

Abstract
Phospholipase A2 (PLA2) hydrolyzes the sn-2 position of cell membrane phospholipids to release fatty acids and lysophospholipids. We have previously reported that group V secretory PLA2 (sPLA2) translocates from the Golgi and recycling endosomes of mouse peritoneal macrophages to newly formed phagosomes and regulates the phagocytosis of zymosan, suggesting a role in innate immunity. Here we report that in macrophages lacking group V sPLA2 phagosome maturation was reduced 50–60% at early time points while the binding of zymosan was unimpaired. The ability of group V sPLA2 to regulate phagocytosis extended to phagocytosis of IgG- and complement-opsonized sheep red blood cells. Moreover, macrophages lacking group V sPLA2 had delays in phagocytosis, phagosome maturation and killing of Candida albicans. Cytokine production and eicosanoid generation were not impaired by the lack of group V sPLA2. Furthermore, in a model of systemic candidiasis, mice lacking group V sPLA2 had an increased fungal burden in the kidney, liver and spleen at day 7 post-infection and increased mortality. Thus, group V sPLA2 regulates phagocytosis through major phagocytic receptors and contributes to the innate immune response against Candida albicans by regulating phagocytosis and killing through a mechanism that is likely dependent on phagolysosome fusion.
Keywords: phagocytosis, macrophages, fungal infection, phagolysosome fusion, Phospholipase A2
Introduction
Phagocytosis, the process whereby phagocytic cells internalize large particles, is a key component of the innate immunity (1). The mechanisms of phagocytosis depend on several variables including the type of phagocytic cell, the nature of the stimulus, the number and types of receptors involved, and the ability of certain microorganisms to subvert the phagocytic process. Nevertheless, whatever the stimulus, phagocytosis involves three well characterized steps, namely, binding of targets, formation of a phagocytic cup with subsequent particle uptake, and maturation of the phagosome by fusion with endosomal and lysosomal components to form the phagolysosome responsible for killing of pathogens (2).
Zymosan is a particle derived from the cell wall of Saccharomyces Cerevisiae that has long been used as a model system to study phagocytosis. Zymosan, is composed of carbohydrate polymers such as mannans and β-glucans and can bind a variety of receptors, such as dectin-1, specific ICAM-3 grabbing non integrin related 1 (SIGNR1), complement receptor 3 (CR3) and the mannose receptor (3, 4). Dectin-1 is the main β-glucan receptor (5) and has been identified in many cell types including monocytes/macrophages, neutrophils, and dendritic cells (3, 6). In macrophages, dectin-1 is essential for binding and uptake of zymosan (7) and for reactive oxygen species (ROS)3 generation (8, 9) while zymosan-induced cytokine production needs the cooperative action of dectin-1 and TLR-2 (10).
Candida albicans (C. albicans) is an opportunistic pathogen that shares many signaling events with zymosan (8, 11). C. albicans is phagocytosed through opsonin and non-opsonin receptors. Upon internalization, it is killed by activation of the respiratory burst and by activation of lysosomal proteins, an oxygen-independent mechanism (12). Its ability to establish a disseminated infection depends on down regulation of both innate and acquired immune responses (13). Nevertheless, resistance to C. albicans infection is determined primarily by the ability of professional phagocytes to ingest and kill C. albicans (14).
Phospholipase A2 hydrolyzes the ester bond in the sn-2 position of cell membrane phospholipids to generate free fatty acids and lysophospholipids. When the free fatty acid is arachidonic acid, it serves as substrate for generation of eicosanoids, potent mediators of inflammation. There are over twenty mammalian species of PLA2. Of these, cytosolic PLA2α (cPLA2α) is absolutely required for arachidonic acid release and subsequent eicosanoid biosynthesis in response to various stimuli (15) such as zymosan (16). We have previously shown that group V secretory PLA2 (sPLA2) augments cPLA2α-dependent eicosanoid generation in freshly isolated mouse peritoneal macrophages after phagocytosis of zymosan (17). Using cultured peritoneal macrophages we showed that group V sPLA2 translocates to the forming phagosomes during phagocytosis of zymosan and regulates phagocytosis. This function was not shared with cPLA2α or with a related member of the sPLA2 family, group IIA sPLA2 (18).
Here we investigate the mechanism through which group V sPLA2 regulates phagocytosis, its contribution to phagocytosis and killing of Candida albicans, and its role in host defense against systemic candidiasis.
Materials and Methods
Materials
Zymosan A, paraformaldehyde, complement C5-deficient human serum, gelatin veronal buffer (GVB), and bovine serum albumin (BSA) were from Sigma (St. Louis, MO). FUN-1 [2-chloro-4-(2,3-dihydro-3-methyl-(benzo-1,3-thiazol-2-yl)-methylidene)-1-phenylquinolinium iodide], and FITC-Zymosan were from Molecular Probes (Invitrogen, Carlsbad, CA). Sabouraud dextrose agar plates were from BD Diagnostic (Franklin Lakes, NJ). Other reagents were rat monoclonal IgG2b anti-dectin-1 (clone 2A11) (Serotec, Raleigh, NC); rat monoclonal IgG2a against lysosome-associated membrane protein (Lamp)-1 (BD Pharmigen, San Jose, CA); goat polyclonal IgG against cathepsin D and control goat IgG (Santa Cruz Biotechnology, Santa Cruz, CA); sheep red blood cells (sRBC), and rabbit IgG anti-sRBC (MP Biomedical, Solon, Ohio); rabbit IgM anti-sRBC (Fitzgerald Industries International, Concord, MA); Cy3-conjugated donkey anti-rabbit IgG and rabbit IgG (Jackon ImmunoResearch, West Grove, PA). Mouse recombinant group V sPLA2 was described previously (19).
Cell culture
Peritoneal Macrophages
Mice with disruption of the gene encoding group V sPLA2 (Pla2g5) (17) and wild type littermates, bred to a C57BL/6 background for 11 generations, were 4–6 months old. The use of mice for this study was reviewed and approved by the Animal Care and Use Committee of the Dana-Farber Cancer Institute (Boston, MA). Peritoneal cavities of mice were flushed with 5 mL of ice-cold RPMI (18). 1x105 cells were plated on sterile glass coverslips in 24-well tissue culture plates in culture medium (RPMI, 10% fetal bovine serum, 2 mM L-glutamine, 100 units/mL penicillin, and 100 μg/mL streptomycin, 10% non-essential amino acids, 10% Hepes, 10% Sodium pyruvate, 50 μM beta-mercaptoethanol, 10μg/mL gentamicin), and incubated overnight at 37°C with 5% CO2. After washing non-adherent cells, macrophages were cultured for an additional 24 hours (for a total of 48 h of culture), then washed, and stimulated.
Binding assay and expression of dectin-1
Peritoneal macrophages were preincubated for 30 min with 4μM cytochalasin D (Sigma) and then stimulated with zymosan at 10 particle per cell (ppc), in culture medium containing 4μM cytochalasin D for 15 min to 1 h. Cells were washed extensively to remove unbound zymosan, stained with Diff-Quick (Dade Behring Inc. Newark, DE) and visualized by light microscopy (18). The binding index was obtained by dividing the number of bound particles by the total number of cells in the field, multiplied by the percentage of cells binding at least two particles.
To analyze dectin-1 expression, peritoneal macrophages were cultured for 48 h on Petri dishes and harvested by incubation with PBS containing lidocaine (4mg/ml)/EDTA 5mM. Cells were fixed with 4% paraformaldehyde and blocked in HBA (Hanks’ balanced salt solution without Mg2+ or Ca2+ (HBSS--) containing 0.1% BSA) containing 10% normal donkey serum (blocking buffer) for 30 min. Macrophages were incubated with rat anti-dectin-1 (20 μg/mL) or control rat IgG (Jackson ImmunoResearch), in blocking buffer for 1 h, washed, and incubated for 1 h with allophycocyanin (APC)-conjugated donkey anti-rat IgG (1:200) (Jackson ImmunoResearch). All the steps were performed at room temperature. Alternatively cells were stained with APC-conjugated rat anti-mouse CD11b (BD Pharmigen) or APC-conjugated IgG2b and FITC-conjugated rat anti-mouse F4/80 or FITC-conjugated IgG2a (eBioeciences, San Diego, CA) (5 μg/mL). Flow cytometric analysis was performed on a FACSCanto flow cytometer, and data were analyzed using the FlowJo software.
Assessment of phagolysosome fusion
Peritoneal macrophages were stimulated with 2.5 ppc FITC-zymosan for 10 min to 1 h, fixed with 2% paraformaldehyde in PBS for 15 min, and permeabilized with 0.025% saponin in PBS for 10 min. Cells were washed, blocked in HBA containing 5% normal donkey serum (blocking buffer) for 1 h and incubated with rat mAb anti-Lamp-1 (2.5 μg/mL), or goat polyclonal anti-cathepsin D (10 μg/mL) in blocking buffer for 2 h. Negative control cells were incubated with rat IgG or goat IgG, respectively. After washing, cells were incubated for 1 h with Cy3-conjugated donkey anti-rat IgG (1:400) or FITC-conjugated donkey anti-goat IgG (Jackson Immunoresearch), washed and mounted in Vectashield™ mounting medium (Vector Laboratories, Burlingame, CA) (18). All the steps were performed at room temperature. In selected experiments macrophages were stimulated with C. albicans, stained for Lamp-1, and counterstained with Calcofluor white M2R (Sigma) (25 μM), a fluorochrome that binds the cell walls of fungi and can be visualized under UV light (20). Cells were imaged using a Nikon C1 confocal system combined with an Eclipse TE2000-U inverted microscope with 60X oil PlanApo NA 1.4 objective. 8–10 Z-stack images were acquired through a small pinhole, and each image (0.05 μm) was saved as a series of independent sections and as a merged stack image using Nikon EZ-C Gold version 3.40 build 691. Conventional Nomarski differential contrast images and epifluorescence images were taken using a SPOT-RT digital camera and data were analyzed using Photoshop software. The fusion index was obtained by analyzing each section individually and as merged images or epifluorescence images for accumulation of Lamp-1 (or cathepsin D) around the phagosomes (21) and by dividing the number of Lamp-1-surrounded phagosomes by the total number of cells in the field, multiplied by the percentage of cells showing at least one phagosome whose membrane was completely stained with Lamp-1.
Erythrocyte phagocytosis assay
For IgG-opsonization, sRBC (10% suspension) were washed in GVB, incubated for 20 min at 37°C with a subagglutinating concentration of rabbit IgG anti-sRBC (10 μg/mL) and washed again. Alternatively, to generate iC3b-coated erythrocytes, sRBC were incubated with rabbit IgM anti- sRBC (10 μg/mL) for 20 min at 37°C, washed and resuspended in GVB containing 10% C5-depleted human serum for 20 min at 37°C. During this incubation C3b binds IgM and is then converted to iC3b, the ligand of CR3 (22). For iC3b-opsonized sRBC, peritoneal macrophages were preincubated for 15 min with RPMI containing 150 nM phorbolmyristate acetate (PMA) without FBS, to activate CR3 on the surface of the macrophages (1, 2). The medium was then substitute by culture medium. IgG- or iC3b-opsonized sRBC (~10 sRBC/cell) were gently pelleted onto macrophages (30xg for 2 min), and incubated at 37°C in culture medium (23). At the indicated time (5 min to 1 h) glass cover-slips were taken off the plate, washed, and non-ingested sRBC were lysed with 0.2% NaCl followed by 1.6% NaCl. Coverslips were dried, fixed and stained with Diff-Quick. Phagosomes containing sRBC were counted by optical microscopy. The phagocytic index was calculated by dividing the number of phagosomes by the total number of cells in a field, which was multiplied by the percent of phagocytosing cells (18).
Phagocytosis and killing of Candida albicans
C. albicans (ATCC 10231) was streaked on Sabouraud agar plates and incubated for 48 h at 37°C. The plates were maintained at 4°C, and the day before the experiment one colony was streaked on a fresh plate and incubated at 37°C. C. albicans yeast were scraped from the plate, washed in endotoxin-free PBS (Sigma), and counted. C. albicans (m.o.i. 5) was gently pelleted (30xg) onto peritoneal macrophages and incubated for 5 min to 1 h at 37°C in 5% CO2. After washing to remove non-phagocytosed microorganisms, internalized yeast were counted as described (18). In selected experiments yeast were stained with FUN-1 (8) in glucose buffer (RPMI containing 2% glucose, 10 mM HEPES, 10μM FUN-1) to allow active metabolism. Macrophages stimulated with C. albicans were fixed with 2% paraformaldehyde for 15 min, washed and mounted in Vectashield™ mounting medium. To distinguish internalized yeast we used Nomarsky images, and to visualize dead organisms we used fluorescence microscopy (488 nm output) (Figure 4B). In another set of experiments time-dependent stimulation with C. albicans was followed by staining with Diff-Quick and visualization by optical microscopy (18).
In addition to using FUN-1, we measured killing of C. albicans using a serial dilution technique (24). Macrophages were challenged with live microorganisms for 15 min as described above, and washed to remove non-ingested C. albicans. Before incubation and after 1, 2, and 3 h of incubation at 37°C in 5% CO2 (20, 24, 25) the wells were scraped with a plastic paddle, and macrophages were lysed with 1 mL of endotoxin-fee water (Sigma) (25). To quantify the number of viable C. albicans, 100 μl of serially diluted samples were spread on Sabouraud plates and incubated at 37°C for 24 h. The percentage of yeast killed by the macrophages was determined as follows: [1-(colony forming units (CFU) after incubation/CFU recovered at the start of incubation)] x100 (24, 25).
Synchronized phagocytosis
In selected experiments to synchronize phagocytosis, peritoneal macrophages were washed in cold RPMI and cooled on ice for 5 min before the addition of C. albicans for 15 min on ice. Unbound C. albicans was removed by washing cells once with warm medium. Cells were then incubated at 37°C for the indicated periods of time, stained for Lamp-1 and cathepsin D and counterstained with Calcofluor white M2R.
Cytokine production and cysteinyl leukotriene generation
Peritoneal macrophages, were stimulated with C. albicans (m.o.i. 0.1 to 10) or zymosan (10 ppc) for 3 h. Supernatants were then harvested and assayed for TNF-α and cysteinyl leukotrienes (Cys-LTs) by commercial immunoassays (TNF-α, R&D Systems Minneapolis, MN; Cys-LTs, GE Healthcare, Piscataway, NJ).
In vivo models of systemic candidiasis
200 μl of 0.9 % NaCl containing 5x105 CFU of C. albicans (to evaluate fungal burden) or 1x106 (for the survival assay) were injected intravenously in Pla2g5-null and wild type mice. Mice were checked daily for 33 days. Subgroups of 3–5 mice were sacrificed 1, 3, and 7 days after injection. Heart, lungs, liver, spleen and left kidney were removed aseptically, weighed, and homogenized in 1 mL endotoxin-fee PBS in a tissue grinder. 100 μl of serially diluted suspensions were then plated on Sabouraud plates. After 24 h growth at 37°C, numbers of C. albicans colonies were counted and expressed as log CFU per gram of tissue (25). The right kidney was fixed with 4% formalin and embedded in paraffin, and serial sections were examined microscopically after staining with a combination of Periodic-Acid Schiff and hematoxylin-eosin (20).
Statistics
We performed two way analysis of variance using the GraphPad Prism 5 software to compare time-dependent phagocytosis between populations of peritoneal macrophages derived from Pla2g5-null mice and their wild type littermate. Differences in C. albicans colonization of tissues were analyzed by Mann-Whitney U test and survival by the log-rank test. Otherwise differences in outcomes were analyzed by Student’s t-test
Results
Reduced phagocytosis in Pla2g5-null macrophages is not due to decreased binding of zymosan
The first step in phagocytosis is the binding of the ligand to its cognate receptor. We first confirmed our previous finding (18) that Pla2g5-null macrophages have a defect in phagocytosis of zymosan (Fig. 1A). To assess whether the decrease in phagocytosis by Pla2g5-null macrophages was due to reduced binding of zymosan particles, we preincubated macrophages with Cytochalasin D to inhibit phagocytosis and then stimulated the cells with zymosan. Fig. 1B shows that Pla2g5-null macrophages bind zymosan as efficiently as wild type macrophages. Because the main phagocytic receptor for zymosan is dectin-1 (3, 7), we also examined dectin-1 expression by flow cytometry and found that Pla2g5-null and wild type peritoneal macrophages, identified by expression of F4/80 and CD11b, have similar expression of dectin-1 (Fig. 1C).
Figure 1.

Binding of zymosan and expression of Dectin-1. (A) Peritoneal macrophages isolated from wild type (black bars) and Pla2g5-null (open bars) mice were stimulated with zymosan (10 ppc) for 5 min to 1 h. Cells were fixed and stained with Diff-Quick. The phagocytic index was obtained as previously described (18). (B) Unopsonized zymosan was added to macrophages pre-treated with cytochalasin D (4μM) for 30 min and incubated at 37°C for 15 min to 1 h. After two washes to remove unbound particles, cells were stained with Diff-Quick and bound particles were counted using an optical microscope. The binding index was obtained as described in “Material and Methods”. (C) Dectin-1 expression was analyzed by immunofluoresence staining and flow cytometry on mouse peritoneal macrophages isolated from wild type (solid line) and Pla2g5-null macrophages (dashed line). Shaded histogram represents isotype control Ab. Data are mean ± SEM of three experiments (B), or representative of two experiments with duplicate samples (A, C).
Critical role of group V sPLA2 in phagosomal maturation
We have previously reported that in mouse peritoneal macrophages group V sPLA2 translocates to the phagocytic cup and newly formed phagosomes within minutes of phagocytosis of zymosan (18). These data suggest that group V sPLA2 may influence phagosome formation and maturation. To assess this hypothesis we studied the maturation of phagosomes containing FITC-zymosan by their ability to fuse over a period of 1 h with Lamp-1, a marker of late endosomes and lysosomes (26, 27). Fusion was quantified by counting zymosan particles fully surrounded by Lamp-1 that visualized by staining with Cy3-conjugated antibody. Phagosomes containing FITC-zymosan merged with lysosomes in wild type peritoneal macrophages more efficiently than in Pla2g5-null macrophages (Fig. 2A). Formal quantification of phagolysosome fusion over 1 h confirmed that Pla2g5-null macrophages had a 50–60% reduction in maturation of phagosomes (Fig. 2B, n=3, p <0.03 by analysis of variance).
Figure 2.

Pla2g5-null macrophages have a delay in phagosome maturation. Indirect immunofluorescence microscopy was used to visualize the distribution of Lamp-1 in macrophages phagocytosing FITC-zymosan (2.5 ppc) for 10 min to 1 h as described under “Material and methods”. (A) Representative images at 1h. Closed arrow: Lamp-1-positive phagosomes. Open arrow: Lamp-1-negative phagosomes (B) The fusion index was calculated for wild type (closed bars) and Pla2g5-null (open bars) macrophages, as described under “Material and Methods”. Data are mean ± SEM of at least three experiments.
Pla2g5-null peritoneal macrophages are defective in phagocytosis of opsonized targets
Opsonization of phagocytic targets with IgG or complement potentiates phagocytosis and contributes to clearance of pathogens. Therefore, we investigated the ability of group V sPLA2 to regulate phagocytosis mediated by two major phagocytic receptors, Fcγ receptors and complement receptor 3 (CR3). The kinetics of phagocytosis of IgG-opsonized sRBC was more rapid than that of zymosan (18) or complement-coated targets perhaps reflecting utilization of different signaling molecules (2), and was attenuated in Pla2g5-null peritoneal macrophages compared with wild type macrophages (Fig. 3A, n=3, p<0.01 by analysis of variance). In Pla2g5-null peritoneal macrophages phagocytosis of iC3b-opsonized sRBC was attenuated ~60% compared to wild-type macrophages at each time point analyzed (Fig. 3B, n=3, p<0.02 by analysis of variance). Thus, the ability of group V sPLA2 to modulate phagocytosis extends to other major phagocytic receptors essential for innate and acquired immune responses against pathogens.
Figure 3.

Phagocytosis of IgG- and iC3b-opsonized sheep red blood cells is impaired in Pla2g5-null peritoneal macrophages. Peritoneal macrophages isolated from Pla2g5-null (open bars) and wild type (black bars) mice were stimulated with IgG-sRBC (A) or iC3b-sRBC (B) (10 ppc) for 5 min to 1 h. Cells were fixed and stained with Diff-Quick. The phagocytic index was obtained as described in “Material and Methods”. Data are mean ± SEM of three experiments.
Group V secretory PLA2 is required for efficient phagocytosis and killing of C. albicans
To examine whether group V sPLA2 modulates phagocytosis of live fungi we analyzed phagocytosis of C. albicans yeast by peritoneal macrophages from 5 min to 1 h, the time of the peak of phagocytosis. Phagocytosis of C. albicans was reduced ~50% in Pla2g5-null peritoneal macrophages compared to wild type macrophages at all time points studied (Fig. 4A, n=3, **p<0.02; *p<0.05 by t test). Phagocytosis of C. albicans by professional phagocytes is followed by killing (14). Therefore we also examined killing of C. albicans by Pla2g5-null macrophages. FUN-1 is a vital dye that stains nucleic acids. Live C. albicans have a diffuse green cytoplasmic fluorescence while dead cells exhibit an extremely bright, diffuse, green-yellow fluorescence (28). In wild type macrophages starting at 1 h the ingested C. albicans exhibited extremely bright, green-yellow fluorescence indicative of death, which was not observed in Pla2g5-null peritoneal macrophages (Fig. 4B). Using a conventional serial dilution technique, we confirmed that Pla2g5-null peritoneal macrophages had a decreased ability to kill C. albicans at 1 h (Fig. 4C; n=4, #p<0.03 by t-test). Nevertheless, killing reached wild type levels by 2 and 3 hrs. Thus, Pla2g5-null macrophages are impaired in their ability to phagocytose C. albicans and have a delay in killing of C. albicans.
Figure 4.

Impaired phagocytosis and killing of C. albicans by Pla2g5-null peritoneal macrophages. (A) Peritoneal macrophages isolated from Pla2g5-null (open bars) and wild type (black bars) mice were incubated with C. albicans from 5 min to 1 h (yeast to cell ratio 5:1). The phagocytic index was obtained as described under “Material and Methods”. (B) C. albicans viability was distinguished by fluorescence staining with FUN-1. Nuclei were stained in blue by Hoechst dye. (C) Viability of ingested C. albicans was assessed by a limiting dilution technique and is expressed as a percentage of viable yeast initially ingested, as described under “Material and Methods”. (D) Indirect immunofluorescence microscopy was used to visualize the distribution of Lamp-1 in macrophages phagocytosing C. albicans (m.o.i. 2) for 30 min. The fusion index was calculated for wild type (closed bars) and Pla2g5-null (open bars) macrophages, as described under “Material and Methods”. (E) Representative images at 30 min. Data are mean ± SEM of three experiments (A), of at least four independent experiments, each with quadruplicate measurement (C), or of two experiments (D).
Group V sPLA2 regulates phagolysosome fusion but not TNF-α or Cys-LTs production in response to fungal particles
Various lines of evidence have shown that oxygen-independent mechanisms, such as lysosomal enzymes of macrophages (14), are important in killing of C. albicans. Because we found a delay in phagolysosome fusion in Pla2g5-null macrophages after phagocytosis of zymosan, we analyzed fusion of C. albicans-containing phagosomes with lysosomes stained by Lamp-1. We found that, as for zymosan, Pla2g5-null macrophages have a defect in phagolysosome fusion after ingestion of C. albicans that was marked at 30 min (Fig. 4, D and E; n=2, p<0.05 by t test).
To more clearly elucidate the delay in phagolysosome fusion in Pla2g5-null macrophages, we synchronized phagocytosis of C. albicans by allowing particles to bind at 4°C, washing away excess unbound particles, and then transferring cells to 37°C to initiate phagocytosis. Macrophages were then immunostained for Lamp-1. Pla2g5-null macrophages had a defect in fusion of phagosomes with Lamp-1 positive organelles that was marked (~70%) at 30 min while most phagosomes were able to acquire Lamp-1 within 1 h (Fig. 5A). To confirm that Pla2g5-null phagosomes were delayed in acquisition of lysosomal markers, we also immunostained phagosomes for cathepsin D, a lysosomal luminal protease (21). Pla2g5-null phagosomes did not recruit cathepsin D as efficiently as wild type phagosomes at 30 min and 1 h, although most phagosomes acquired cathepsin D within 2 h (Fig. 5B). Synchronized phagocytosis also showed that Pla2g5-null macrophages have a defect in phagocytosis at 30 min but not at later time points (Fig. 5C). Thus the absence of group V sPLA2 delays fusion of phagosomes with lysosomes and slows the phagocytic process (Fig. 5; n=2, *p<0.05; **p<0.02 by t test).
Figure 5.

Delayed phagosome-lysosome fusion in Pla2g5-null peritoneal macrophages. Peritoneal macrophages isolated from wild type (black bars) and Pla2g5-null (open bars) mice were incubated with C. albicans from 30 min to 2 h (m.o.i. 2). To synchronize phagocytosis, particles were first allowed to bind at 4°C, excess unbound particles were washed away then cell transferred at 37°C. Indirect immunofluorescence microscopy was used to visualize the distribution of Lamp-1 (A) or cathepsin D (B). The fusion index (A, B) and phagocytic index (C) were calculated as described in “Materials and Methods”. Data are mean ± SEM of two experiments.
TNF-α is a key mediator in protecting against disseminated candidiasis as demonstrated by the fact that the lack of TNF-α worsens the course of disseminated candidiasis (25). Pla2g5-null macrophages produced similar amount of TNF-α compared to wild type macrophages after phagocytosis of either C. albicans or zymosan for 3 h (Tab. I), indicating that group V sPLA2 does not regulate phagocytosis and killing through generation of TNF-α.
Table I.
Production of TNF-α and Cys-LTs by peritoneal macrophages stimulated with C. albicans or zymosan
| Mediator | Mouse strain |
C. albicans (moi) |
Zymosan (ppc) | ||
|---|---|---|---|---|---|
| 0.1 | 1 | 10 | 10 | ||
| TNF-α, ng/mL | Wild type | 1.58 ± 0.05 | 7.33 ± 0.43 | 9.63 ± 0.09 | 15.61 ± 0.22 |
| Pla2g5-null | 1.58 ± 0.21 | 8.46 ± 0.68 | 10.90 ± 0.42 | 19.07 ± 2.39 | |
| Cys-LTs, ng/106 cells | Wild type | 3.62 ± 14.11 | 19.87 ± 27.15 | 19.57 ± 0.00 | 4.24 ± 32.56 |
| Pla2g5-null | 4.85 ± 56.77 | 22.01 ± 20.37 | 19.88 ± 38.04 | 3.79 ± 11.97 | |
Data represent mean ± SEM of 2 individual mice per strain.
It has been previously shown that zymosan induces eicosanoid generation by monocytes (29), and macrophages (17), in a beta-glucan dependent fashion (16). Leukotrienes have also been implicated in phagocytosis and killing of various microorganisms (30). We found no difference in the amount of Cys-LTs produced after phagocytosis of either zymosan or C. albicans by Pla2g5-null macrophages compared to the wild type macrophages (Tab. I) indicating that group V sPLA2 does not regulate phagocytosis through generation of Cys-LTs.
Pla2g5-null mice are more susceptible than wild type mice to disseminated candidiasis
To examine the function of group V sPLA2 in vivo, we next infected mice intravenously with C. albicans as a model of systemic candidiasis and evaluated the fungal burden and survival of the mice. The kidney is the major target organ in systemic candidiasis (31), hence the fungal burden in the kidney was 10- to 100-fold greater than that seen in the liver and spleen (Fig. 6). One and 3 days after intravenous injection of C. albicans (5x105 CFU/mouse) the number of CFU recovered from the kidney, liver, and spleen was similar in wild type and Pla2g5-null mice. Although at day 7 the increased number of CFU recovered from the kidney of pla2g5-null mice did not achieve statistical significance (Fig. 6A; p<0.07 by Mann-Whitney test), the number of CFU recovered from the liver and spleen was significantly greater in Pla2g5-null mice compared to wild type littermates (Fig. 6, B and C; **p< 0.02; *p<0.03 respectively by Mann-Whitney test). C. albicans recovered from the heart and lung only 24 hours after intravenous infection, was lower than from other organs (<800 CFU/g and <300 CFU/g, respectively), was not significantly different in Pla2g5-null and wild-type mice and had been cleared by day 3.
Figure 6.
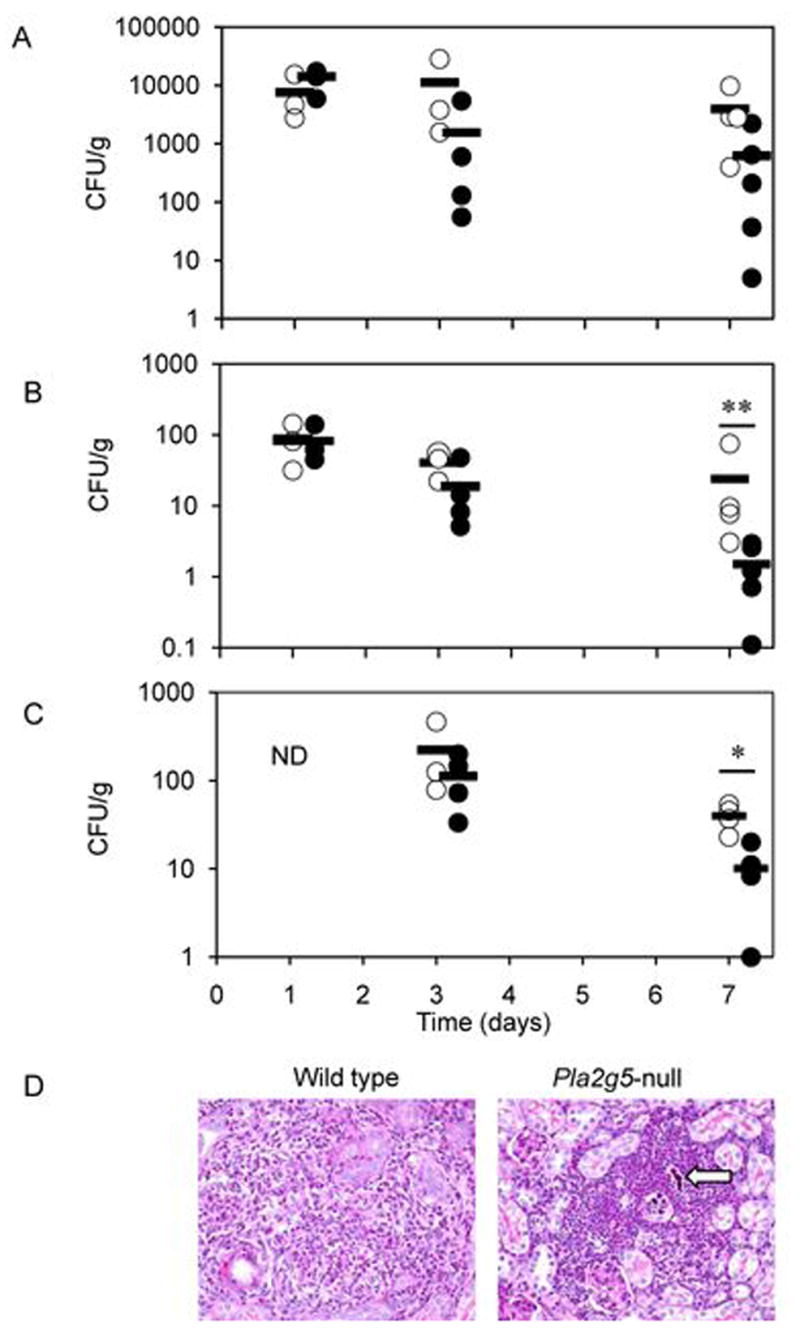
Pla2g5-null mice are more susceptible than wild type mice to live C. albicans infection. Mice were infected with C. albicans by the intravenous injection of 200 μl of 0.9% NaCl containing 5x105 CFU. To assess outgrowth of the microorganism and its ability to invade organs, subgroups of 3 to 5 Pla2g5-null (open circles) and wild type (closed circles) mice were sacrificed 1, 3, and 7 days after injection. The outgrowth of C. albicans was measured by serial dilution and expressed as CFU per gram of tissue. Individual data points are shown for kidney (A), liver (B) and spleen (C); means are indicated by horizontal bars. ND, not determined. (D) Histopathology of the kidney of C. albicans-infected mice. Histological sections of representative wild type and Pla2g5-null kidneys 7 days after infection are stained with Periodic-Acid Schiff. Arrow points to pseudohyphae. Magnification 40X for both panels.
Because the kidney is the major target organ in systemic candidiasis, we examined the microscopic appearance of the kidneys. Kidneys of both wild type and Pla2g5-null mice showed scattered foci of neutrophils infiltrating the renal cortex and medulla on days 1 and 3 after infection with C. albicans. Kidneys of wild type mice were infiltrated with small numbers of yeast and hyphae which appeared to be more extensive in kidneys of Pla2g5-null mice with formation of hyphae and pseudohyphae (data not shown). On day 7 after infection, the kidneys of the wild type mice appeared to be healing with regeneration of interstitial tissue and diffuse infiltration of macrophages, fibroblasts and fibrocytes; fewer neutrophils were present, there were occasional yeasts, but there was no hyphal formation (Fig. 6D). In contrast, on day 7 in the kidneys of Pla2g5-null mice (Fig. 6D) there were dense foci of inflammation with C. albicans yeast and hyphae still present within neutrophilic infiltrates.
To further assess the susceptibility of Pla2g5-null mice to C. albicans infection, mice were injected intravenously with 1x106 C. albicans CFU/mouse. Over the subsequent month 50% of the Pla2g5-null mice died compared to 12.5 % of the wild type mice (Fig. 7; p<0.1 by log-rank test), suggesting that defective clearance of C. albicans in Pla2g5-null mice leads to increased mortality.
Figure 7.
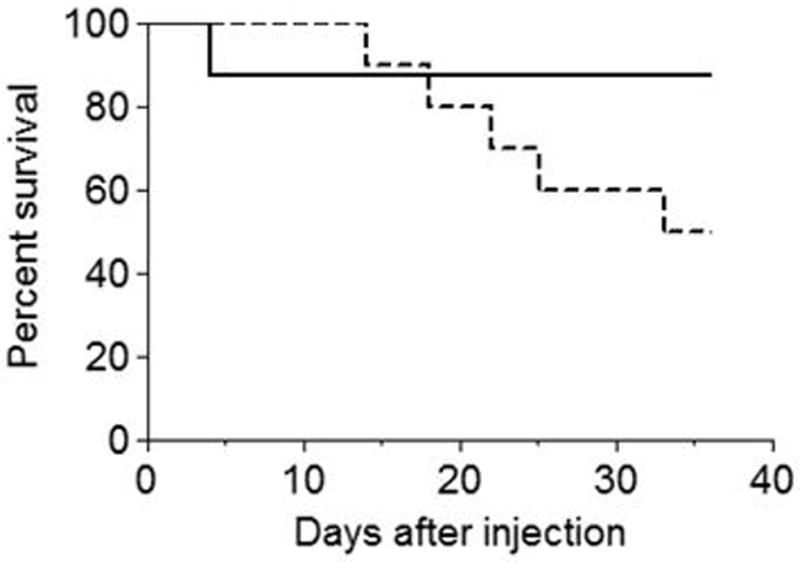
Survival of mice infected with C. albicans. Wild type (filled line) and Pla2g5-null (dotted line) mice were infected i.v. with 1x106 CFU/mouse and followed for 33 days. Data are from one experiment using 11 Pla2g5-null and 9 wild type mice and are expressed as a Kaplan Meier Plot. Similar data were reproduced in a second experiment.
Discussion
There are over twenty mammalian phospholipases A2 (32). Yet it is likely, given the diversity of their biochemical properties and cellular and subcellular distribution that PLA2 enzymes serve a diversity of biological functions independent of their ability to generate eicosanoids. We have previously reported that group V secretory PLA2 regulates phagocytosis of zymosan and translocates to the forming phagosomes (18). In the present study we demonstrated that the defect in phagocytosis by Pla2g5-null macrophages is not due to impaired expression of dectin-1 or to impaired binding of zymosan. However, phagolysosome fusion was defective in Pla2g5-null macrophages. Thus, group V sPLA2 regulates phagocytosis by influencing the rate of maturation of the phagosomes with functional consequences for phagocytosis and killing of live pathogens.
Zymosan and C. albicans share similar signaling pathways, including activation of dectin-1 and TLRs on phagocytes (11, 33). Indeed, Pla2g5-null macrophages were impaired in their ability to engulf C. albicans (Fig. 4A) as well as zymosan ((18) and Fig. 1A). Pla2g5-null macrophages also had a delayed ability to kill C. albicans (Fig. 4, B and C). Indeed, while 1 h after ingestion wild type macrophages were able to kill about 50% of the ingested C. albicans, Pla2g5-null macrophages were able to kill only about 15 % of the yeast (Fig. 4C) and by 2 h there was no difference. It has been previously reported that among sPLA2s group IIA, group V, and group X sPLA2 have antibacterial activity (34, 35) though there are no data on their ability to kill fungi. Addition of mouse recombinant group V sPLA2 directly to C. albicans, did not affect the survival of the yeast (data not shown) suggesting that the delay in killing in Pla2g5-null macrophages is not due to direct anti-fungal actions of the enzyme.
Macrophages are essential in protection against candidiasis (12) relying on both non-oxidative and oxidative mechanisms to kill the organism (14). The non oxidative mechanisms of killing used by macrophages include lysosomal enzymes activated when the phagosome matures to form a microbicidal organelle (12). The defect in phagocytosis of C. albicans, and the delay in killing of C. albicans showed by Pla2g5-null macrophages, suggests a role for the enzyme in maturation of phagosomes containing C. albicans. Indeed, using C. albicans stained with Calcofluor white we showed impaired fusion of LAMP-1 positive organelles with phagosomes in Pla2g5-null macrophages (Fig. 4D), a defect similar to that seen in zymosan-containing phagosomes. For this set of experiments we used centrifugation to synchronize the onset of phagocytosis. This protocol did not remove unbound targets thereby allowing ongoing phagocytosis as occurs during the course of infection, but potentially masking the impact of the lack of group V sPLA2 on the rate of phagocytosis and phagolysosome fusion. We therefore used a temperature-dependent technique to allow binding of targets to the macrophage at 4°C, washing to remove unbound particles, and then initiating phagocytosis by rapidly bringing the temperature to 37°C. Using this protocol to synchronize the onset of phagocytosis we showed a very marked defect (~70–80%) in acquisition by the phagosome of two lysosomal markers, Lamp-1 (a late endosome and lysosome marker) and cathepsin D (a lysosome marker), in Pla2g5-null macrophages at 30 min, an effect that was less marked at 1 h and 2 h. We also showed that the absence of group V sPLA2 slows the phagocytic process (Fig. 5). The oxidative mechanisms of killing include generation of reactive nitrogen and oxygen species and have been shown to be mainly dependent on dectin-1 (8). Because resident peritoneal macrophages are poor producers of ROS (12) we used thioglycollate-elicited macrophages to investigate production of ROS but found that they did not produce a significant amount after stimulation with C. albicans. However Pla2g5-null thioglycollate-elicited macrophages stimulated with zymosan, a more potent stimulus for ROS production, showed a diminished capacity to generate ROS compared with wild type macrophages (data not shown). Hence, our major finding is the delay in phagosome fusion in Pla2g5-null macrophages in response to C. albicans, and any defect in oxidative mechanisms remains to be determined.
Proinflammatory cytokines such as TNF-α, IL-1, IL-6 and IFN-γ activate professional phagocytes to phagocytose and kill the yeast. In particular, TNF-α production is critical for the host response against the fungus as demonstrated by increased mortality of mice lacking cytokines of the TNF family (25, 36). Lack of group V sPLA2 had no effect on TNF-α generation after phagocytosis of zymosan or C. albicans (Tab. I). Thus, group V sPLA2 does not regulate phagocytosis and killing of C. albicans through regulation of TNF-α production. This is consistent with previous data reporting that binding of zymosan in the absence of phagocytosis is sufficient for proinflammatory cytokine production (37). Therefore a defect in phagocytosis and phagosome maturation would not necessarily lead to diminished generation of TNF-α.
It has been previously shown that zymosan and C. albicans induce leukotriene generation by inflammatory cells (16, 17, 29). However, lack of group V sPLA2 did not affect leukotriene generation in response to zymosan or C. albicans (Tab. I). These data may seem in conflict with the previously reported role of group V sPLA2 in regulating leukotriene generation after phagocytosis of zymosan (17). However, the peritoneal macrophages used in the present study were cultured for 2 days whereas in our previous study macrophages were cultured for just a few hours. Indeed the ability of macrophages to produce eicosanoids depends on the state of activation/maturation of the macrophages (38, 39) and prolonged culture gives rise to a different phenotype of macrophage with attenuated ability to produce leukotrienes compared to freshly isolated peritoneal macrophages; 5ng per 106 cells (Tab. I) compared to 42ng per 106 cells (17), respectively, after phagocytosis of zymosan. Our current data also support the hypothesis that the role of group V sPLA2 in regulating phagocytosis is independent of its role in eicosanoid generation and is consistent with the observation that phagocytosis of zymosan is intact in cPLA2α-deficient macrophages (18) and is not impaired by cPLA2α inhibitors (40). Furthermore, it has been previously shown that although binding of beta-glucan induces cPLA2α activation and AA release (16), the internalization of yeast particles was not essential for cPLA2α activation although AA release was enhanced after C. albicans internalization (16). These data emphasize the concept of non-redundant functions of different types of PLA2 (41) and support the hypothesis that the ability of group V sPLA2 to modulate phagocytosis of yeast particles is related to the regulation of phagocytic events that require formation and/or maturation of the phagosome as opposed to cellular responses triggered only by binding to specific receptors.
Our finding that group V sPLA2 modulates phagocytosis and killing of C. albicans, prompted us to extend the study to an in vivo model of systemic candidiasis (42). While the early fungal burden up to day 3 was equivalent in wild type and Pla2g5-null mice, by day 7 these mice showed an impaired ability to clear C. albicans from spleen and liver and kidney (Fig. 6) (31). Depending on the dose and strain of C. albicans, and the strain of mice, lesions in the kidney may vary from white macroscopic abscesses to lesions with a diffuse inflammatory reaction containing polymorphonuclear cells and/or macrophages (42, 43). In our model, histological examination of the kidney of wild type mice at day 7 (Fig. 6D) showed an inflammatory response with residual neutrophilic infiltration replaced by mononuclear cells, fibroblasts, and occasional yeast. However, kidneys from mice lacking group V sPLA2 (Fig. 6D) showed persistent neutrophilic infiltrates with yeast and hyphae, suggestive of an impaired ability to clear the infection and actively proliferating C. albicans. These data indicate that the distribution of C. albicans into the organs in the early phase of infection is similar between Pla2g5-null and wild type mice. As the infection proceeds and C. albicans localizes in liver and spleen and increasingly in the kidney (42), Pla2g5-null mice demonstrate a defect in clearance of the fungal infection compared to wild type mice, most likely due to impaired phagocytosis and killing. At a higher dose of C. albicans mice lacking group V sPLA2 showed increased mortality (Fig. 7). Thus our data are consistent with the essential role of phagocytosis in the host immune response to fungal pathogens and consistent with those of other groups showing that a defect in phagocytosis can lead to enhanced fungal dissemination and increased lethality in experimental animals (14, 44) and humans (45).
Although the resistance to C. albicans infection is determined initially by the innate immune system, the adaptive immune system also contributes to protection against fungal infections (13). Several studies have shown the protective role of Th-1 rather than Th-2 cytokines (46), while the function of antibodies in fungal infection is still controversial (13). Yet the role of opsonic receptors in mediating phagocytosis of C. albicans is well established and therefore the modulation of phagocytosis through opsonin receptors by group V sPLA2 (Fig. 3) could also contribute to the decreased survival of Pla2g5-null mice.
The present study has extended our early data on the role of group V sPLA2 in regulating phagocytosis of zymosan to show that its role extends to regulation of ingestion and killing of C. albicans and phagolysome maturation. The ability of group V sPLA2 to modulate phagocytosis through several receptors renders this molecule a key regulator of signaling generated by multiple ligands and likely multiple pathogens. These signals likely converge to regulate one or more fundamental steps in the phagocytic process thereby contributing to an efficient innate and adaptive immune response. The relevance of the in vitro findings is indicated by the impaired clearance of C. albicans in mice lacking group V sPLA2 with associated higher mortality, providing the first demonstration that group V sPLA2 plays an important role in the host response to fungal pathogens in vivo.
Acknowledgments
We thank Dr. K. Frank Austen for critically reviewing the manuscript. We thank Emanuela Pandeli and Sangita Banerjee for technical assistance.
Footnotes
Disclosures
The authors have no financial conflict of interest.
This work was supported by a grant from the National Institute of Allergy And Infectious Diseases AI064226 (B.B.); by bridge grants from the American Academy of Allergy Asthma and Immunology, and the Brigham and Women’s Hospital Biomedical Research Institute; and NIH grants HL070946 and HL036110 (J.P.A.); AI041144 and HL036110 (H.R.K.); and R37HL36235 (M.H.G.).
The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institute of Allergy And Infectious Diseases or the National Institutes of Health.
Abbreviations used in this paper: ROS, reactive oxygen species; C. albicans, candida albicans; cPLA2, cytosolic phospholipase A2; sPLA2, secretory phospholipase A2; sRBC, sheep red blood cells; Pla2g5, the gene encoding group V sPLA2; ppc, particles per cell; CFU, colony forming units; Cys-LTs, cysteinyl-leukotrienes.
References
- 1.Aderem A, Underhill DM. Mechanisms of phagocytosis in macrophages. Annu Rev Immunol. 1999;17:593–623. doi: 10.1146/annurev.immunol.17.1.593. [DOI] [PubMed] [Google Scholar]
- 2.Underhill DM, Ozinsky A. Phagocytosis of microbes: complexity in action. Annu Rev Immunol. 2002;20:825–852. doi: 10.1146/annurev.immunol.20.103001.114744. [DOI] [PubMed] [Google Scholar]
- 3.Brown GD, Taylor PR, Reid DM, Willment JA, Williams DL, Martinez-Pomares L, Wong SY, Gordon S. Dectin-1 is a major beta-glucan receptor on macrophages. J Exp Med. 2002;196:407–412. doi: 10.1084/jem.20020470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Taylor PR, Brown GD, Herre J, Williams DL, Willment JA, Gordon S. The role of SIGNR1 and the beta-glucan receptor (dectin-1) in the nonopsonic recognition of yeast by specific macrophages. J Immunol. 2004;172:1157–1162. doi: 10.4049/jimmunol.172.2.1157. [DOI] [PubMed] [Google Scholar]
- 5.Brown GD, Gordon S. Immune recognition. A new receptor for beta-glucans. Nature. 2001;413:36–37. doi: 10.1038/35092620. [DOI] [PubMed] [Google Scholar]
- 6.Taylor PR, Brown GD, Reid DM, Willment JA, Martinez-Pomares L, Gordon S, Wong SY. The beta-glucan receptor, dectin-1, is predominantly expressed on the surface of cells of the monocyte/macrophage and neutrophil lineages. J Immunol. 2002;169:3876–3882. doi: 10.4049/jimmunol.169.7.3876. [DOI] [PubMed] [Google Scholar]
- 7.Herre J, Marshall AS, Caron E, Edwards AD, Williams DL, Schweighoffer E, Tybulewicz V, Reis e Sousa C, Gordon S, Brown GD. Dectin-1 uses novel mechanisms for yeast phagocytosis in macrophages. Blood. 2004;104:4038–4045. doi: 10.1182/blood-2004-03-1140. [DOI] [PubMed] [Google Scholar]
- 8.Gantner BN, Simmons RM, Underhill DM. Dectin-1 mediates macrophage recognition of Candida albicans yeast but not filaments. Embo J. 2005;24:1277–1286. doi: 10.1038/sj.emboj.7600594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Underhill DM, Rossnagle E, Lowell CA, Simmons RM. Dectin-1 activates Syk tyrosine kinase in a dynamic subset of macrophages for reactive oxygen production. Blood. 2005;106:2543–2550. doi: 10.1182/blood-2005-03-1239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gantner BN, Simmons RM, Canavera SJ, Akira S, Underhill DM. Collaborative induction of inflammatory responses by dectin-1 and Toll-like receptor 2. J Exp Med. 2003;197:1107–1117. doi: 10.1084/jem.20021787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Netea MG, Gow NA, Munro CA, Bates S, Collins C, Ferwerda G, Hobson RP, Bertram G, Hughes HB, Jansen T, Jacobs L, Buurman ET, Gijzen K, Williams DL, Torensma R, McKinnon A, MacCallum DM, Odds FC, Van der Meer JW, Brown AJ, Kullberg BJ. Immune sensing of Candida albicans requires cooperative recognition of mannans and glucans by lectin and Toll-like receptors. J Clin Invest. 2006;116:1642–1650. doi: 10.1172/JCI27114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Vazquez-Torres A, Balish E. Macrophages in resistance to candidiasis. Microbiol Mol Biol Rev. 1997;61:170–192. doi: 10.1128/mmbr.61.2.170-192.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Romani L. Immunity to fungal infections. Nat Rev Immunol. 2004;4:1–23. doi: 10.1038/nri1255. [DOI] [PubMed] [Google Scholar]
- 14.Odds FC. Candida and candidosis. Bailliaere Tindall; London: 1988. [Google Scholar]
- 15.Gijon MA, Spencer DM, Siddiqi AR, Bonventre JV, Leslie CC. Cytosolic phospholipase A2 is required for macrophage arachidonic acid release by agonists that Do and Do not mobilize calcium. Novel role of mitogen-activated protein kinase pathways in cytosolic phospholipase A2 regulation. J Biol Chem. 2000;275:20146–20156. doi: 10.1074/jbc.M908941199. [DOI] [PubMed] [Google Scholar]
- 16.Suram S, Brown GD, Ghosh M, Gordon S, Loper R, Taylor PR, Akira S, Uematsu S, Williams DL, Leslie CC. Regulation of cytosolic phospholipase A2 activation and cyclooxygenase 2 expression in macrophages by the beta-glucan receptor. J Biol Chem. 2006;281:5506–5514. doi: 10.1074/jbc.M509824200. [DOI] [PubMed] [Google Scholar]
- 17.Satake Y, Diaz BL, Balestrieri B, Lam BK, Kanaoka Y, Grusby MJ, Arm JP. Role of group V phospholipase A2 in zymosan-induced eicosanoid generation and vascular permeability revealed by targeted gene disruption. J Biol Chem. 2004;279:16488–16494. doi: 10.1074/jbc.M313748200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Balestrieri B, Hsu VW, Gilbert H, Leslie CC, Han WK, Bonventre JV, Arm JP. Group V secretory phospholipase A2 translocates to the phagosome after zymosan stimulation of mouse peritoneal macrophages and regulates phagocytosis. J Biol Chem. 2006;281:6691–6698. doi: 10.1074/jbc.M508314200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Rouault M, Le Calvez C, Boilard E, Surrel F, Singer A, Ghomashchi F, Bezzine S, Scarzello S, Bollinger J, Gelb MH, Lambeau G. Recombinant production and properties of binding of the full set of mouse secreted phospholipases A2 to the mouse M-type receptor. Biochemistry. 2007;46:1647–1662. doi: 10.1021/bi062119b. [DOI] [PubMed] [Google Scholar]
- 20.Marr KA, Balajee SA, Hawn TR, Ozinsky A, Pham U, Akira S, Aderem A, Liles WC. Differential role of MyD88 in macrophage-mediated responses to opportunistic fungal pathogens. Infect Immun. 2003;71:5280–5286. doi: 10.1128/IAI.71.9.5280-5286.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Huynh KK, Eskelinen EL, Scott CC, Malevanets A, Saftig P, Grinstein S. LAMP proteins are required for fusion of lysosomes with phagosomes. EMBO J. 2007;26:313–324. doi: 10.1038/sj.emboj.7601511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ehlers MR. CR3: a general purpose adhesion-recognition receptor essential for innate immunity. Microbes Infect. 2000;2:289–294. doi: 10.1016/s1286-4579(00)00299-9. [DOI] [PubMed] [Google Scholar]
- 23.Blander JM, Medzhitov R. Regulation of phagosome maturation by signals from toll-like receptors. Science. 2004;304:1014–1018. doi: 10.1126/science.1096158. [DOI] [PubMed] [Google Scholar]
- 24.Netea MG, Van Der Graaf CA, Vonk AG, Verschueren I, Van Der Meer JW, Kullberg BJ. The role of toll-like receptor (TLR) 2 and TLR4 in the host defense against disseminated candidiasis. J Infect Dis. 2002;185:1483–1489. doi: 10.1086/340511. [DOI] [PubMed] [Google Scholar]
- 25.Netea MG, van Tits LJ, Curfs JH, Amiot F, Meis JF, van der Meer JW, Kullberg BJ. Increased susceptibility of TNF-alpha lymphotoxin-alpha double knockout mice to systemic candidiasis through impaired recruitment of neutrophils and phagocytosis of Candida albicans. J Immunol. 1999;163:1498–1505. [PubMed] [Google Scholar]
- 26.Scott CC, Botelho RJ, Grinstein S. Phagosome maturation: a few bugs in the system. J Membr Biol. 2003;193:137–152. doi: 10.1007/s00232-002-2008-2. [DOI] [PubMed] [Google Scholar]
- 27.Vieira OV, Botelho RJ, Grinstein S. Phagosome maturation: aging gracefully. Biochem J. 2002;366:689–704. doi: 10.1042/BJ20020691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Henry-Stanley MJ, Garni RM, Wells CL. Adaptation of FUN-1 and Calcofluor white stains to assess the ability of viable and nonviable yeast to adhere to and be internalized by cultured mammalian cells. J Microbiol Methods. 2004;59:289–292. doi: 10.1016/j.mimet.2004.07.001. [DOI] [PubMed] [Google Scholar]
- 29.Czop JK, Austen KF. A beta-glucan inhibitable receptor on human monocytes: its identity with the phagocytic receptor for particulate activators of the alternative complement pathway. J Immunol. 1985;134:2588–2593. [PubMed] [Google Scholar]
- 30.Peters-Golden M, Canetti C, Mancuso P, Coffey MJ. Leukotrienes: underappreciated mediators of innate immune responses. J Immunol. 2005;174:589–594. doi: 10.4049/jimmunol.174.2.589. [DOI] [PubMed] [Google Scholar]
- 31.Ashman RB, Fulurija A, Papadimitriou JM. Strain-dependent differences in host response to Candida albicans infection in mice are related to organ susceptibility and infectious load. Infect Immun. 1996;64:1866–1869. doi: 10.1128/iai.64.5.1866-1869.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Schaloske RH, Dennis EA. The phospholipase A2 superfamily and its group numbering system. Biochim Biophys Acta. 2006;1761:1246–1259. doi: 10.1016/j.bbalip.2006.07.011. [DOI] [PubMed] [Google Scholar]
- 33.Brown GD. Dectin-1: a signalling non-TLR pattern-recognition receptor. Nat Rev Immunol. 2006;6:33–43. doi: 10.1038/nri1745. [DOI] [PubMed] [Google Scholar]
- 34.Beers SA, Buckland AG, Koduri RS, Cho W, Gelb MH, Wilton DC. The antibacterial properties of secreted phospholipases A2: a major physiological role for the group IIA enzyme that depends on the very high pI of the enzyme to allow penetration of the bacterial cell wall. J Biol Chem. 2002;277:1788–1793. doi: 10.1074/jbc.M109777200. [DOI] [PubMed] [Google Scholar]
- 35.Koduri RS, Gronroos JO, Laine VJ, Le Calvez C, Lambeau G, Nevalainen TJ, Gelb MH. Bactericidal properties of human and murine groups I, II, V, X, and XII secreted phospholipases A. J Biol Chem. 2002;277(2):5849–5857. doi: 10.1074/jbc.M109699200. [DOI] [PubMed] [Google Scholar]
- 36.Steinshamn S, Bemelmans MH, van Tits LJ, Bergh K, Buurman WA, Waage A. TNF receptors in murine Candida albicans infection: evidence for an important role of TNF receptor p55 in antifungal defense. J Immunol. 1996;157:2155–2159. [PubMed] [Google Scholar]
- 37.Mukhopadhyay S, Herre J, Brown GD, Gordon S. The potential for Toll-like receptors to collaborate with other innate immune receptors. Immunology. 2004;112:521–530. doi: 10.1111/j.1365-2567.2004.01941.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Coffey MJ, Wilcoxen SE, Sporn PH, Peters-Golden M. Regulation of 5-lipoxygenase activity in mononuclear phagocytes: characterization of an endogenous cytosolic inhibitor. Prostaglandins Other Lipid Mediat. 1998;56:103–117. doi: 10.1016/s0090-6980(98)00046-x. [DOI] [PubMed] [Google Scholar]
- 39.Humes JL, Burger S, Galavage M, Kuehl FA, Jr, Wightman PD, Dahlgren ME, Davies P, Bonney RJ. The diminished production of arachidonic acid oxygenation products by elicited mouse peritoneal macrophages: possible mechanisms. J Immunol. 1980;124:2110–2116. [PubMed] [Google Scholar]
- 40.Girotti M, Evans JH, Burke D, Leslie CC. Cytosolic phospholipase A2 translocates to forming phagosomes during phagocytosis of zymosan in macrophages. J Biol Chem. 2004;279:19113–19121. doi: 10.1074/jbc.M313867200. [DOI] [PubMed] [Google Scholar]
- 41.Balestrieri B, Arm JP. Group V sPLA2: classical and novel functions. Biochim Biophys Acta. 2006;1761:1280–1288. doi: 10.1016/j.bbalip.2006.07.008. [DOI] [PubMed] [Google Scholar]
- 42.Tuite A, Mullick A, Gros P. Genetic analysis of innate immunity in resistance to Candida albicans. Genes Immun. 2004;5:576–587. doi: 10.1038/sj.gene.6364130. [DOI] [PubMed] [Google Scholar]
- 43.Odds FC, Van Nuffel L, Gow NA. Survival in experimental Candida albicans infections depends on inoculum growth conditions as well as animal host. Microbiology. 2000;146(Pt 8):1881–1889. doi: 10.1099/00221287-146-8-1881. [DOI] [PubMed] [Google Scholar]
- 44.Ashman RB. Candida albicans: pathogenesis, immunity and host defence. Res Immunol. 1998;149:281–288. doi: 10.1016/s0923-2494(98)80752-9. discussion 494–286. [DOI] [PubMed] [Google Scholar]
- 45.Crowe SM, Vardaxis NJ, Kent SJ, Maerz AL, Hewish MJ, McGrath MS, Mills J. HIV infection of monocyte-derived macrophages in vitro reduces phagocytosis of Candida albicans. J Leukoc Biol. 1994;56:318–327. doi: 10.1002/jlb.56.3.318. [DOI] [PubMed] [Google Scholar]
- 46.Mencacci A, Cenci E, Del Sero G, d’Ostiani CF, Montagnoli C, Bacci A, Bistoni F, Romani L. Innate and adaptive immunity to Candida albicans: A new view of an old paradigm. Rev Iberoam Micol. 1999;16:4–7. [PubMed] [Google Scholar]