 2005 and January 31, 2006. The controls were age matched with the cases such that the controls were within 5 years of age of their matched case. If a control older than the case was available, it was selected over controls younger than the case. As a result, 13 of the 16 controls were the same age or older than their matched case. When possible, the controls were also gender matched with the cases, such that 14 of the 16 controls were gender matched with their paired case.
KRAS Mutational Analysis
Selected PanINs and IPMNs were microdissected from formalin-fixed, paraffin-embedded tissue sections as has been described.18 The neoplastic cellularity of the microdissected PanINs was ~80%. DNA was isolated using the QiAamp DNeasy Tissue Kit (Qiagen, Valencia, CA). KRAS gene sequencing was performed using BigDye 1.0 and a 377XL capillary sequencer (ABI, Applied Biosystems, Foster City, CA).
Statistical Analyses
The number of PanIN profiles were compared between individuals who underwent whipple resection and those who underwent distal pancreatectomy using the Mann-Whitney rank sum test, and the percentage of ducts involved by PanINs in the cases was compared with the percentage in controls using Wilcoxon-rank sum. The Spearman rank correlation was used to determine the relationship between the percentage of ducts with PanIN lesions and the EUS score. Analyses were performed using STATA version 8.
RESULTS
Patients
The cases included 5 males and 3 females. They ranged in age from 38 to 73 years (mean 54.5 y). They had between 3 and 5 family members previously diagnosed with pancreatic cancer, and 3 of the 8 had been diagnosed clinically with diabetes mellitus. The EUS findings are summarized in Table 1. The controls included 8 males and 8 females. They ranged in age from 38 to 78 years (mean 56.3 y).
IPMNs
IPMNs were identified in 4 of the 8 cases. Two of the cases had 2 IPMNs each, and 2 of the cases had a single IPMN. These 6 IPMNs were all IPMN-adenoma (low-grade dysplasia) and none were associated with an invasive cancer. The 6 IPMNs ranged in size from 6mm to 1.5 cm. All 6 IPMNs were associated with adjacent areas of parenchymal atrophy. These areas were upstream from the IPMNs and characterized by acinar dropout, islet aggregation, fibrosis, and fatty replacement of the pancreatic parenchyma. These areas were not further evaluated in the studies correlating PanIN lesions with parenchymal changes.
PanIN
PanIN was identified in all 8 cases (Table 2). In the cases, a mean of 34 PanIN lesions (profiles) was identified per resected portion of pancreas. By contrast, the 16 controls included in the current study harbored a mean of only 1.9 PanIN profiles per surgically resected portion of pancreas. Similarly, in a previous study of 122 surgically resected pancreata with chronic pancreatitis (mean age 55 y) we found a mean of 3.3 PanIN profiles per surgically resected portion of pancreas.47
TABLE 2.
Pathologic Features
| Case | Surgery | IPMN | No. Duct Profiles | Total No PanIN Profiles (% of Total) | PanIN-1A | PanIN-1B | PanIN-2 | PanIN-3 | Relative Degree of Lobular Atrophy | Other Findings |
|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Whipple | IPMN-adenoma | 419 | 4 (1.0%) | 3 | 0 | 1 | 0 | Rare | Amyloid in islets |
| 2 | Distal panc. | — | 359 | 8 (2.2%) | 6 | 2 | 0 | 0 | None | Lymphocytic pancreatitis |
| 3 | Whipple | — | 282 | 9 (3.2%) | 4 | 3 | 2 | 0 | Rare | Serous cystadenoma, amyloid in islets |
| 4 | Distal panc. | — | 246 | 39 (15.9%) | 23 | 14 | 2 | 0 | Moderate numbers | Amyloid in islets, acinar nodules |
| 5 | Distal panc. | — | 385 | 40 (11.1%) | 30 | 8 | 2 | 0 | Multifocal, associated with PanINs | Acinar nodules |
| 6 | Distal panc. | Two IPMN-adenomas | 165 | 45 (27.3%) | 14 | 5 | 12 | 14 | Multifocal, associated with PanINs | Possible focus of microinvasion |
| 7 | Distal panc. | IPMN-adenoma | 328 | 47 (14.3%) | 14 | 32 | 1 | 0 | Multifocal, associated with PanINs | |
| 8 | Whipple | Two IPMN-adenomas | 356 | 81 (22.8%) | 23 | 50 | 8 | 0 | Multifocal, associated with PanINs | Atrophy, also associated with IPMNs |
panc. Indicates pancreatectomy.
The PanIN lesions identified in the 8 cases in the current study were mostly PanIN-1 and PanIN-2 lesions, but in 1 of the 8 pancreata the lesions reached the level of PanIN-3. The PanIN lesions in the cases were remarkably extensive, involving as many as 27.3% of the duct profiles in 1 of the pancreata (mean 10.7%, range 1.0% to 27.3%). There was no difference in the number of PanIN profiles in the head of the gland (mean 31.3 per resection specimen) as compared with the number in the tail of the gland (mean 35.8 per resection specimen) (P=0.65). The number of PanIN profiles in the cases increased with patient age (r=0.81, P<0.015).
PanIN lesions in the 16 age-matched controls were limited to PanIN-1 and PanIN-2 lesions and involved a mean of 1.9% of the duct profiles (range 0% to 9.2%). The percentage of ducts involved by PanIN lesions in the controls was significantly lower than in the 8 cases (P<0.01). No IPMNs were identified in the 16 controls.
KRAS Gene Analysis
Activating point mutations in the KRAS gene are early events in the development of pancreatic neoplasia.14 We microdissected precursor lesions (PanINs and IPMNs) from 6 of the pancreata. These were low-grade precursor lesions, including genotyped PanIN-1, PanIN-2, and IPMN-adenoma. In 2 of the 6 cases (cases 7 and 8) different KRAS gene mutations were found in different precursor lesions (Table 3). In 2 cases, 1 mutation was identified (cases 1 and 6). In case 4, two lesions were analyzable and both were wild type, and in case 5, only 1 lesion was analyzable and it was wild type. The presence of different KRAS gene mutations in different precursor lesions microdissected from the same pancreas suggests that these lesions are the product of multifocal disease, and not caused by a single large branching precursor lesion involving multiple ducts.
TABLE 3.
KRASGene Analyses
| Case | No. Foci Microdissected | No. Amplifiable Foci | IPMN-Adenoma | PanIN |
|---|---|---|---|---|
| 1 | 10 | 6 | MutAX3 | MutA(x2), wt(x1) |
| 4 | 5 | 2 | — | Wt(x2) |
| 5 | 6 | 1 | — | Wt(x1) |
| 6 | 11 | 8 | wtX2 | MutB, wt(x4) |
| 7 | 8 | 7 | — | MutC, MutD(x2), wt(x4) |
| 8 | 13 | 10 | MutE, wtX2 | MutE, MutC(x3), wt(x3) |
A indicates codon 13 GGC→GAC; B, codon 12 GGT→GCT; C, codon 12 GGT→CGT; D, codon 13 GGC→GTC/GCC; E, codon 12 GGT→GAT; Mut, mutant; wt, wild type at both codons 12 and 13; x, number of cases.
Parenchymal Changes Associated With PanINs
The pancreatic parenchyma not associated with PanINs or IPMNs was histologically unremarkable (Fig. 1A). The lobular units were well defined and generally free of inflammation. The acinar cells were of normal size with abundant apical granular cytoplasm, and they formed barely perceptible lumina.
FIGURE 1.

Normal pancreatic lobule (A) and progressive lobular atrophy (B–F). The earliest changes included a mild dilatation of the acinar lumina associated with a thinning of the acinar cells and a subtle loss of acinar granularity (B). More significant lobular atrophy included a considerable loss of acinar cells such that only a few cells with granular eosinophilic cytoplasm remained (C–E). Eventually no acinar cells, and in some lobules no ductal cells, remained (F).
A spectrum of parenchymal atrophy was associated with the PanINs in the cases (Figs. 1B–F). These atrophic changes had a remarkable predisposition to involve lobular units such that the changes in a single lobular unit were uniform, and a lobule with marked atrophy could be adjacent to a histologically normal lobule. The subtlest changes included a thinning of the acinar cells with a loss of the apical granular cytoplasm and slight dilatation of the acinar lumina (Fig. 1B). Other lobular units had a more significant loss of acinar cells such that only a few cells with granular eosinophilic cytoplasm remained (Figs. 1C, D). These were usually in continuity with small ducts such that it was difficult to distinguish between ductal cells and acinar cells. The small ducts themselves were often prominent and occasionally formed tortuous cords of small ducts toward the center of the residual lobular unit (Fig. 1E).
The most dramatic lobular changes in the cases included marked acinar dropout such that essentially no acinar cells remained (Fig. 1F). These lobular units were instead composed of a central, slightly dilated duct surrounded by aggregates of islets of Langerhans embedded in fibro-fatty connective tissue.
These atrophic changes were often associated with the PanINs (Figs. 2A–D). In 4 of the 8 pancreata (cases 5, 6, 7, and 8) there was an almost one-to-one association between PanIN lesions and atrophy of the lobular unit surrounding the duct with the PanIN (Figs. 2A–D). This association was present, but not as striking as in another pancreas (case 4) and only rarely observed in the 3 pancreata with the less than 10 PanIN profiles (cases 1, 2, and 3). The association of lobular atrophy with PanINs was even seen with flat PanINs without significant cytologic or architectural atypia (PanIN-1A).
FIGURE 2.

In most of the pancreata there was a dramatic association between PanIN and the lobular atrophy (A–D). For example, in (A) notice the normal lobules associated with a normal duct (top) contrasting with the dramatic atrophy of the 3 lobular units associated with PanIN lesions involving 3 branching ducts toward the center and lower half of the field. Similar associations between PanINs and lobular atrophy can be appreciated in (B–D).
A similar, albeit less dramatic, association was noted in the 16 controls between PanIN lesions and lobular parenchymal atrophy (Fig. 3). The association between nonfamilial PanINs and lobular parenchymal atrophy has been reported previously13 and was seen even with PanIN-1A lesions (Fig. 3).
FIGURE 3.

Low-magnification image of a PanIN, in a patient without a family history, associated with lobular parenchymal atrophy (A). High-magnification view of a PanIN-1A lesion in a control associated with lobular atrophy (B). Note the few remaining acini associated with the duct, the fibrosis, and the aggregation of islets. Low-magnification and high-magnification views of a PanIN-1A lesion in a control associated with lobular atrophy (C and D).
Other Changes in the Cases
Amyloid deposition in the islets of Langerhans was seen in 3 of the 8 cases, 2 of whom were clinically diagnosed with type II diabetes mellitus before surgery. Two cases had acinar nodules in the pancreas, 1 case had a small abscess presumably from a previous biopsy, and 1 pancreas had a mixed inflammatory cell infiltrate. This mixed inflammatory cell infiltrate, in contrast to the atrophic changes described earlier, did not follow a lobular pattern, but instead, inflammatory cells extended across lobules. Inflammatory pancreatitis was not present or, at most, was only minimal in the other cases.
PanIN Versus EUS
As previously reported,8 EUS changes were graded from 0 to 9, with 1 point each given for echogenic foci, echogenic strands, lobularity, main ductal dilation, visible side branches, echogenic duct walls, duct irregularity, cysts, and stones.9,20,48,53 The Spearman correlation between the percentage of ducts with PanIN lesions in the 8 cases (Table 2) and the EUS score (Table 1) was 0.85 (P<0.007). These results are shown in Figure 4.
FIGURE 4.
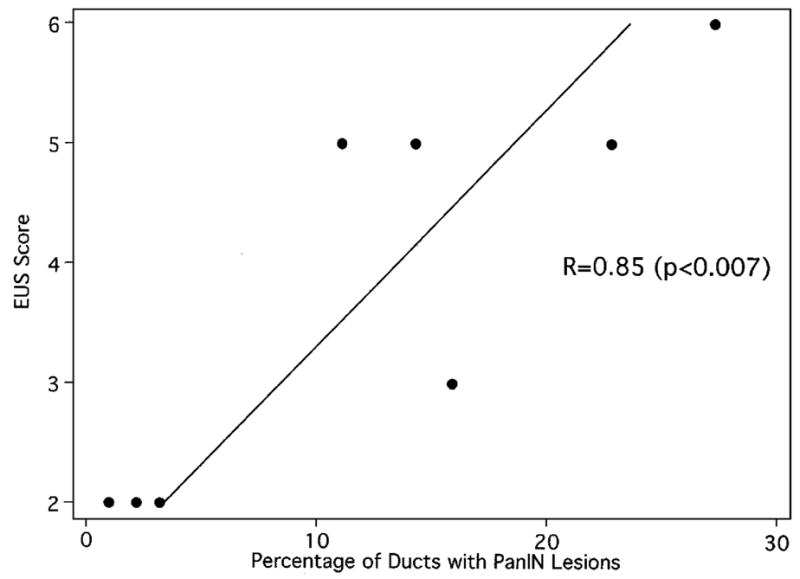
Percentage of ducts with PanIN lesions versus the EUS score for the 8 cases included in this analysis.
DISCUSSION
We describe 2 striking histologic changes in pancreata surgically resected from patients with a strong family history of pancreatic cancer. First, these pancreata are remarkable for multiple precursor lesions, including PanIN lesions, involving as many as 27.3% of the duct profiles in a single distal pancreatectomy. Second, the precursor lesions in these pancreata, even the low-grade PanIN-1 lesions, were often directly associated with lobular atrophy of the surrounding pancreatic parenchyma. The latter association was also present, although much more focally, in the controls.
The number of PanINs identified in the cases with a strong family history of pancreatic cancer was remarkably high. All 8 cases contained PanINs, a total of 273 PanIN profiles were identified in the cases (mean 34 PanIN profiles per resected portion of pancreas), and the density of PanIN profiles in the cases was significantly greater than that observed in age-matched controls (P<0.01). These multiple PanIN profiles represent multifocal disease as demonstrated by the distinct KRAS gene mutations identified in separately microdissected precursor lesions from 2 of the patients.
The finding of such a high density of multifocal precursor lesions in a single pancreas suggests that the portion of the gland not resected in these individuals retains a significantly increased risk of developing an invasive pancreatic cancer. Although one might argue that these patients would benefit from a total pancreatectomy, the benefit of such surgery is unproven, whereas the risks are real. A patient has been reported who died after a prophylactic total pancreatectomy, and all patients who undergo a total pancreatectomy develop brittle diabetes mellitus.6 Therefore, short of total pancreatectomy, our recommendation for the patients in this series with multiple precursor lesions has been continued surveillance of the pancreatic remnant, using EUS and CT or magnetic resonance cholangiopancreatography.
The distinctive pattern of lobular atrophy associated with PanINs observed in the pancreata in this study has also been reported focally in elderly patients by Detlefsen et al13 and diffusely in patients with a strong family history of pancreatic cancer by Meckler et al.41 Meckler et al41 concluded that the “dysplasia appeared to develop within the microcystically dilated intralobular glands and ducts.” This conclusion suggested that the familial pancreatic cancer gene could be a pancreatitis-inducing or cyst-inducing gene. When compared to findings in animal models, however, the morphology of lobular atrophy observed in all 3 series of patients suggests that it is the epithelial precursor lesions that are causing the parenchymal atrophy and duct dilatation. 13 Surgical obstruction of the pancreatic duct in animal models causes a stereotypic series of changes in the pancreas and these are the very same alterations identified in the pancreata in all 3 series of patients.2,4,5,10,30,35,44,52,54
The first change after experimental ligation of the pancreatic duct in animal models is a thinning of the acinar cells with a loss of the apical granular cytoplasm and retention of the basal basophilic cytoplasm. 10,44,54 This acinar cell thinning is associated with slight dilatation of the acinar lumina (compare with Fig. 1B). The acinar cells then die through the processes of necrosis and apoptosis.19,52 Acinar cell loss progresses over time until only a few cells with granular eosinophilic cytoplasm remain, and eventually it is difficult to distinguish between ductal cells and acinar cells (compare with Figs. 1C–E). In the end stages, there is a marked acinar dropout such that essentially no acinar cells remain (compare with Fig. 1F). The lobular units are then composed of a central, slightly dilated duct surrounded by aggregates of islets of Langerhans and embedded in fibro-fatty connective tissue.30 The histologic changes reported here (Fig. 1), by Detlefsen et al, and by Meckler et al are essentially phenocopies of these features seen in animal models of pancreatic duct ligation.41 It therefore seems that it is not the atrophy that is causing the PanINs to develop, but rather that it is the PanINs which develop first, producing multiple foci of small duct obstruction, in turn progressing to multifocal lobulocentric atrophy.13
The initial event in some forms of familial pancreatic cancer therefore seems to be the development of multiple intraductal neoplasms (PanINs and IPMNs). By analogy with FAP, this pattern of epithelial neoplasia suggests that some cases of familial pancreatic cancer are caused by mutations in a gatekeeper gene.27,28 Gatekeeper genes are genes that directly regulate the emergence of incipient neoplasms by exerting control over stem cell numbers,34 promoting senescence, or perhaps by inhibiting cell proliferation or promoting cell death. In the original gatekeeper models of neoplasia,24,25,49 only one mutation, in addition to the germline mutation, is needed to initiate neoplasia. If an inherited mutation in a gatekeeper gene is recessive, the lesions are focal, rather than diffuse, and the second hit occurs in the second allele of the gatekeeper gene. Examples of gatekeeper genes include the RB, VHL, NF1, MEN1, and APC genes. Inactivation of a gatekeeper gene tends to lead to a very specific tissue distribution of cancer, and individuals with an inherited mutation in a gatekeeper gene have as much as a 103 increased risk of developing cancer.27,28 The cases in this study with multiple precursor lesions thus reasonably fit the characteristics of inherited mutations in a recessive gatekeeper gene.
The striking association we observed between precursor lesions (PanINs and IPMNs) and obstructive changes in the pancreatic parenchyma correlates with chronic pancreatitis-like changes observed radiologically in these patients (Fig. 4). Chronic pancreatitis-like changes, including chronic pancreatitis-like abnormalities of the ducts (ectasia, irregularity, saccules) and parenchyma (heterogeneity, lobularity), were observed by EUS and by endoscopic retrograde cholangiopancreatography (ERCP) in two-thirds of the 109 patients screened in CAPS 1 and CAPS 2.7,8 These changes correlate with markers of neoplasia including abnormal DNA methylation in the pancreatic juice,40 and were significantly more common and more severe in individuals from familial pancreatic cancer kindreds than in controls, even after adjusting for age and alcohol exposure.8 Similar changes have also been reported by others in patients with a strong family history of pancreatic cancer.6 Brentnall et al6 screened 14 patients with a strong family history of pancreatic cancer using a combination of EUS, ERCP, and spiral computed tomography. Nine of these 14 were found to have heterogenous pancreatic parenchyma on EUS and 8 had ERCP changes including irregular and poor duct filling, and ectactic ducts with saccules. The histologic changes of lobular parenchymal atrophy we describe correlate with the heterogenous pancreatic parenchyma seen on EUS, whereas the precursor lesions themselves, particularly the small IPMNs, explain the irregular and poor duct filling, and the ectactic ducts seen by ERCP.
These observations therefore provide a morphologic basis to an approach to screening for early pancreatic neoplasia, particularly multifocal intraductal precursors of the type seen in familial pancreatic cancer. Multifocal PanINs and IPMNs will produce multiple foci of atrophy and cyst formation scattered in a background of intact pancreatic parenchyma. This heterogeneous pattern of atrophy will produce, as observed in the current study, and in the patients reported by Brentnall et al, a heterogeneous pattern on EUS. This heterogeneity can be quantified using the standard deviation of the gray-scale distribution (pixel densities) of the B-mode sonographic image.43 Indeed, Morita et al reported that the mean brightness and standard deviation derived from the gray-scale histogram increase after pancreatic duct ligation in dogs.43 Simply put, although the precursor lesions may be too small to visualize by currently available imaging technologies, the effects they produce on the pancreatic parenchyma can be detected and quantified.
Although the data do not allow us to draw conclusions about the optimal age at which to begin screening high-risk patients for pancreatic cancer, the observation that the number of PanIN profiles in the cases increased significantly with patient age (r=0.81, P<0.015) suggests that age should be strongly factored into any screening program.
At first glance it would seem that intraductal precursor lesions produce the lobular parenchymal atrophy by physically obstructing the affected duct. The atrophic changes were, however, observed even in association with flat, low-grade lesions (Fig. 3). The epithelial cells of the lesions clearly do not obstruct the lumen, suggesting that other mechanisms may contribute to the atrophy observed. For example, mucins and aquaporins are known to be abnormally expressed in PanINs, and their expression could alter the viscosity of intraluminal secretions causing a relative reduction in their flow.26,37,45,46 Alternatively, the precursor lesions could aberrantly express proteins which prematurely activate proteases such as trypsinogen.45 The premature activation of trypsinogen has been shown to play a role in the development of obstructive atrophy in the pancreas,42 and PanINs are known to ectopically express a number of proteins.45 For example, the expression of even a small amount of enterokinase on the surface of PanINs could prematurely activate trypsinogen, initiating a cascade of enzyme activation that leads to atrophy.
Finally, our findings suggest that a vicious cycle may develop in the pancreata of patients with a strong family history of pancreatic cancer. We have shown that the precursor lesions cause lobular chronic pancreatitis. The resultant episodes of tissue injury and repair may disrupt homeostatic mechanisms that govern stem cell regulation, furthering neoplastic progression, which would in turn produce more lobular chronic pancreatitis, continuing the vicious cycle.3,11,23
In summary, some patients with a strong family history of pancreatic cancer develop numerous noninvasive epithelial precursor lesions including PanINs and IPMNs. These lesions are associated with lobular atrophy of the pancreatic parenchyma. The multifocality of the precursor lesions suggests that an inherited mutation of a gatekeeper gene is responsible for some cases of familial pancreatic cancer. The lobular atrophy associated with these precursor lesions provides an explanation for the chronic pancreatitis-like changes seen in these pancreata, and the patchy nature of this atrophy suggests an approach for screening for early pancreatic neoplasia in at-risk individuals.
Acknowledgments
Supported by the NIH SPORE (Specialized Programs of Research Excellence) in Gastrointestinal Cancer Grant CA62924, a grant from the National Cancer Institute (R01 CA97075), the Michael Rolfe Foundation for Pancreatic Cancer Research, and the Goldman Family.
References
- 1.Amundadottir LT, Thorvaldsson S, Gudbjartsson DF, et al. Cancer as a complex phenotype: pattern of cancer distribution within and beyond the nuclear family. PLoS Med. 2004;1:e65. doi: 10.1371/journal.pmed.0010065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Arnozan V. Contribution a l’Etude du Pancreas du Lapin. Lesions provoquees par la ligature du canal de Wirsung. Arch Physiol Norm Pathol. 1884;3:287–316. [Google Scholar]
- 3.Beachy PA, Karhadkar SS, Berman DM. Tissue repair and stem cell renewal in carcinogenesis. Nature. 2004;432:324–331. doi: 10.1038/nature03100. [DOI] [PubMed] [Google Scholar]
- 4.Boerma D, Straatsburg IH, Offerhaus GJ, et al. Experimental model of obstructive, chronic pancreatitis in pigs. Dig Surg. 2003;20:520–526. doi: 10.1159/000073688. [DOI] [PubMed] [Google Scholar]
- 5.Boquist L, Edström C. Ultrastructure of pancreatic acinar and islet parenchyma in rats at various intervals after duct ligation. Virchows Arch Abt A Path Anat. 1970;349:69–79. doi: 10.1007/BF00548522. [DOI] [PubMed] [Google Scholar]
- 6.Brentnall TA, Bronner MP, Byrd DR, et al. Early diagnosis and treatment of pancreatic dysplasia in patients with a family history of pancreatic cancer. Ann Intern Med. 1999;131:247–255. doi: 10.7326/0003-4819-131-4-199908170-00003. [DOI] [PubMed] [Google Scholar]
- 7.Canto MI, Goggins M, Yeo CJ, et al. Screening for pancreatic neoplasia in high-risk individuals: an EUS-based approach. Clin Gastroenterol Hepatol. 2004;2:606–621. doi: 10.1016/s1542-3565(04)00244-7. [DOI] [PubMed] [Google Scholar]
- 8.Canto MI, Goggins M, Hruban RH, et al. Screening for early pancreatic neoplasia in high-risk individuals: a prospective controlled study. Clin Gastroenterol Hepatol. 2006 doi: 10.1016/j.cgh.2006.02.005. (Epub ahead of print) [DOI] [PubMed] [Google Scholar]
- 9.Catalano MF, Lahoti S, Geenen JE, et al. Prospective evaluation of endoscopic ultrasonography, endoscopic retrograde pancreatography, and secretin test in the diagnosis of chronic pancreatitis. Gastrointest Endosc. 1998;48:11–17. doi: 10.1016/s0016-5107(98)70122-1. [DOI] [PubMed] [Google Scholar]
- 10.Churg A, Richter WR. Early changes in the exocrine pancreas of the dog and rat after ligation of the pancreatic duct. A light and electron microscopic study. Am J Pathol. 1971;63:521–546. [PMC free article] [PubMed] [Google Scholar]
- 11.DeMarzo AM, DeWeese TL, Platz EA, et al. Pathological and molecular mechanisms of prostate carcinogenesis: implications for diagnosis, detection, prevention, and treatment. J Cell Biochem. 2004;91:459–477. doi: 10.1002/jcb.10747. [DOI] [PubMed] [Google Scholar]
- 12.Dershaw DD, Abramson A, Kinne DW. Ductal carcinoma in situ: mammographic findings and clinical implications. Radiology. 1989;170:411–415. doi: 10.1148/radiology.170.2.2536185. [DOI] [PubMed] [Google Scholar]
- 13.Detlefsen S, Sipos B, Feyerabend B, et al. Pancreatic fibrosis associated with age and ductal papillary hyperplasia. Virchows Arch. 2005;447:800–805. doi: 10.1007/s00428-005-0032-1. [DOI] [PubMed] [Google Scholar]
- 14.DiGiuseppe JA, Hruban RH, Offerhaus GJ, et al. Detection of K-ras mutations in mucinous pancreatic duct hyperplasia from a patient with a family history of pancreatic carcinoma. Am J Pathol. 1994;144:889–895. [PMC free article] [PubMed] [Google Scholar]
- 15.Elkin EB, Hudis C, Begg CB, et al. The effect of changes in tumor size on breast carcinoma survival in the U.S.: 1975–1999. Cancer. 2005;104:1149–1157. doi: 10.1002/cncr.21285. [DOI] [PubMed] [Google Scholar]
- 16.Giardiello FM, Welsh SB, Hamilton SR, et al. Increased risk of cancer in the Peutz-Jeghers syndrome. N Engl J Med. 1987;316:1511–1514. doi: 10.1056/NEJM198706113162404. [DOI] [PubMed] [Google Scholar]
- 17.Giardiello FM, Brensinger JD, Tersmette AC, et al. Very high risk of cancer in familial Peutz-Jeghers Syndrome. Gastroenterology. 2000;119:1447–1153. doi: 10.1053/gast.2000.20228. [DOI] [PubMed] [Google Scholar]
- 18.Goggins M, Hruban RH, Kern SE. BRCA2 is inactivated late in the development of pancreatic intraepithelial neoplasia: evidence and implications. Am J Pathol. 2000;156:1767–1771. doi: 10.1016/S0002-9440(10)65047-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gukovskaya AS, Perkins P, Zaninovic V, et al. Mechanisms of cell death after pancreatic duct obstruction in the opossum and the rat. Gastroenterology. 1996;110:875–884. doi: 10.1053/gast.1996.v110.pm8608898. [DOI] [PubMed] [Google Scholar]
- 20.Hollerbach S, Klamann A, Topalidis T, et al. Endoscopic ultrasonography (EUS) and fine-needle aspiration (FNA) cytology for diagnosis of chronic pancreatitis. Endoscopy. 2001;33:824–831. doi: 10.1055/s-2001-17337. [DOI] [PubMed] [Google Scholar]
- 21.Hruban RH, Adsay NV, Albores-Saavedra J, et al. Pancreatic intraepithelial neoplasia: a new nomenclature and classification system for pancreatic duct lesions. Am J Surg Pathol. 2001;25:579–586. doi: 10.1097/00000478-200105000-00003. [DOI] [PubMed] [Google Scholar]
- 22.Hruban RH, Takaori K, Klimstra DS, et al. An illustrated consensus on the classification of pancreatic intraepithelial neoplasia and intraductal papillary mucinous neoplasms. Am J Surg Pathol. 2004;28:977–987. doi: 10.1097/01.pas.0000126675.59108.80. [DOI] [PubMed] [Google Scholar]
- 23.Jensen JN, Cameron E, Garay MV, et al. Recapitulation of elements of embryonic development in adult mouse pancreatic regeneration. Gastroenterology. 2005;128:728–741. doi: 10.1053/j.gastro.2004.12.008. [DOI] [PubMed] [Google Scholar]
- 24.Kern SE. Clonality: more than just a tumor-progression model. J Natl Cancer Inst. 1993;85:1020–1021. doi: 10.1093/jnci/85.13.1020. [DOI] [PubMed] [Google Scholar]
- 25.Kern SE. Whose hypothesis? Ciphering, sectorials, D lesions, freckles and the operation of Stigler’s Law. Cancer Biol Ther. 2002;1:571–581. doi: 10.4161/cbt.1.5.225. [DOI] [PubMed] [Google Scholar]
- 26.Kim GE, Bae HI, Park HU, et al. Aberrant expression of MUC5AC and MUC6 gastric mucins and sialyl Tn antigen in intraepithelial neoplasms of the pancreas. Gastroenterology. 2002;123:1052–1060. doi: 10.1053/gast.2002.36018. [DOI] [PubMed] [Google Scholar]
- 27.Kinzler KW, Vogelstein B. Gatekeepers and caretakers. Nature. 1997;386:661–669. doi: 10.1038/386761a0. [DOI] [PubMed] [Google Scholar]
- 28.Kinzler KW, Vogelstein B. Familial cancer syndromes: the role of caretakers and gatekeepers. In: Vogelstein B, Kinzler KW, editors. The Genetic Basis of Human Cancer. New York: McGraw-Hill; 1998. pp. 241–242. [Google Scholar]
- 29.Kinzler KW, Nilbert MC, Su LK, et al. Identification of FAP locus genes from chromosome 5q21. Science. 1991;253:661–669. doi: 10.1126/science.1651562. [DOI] [PubMed] [Google Scholar]
- 30.Kirkbride MB. The Islands of Langerhans after ligation of the pancreatic ducts. J Exp Med. 1912;15:101–105. doi: 10.1084/jem.15.1.101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Klein AP, Hruban RH, Brune KA, et al. Familial pancreatic cancer. Cancer J. 2001;7:266–273. [PubMed] [Google Scholar]
- 32.Klein AP, Beaty TH, Bailey-Wilson JE, et al. Evidence for a major gene influencing risk of pancreatic cancer. Genet Epidemiol. 2002;23:133–149. doi: 10.1002/gepi.1102. [DOI] [PubMed] [Google Scholar]
- 33.Klein AP, Brune KA, Petersen GM, et al. Prospective risk of pancreatic cancer in familial pancreatic cancer kindreds. Cancer Res. 2004;64:2634–2638. doi: 10.1158/0008-5472.can-03-3823. [DOI] [PubMed] [Google Scholar]
- 34.Korinek V, Barker N, Moerer P, et al. Depletion of epithelial stem-cell compartments in the small intestine of mice lacking Tcf-4. Nat Genet. 1998;19:379–383. doi: 10.1038/1270. [DOI] [PubMed] [Google Scholar]
- 35.Laguesse MME, Gontier de la Roche A. Les Ilots de Langerhans dans Pancreas. CR Soc Biol. 1902;54:854–857. [Google Scholar]
- 36.Leach FS, Nicolaides NC, Papadopoulos N, et al. Mutations of a mutS homolog in hereditary nonpolyposis colorectal cancer. Cell. 1993;75:1215–1236. doi: 10.1016/0092-8674(93)90330-s. [DOI] [PubMed] [Google Scholar]
- 37.Levi E, Klimstra DS, Andea A, et al. MUC1 and MUC2 in pancreatic neoplasia. J Clin Pathol. 2004;57:456–462. doi: 10.1136/jcp.2003.013292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Longnecker DS, Adler G, Hruban RH, et al. Intraductal papillary-mucinous neoplasms of the pancreas. In: Hamilton SR, Aaltonen LA, editors. World Health Organization Classification of Tumours. Pathology and Genetics of Tumours of the Digestive System. Lyon: IARC Press; 2000. pp. 237–240. [Google Scholar]
- 39.Lynch HT, Smyrk T, Kern SE, et al. Familial pancreatic cancer: a review. Semin Oncol. 1996;23:251–275. [PubMed] [Google Scholar]
- 40.Matsubayashi H, Canto M, Sato N, et al. DNA methylation alterations in the pancreatic juice of patients with suspected pancreatic disease. Gastroenterology. 2005;66:1208–1217. doi: 10.1158/0008-5472.CAN-05-2664. [DOI] [PubMed] [Google Scholar]
- 41.Meckler KA, Brentnall TA, Haggitt RC, et al. Familial fibrocystic pancreatic atrophy with endocrine cell hyperplasia and pancreatic carcinoma. Am J Surg Pathol. 2001;25:1047–1053. doi: 10.1097/00000478-200108000-00009. [DOI] [PubMed] [Google Scholar]
- 42.Mooren FC, Hlouschek V, Finkes T, et al. Early changes in pancreatic acinar cell calcium signaling after pancreatic duct obstruction. J Biol Chem. 2003;278:9361–9369. doi: 10.1074/jbc.M207454200. [DOI] [PubMed] [Google Scholar]
- 43.Morita Y, Takiguchi M, Yasuda J, et al. Endoscopic ultrasonographic findings of the pancreas after pancreatic duct ligation in the dog. Vet Radiol Ultrasound. 1998;39:557–562. doi: 10.1111/j.1740-8261.1998.tb01651.x. [DOI] [PubMed] [Google Scholar]
- 44.Pound AW, Walker NI. Involution of the pancreas after ligation of the pancreatic ducts. I: a histological study. Br J Exp Pathol. 1981;62:547–558. [PMC free article] [PubMed] [Google Scholar]
- 45.Prasad NB, Biankin AV, Fukushima N, et al. Gene expression profiles in pancreatic intraepithelial neoplasia reflect the effects of Hedgehog signaling on pancreatic ductal epithelial cells. Cancer Res. 2005;65:1619–1626. doi: 10.1158/0008-5472.CAN-04-1413. [DOI] [PubMed] [Google Scholar]
- 46.Ringel J, Lohr M. The MUC gene family: their role in diagnosis and early detection of pancreatic cancer. Mol Cancer. 2003;2:9. doi: 10.1186/1476-4598-2-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rosty C, Geradts J, Sato N, et al. p16 Inactivation in pancreatic intraepithelial neoplasias (PanINs) arising in patients with chronic pancreatitis. Am J Surg Pathol. 2003;27:1495–1501. doi: 10.1097/00000478-200312000-00001. [DOI] [PubMed] [Google Scholar]
- 48.Sahai AV, Zimmerman M, Aabakken L, et al. Prospective assessment of the ability of endoscopic ultrasound to diagnose, exclude, or establish the severity of chronic pancreatitis found by endoscopic retrograde cholangiopancreatography. Gastrointest Endosc. 1998;48:18–25. doi: 10.1016/s0016-5107(98)70123-3. [DOI] [PubMed] [Google Scholar]
- 49.Sidransky D. Is human patched the gatekeeper of common skin cancers? Nat Genet. 1996;14:7–8. doi: 10.1038/ng0996-7. [DOI] [PubMed] [Google Scholar]
- 50.Tabar L, Vitak B, Chen HH, et al. The Swedish Two-County Trial twenty years later. Updated mortality results and new insights from long-term follow-up. Radiol Clin North Am. 2000;38:625–651. doi: 10.1016/s0033-8389(05)70191-3. [DOI] [PubMed] [Google Scholar]
- 51.Tersmette AC, Petersen GM, Offerhaus GJ, et al. Increased risk of incident pancreatic cancer among first-degree relatives of patients with familial pancreatic cancer. Clin Cancer Res. 2001;7:738–744. [PubMed] [Google Scholar]
- 52.Walker NI. Ultrastructure of the rat pancreas after experimental duct ligation. I. The role of apoptosis and intraepithelial macrophages in acinar cell deletion. Am J Pathol. 1987;126:439–451. [PMC free article] [PubMed] [Google Scholar]
- 53.Wiersema MJ, Hawes RH, Lehman GA, et al. Prospective evaluation of endoscopic ultrasonography and endoscopic retrograde cholangiopancreatography in patients with chronic abdominal pain of suspected pancreatic origin. Endoscopy. 1993;25:555–564. doi: 10.1055/s-2007-1010405. [DOI] [PubMed] [Google Scholar]
- 54.Zeligs JD, Jano A, Dumont AE. The course and nature of acinar cell death following pancreatic ligation in the guinea pig. Am J Pathol. 1975;80:203–226. [PMC free article] [PubMed] [Google Scholar]