

Abstract
Background
The use of non-autologous stem cells isolated from healthy donors for stem cell therapy is an attractive approach since the stem cells can be culture expanded in advance, thoroughly tested, and formulated into off-the-shelf medicine. However, HLA compatibility and related immunosuppressive protocols can compromise therapeutic efficacy and cause unwanted side effects.
Methods
Mesenchymal stem cells (MSCs) have been postulated to possess unique immune regulatory function. We explored the immunomodulatory property of human and porcine MSCs for the treatment of δ-sarcoglycan-deficient dystrophic hamster muscle without immunosuppression. Circulating as well as tissue markers of inflammation were analyzed. Muscle regeneration and stem cell fate were characterized.
Results
Total white blood cell counts and leukocyte distribution profiles were similar among the saline- and MSC-injected dystrophic hamsters one month post treatment. Circulating levels of immunoglobulin A, vascular cell adhesion molecule-1, myeloperoxidase, and major cytokines involved in inflammatory response were not elevated by MSCs, nor were expression of the leukocyte common antigen CD45 and the cytokine transcriptional activator NF-κB in the injected muscle. Treated muscles exhibited increased cell cycle activity and attenuated oxidative stress. Injected MSCs were found to be trapped in the musculature, and contribute to both preexisting and new muscle fibers, and mediates capillary formation.
Conclusions
Intramuscular injection of non-autologous MSCs can be safely used for the treatment of dystrophic muscle in immunocompetent hosts without inflaming the host immune system.
Keywords: muscular dystrophy, δ-sarcoglycan, regeneration, mesenchymal stem cell
Introduction
Although stem cell therapies have entered the realm of clinical trials, more studies are needed to determine the most effective cell type for various therapeutic regiments. Obtaining sufficient amounts of stem cells required for clinical applications can be a daunting task. The use of autologous stem cells is not always desirable since aged patients often exhibit declined stem cell quality and/or quantity (1-3). Stem cell function may also be pathologically impaired as demonstrated in diabetes and heart disease (4-7). Certain disease-causing genotypes may preclude therapeutic use of autologous stem cells due to the inherent genetic defects (8, 9). From a logistic viewpoint, the use of non-autologous stem cells isolated from healthy donors offers a major advantage since these stem cells can be thoroughly tested and formulated into off-the-shelf medicine in advance. However, the issues of HLA compatibility and related immunosuppression can potentially compromise therapeutic efficacy and cause unwanted side effects.
Bone marrow mesenchymal stem cells (MSCs) have been postulated to possess unique immunomodulatory properties that can be explored for non-autologous stem cell-based therapeutics (10, 11). The immune phenotype of culture-expanded MSCs is widely described as MHC Class I+, MHC Class II-, CD40-, CD80-, and CD86-, which is regarded as non-immunogenic, suggesting that MSCs may even trespass species defense barriers (10-12). This feature of MSCs together with the ease of MSC expansion in culture has been explored to facilitate bone marrow transplantation since conventional myeloablative bone marrow transplantation generally achieves minimal donor stromal engraftment (13). The use of MSCs in allogeneic and xenogeneic transplantation has been reported to reduce the incidence and severity of graft-vs-host disease (GVHD), indicating an unique immunomodulatory property of MSCs (14-16). On the other hand, intracardiac injection of MSCs was found to induce immune responses (17). Clearly, in vivo studies using appropriate pathogenetic or pathophysiologic animal models are needed to address the safety and efficacy of non-autologous MSCs in immunocompetent hosts.
Muscular dystrophies are heterogeneous disorders characterized by tissue inflammation and progressive degeneration of skeletal muscle with or without cardiac complication (18). Muscular dystrophy can be caused by mutations in genes encoding components of the dystrophin glycoprotein complex, which links laminin-α2 in the basal lamina to the actin cytoskeletal network and can serve as a dynamic scaffold for diverse signaling molecules (19). In skeletal muscle, the transmembrane components of the dystrophin complex include α, β, γ, and δ sarcoglycans, and mutations or deletions in each of the four sarcoglycan genes can cause muscular dystrophy in humans and rodents (20). Although both gene and cell therapies have been attempted to treat muscular dystrophy, host immune reactions against the expressed gene product and transplanted cells limit the efficacy of these therapeutic approaches and more efficient immunosuppression is often deemed necessary (21-24). In this aspect, the immunomodulatory property of MSCs may be particularly well suited for tackling the inflammatory nature of dystrophic muscle. We here report that human and porcine MSCs upon intramuscular injection are well tolerated by δ-sarcoglycan-deficient dystrophic hamsters, and the MSC treatment leads to prominent muscle regeneration and attenuated oxidative stress without inflaming the host immune system.
Materials and Methods
Animals
Bio-F1B (normal control) and Bio-TO2 (δ-sarcoglycan null) male hamsters were obtained from BioBreeders (Watertown, MA, USA). Animals were sacrificed by CO2 narcosis. Procedures and protocols conformed to institutional guidelines for the care and use of animals in research.
MSC culture
Fresh human bone marrows were purchased from Lonza Walkersville (Gaithersburg, MD, USA). Human mesenchymal stem cells (HMSCs) were prepared and expanded as described for porcine MSCs (25, 26). HMSCs and PMSCs were culture-expanded in MesenPro RS medium (Invitrogen, Grand Island, NY, USA) and DMEM/F12 medium containing 10% fetal bovine serum, respectively. Both media were also supplemented with 2 mM glutamine, 50 μg/ml gentamycin, and 0.125 μg/ml Fungizone. The amplified MSCs were found to express multiple surface markers such as CD29, CD49c, CD49e, CD51, CD73, CD90, CD105, and CD106, and were negative for CD31, CD34, CD45, CD133, and VEGF-R2. These MSCs were also found to express a broad spectrum of trophic factors such as angiopoietin-1, brain-derived neurotrophic factor (BDNF), bone morphogenetic protein-7 (BMP-7), fibroblast growth factor-1 (FGF-1), FGF-2, FGF-5, FGF-7, FGF-9, granulocyte-colony stimulating factor (G-CSF), growth and differentiation factor-9 (GDF-9), hepatocyte growth factor (HGF), insulin-like growth factor-1 (IGF-1), IGF-2, interleukin-6 (IL-6), leukemia inhibitory factor (LIF), monocyte chemotactic protein-1 (MCP-1), macrophage-colony stimulating factor (M-CSF), nerve growth factor-β (NGF-β), stem cell factor (SCF), stromal derived factor-1 (SDF-1), transforming growth factor-β1 (TGF-β1), and vascular endothelial growth factor-A (VEGF-A). Only MSC cultures exhibiting robust growth capacity (typically less than 5 passages) were used for cell transplantation. Live cell labeling using the nuclear affinity dye DAPI (4′,6′-diamidino-2-phenylindole) was described in our recent work (27).
Intramuscular MSC implantation
Approximately 0.5-1 million trypsinized MSCs resuspended in 0.15 ml of HBSS were administered via direct injection into each left and right hamstring muscle of 4-month old hamsters. Animals received a second intramuscular MSC implantation two weeks later. Injected muscles were harvested 2-3 weeks after the second injection, and processed for biochemical and histological analyses. Control TO2 hamsters received the same volume of HBSS.
Quantitative reverse transcription-polymerase chain reaction (qRT-PCR)
Protocols for RNA preparation and qRT-PCR analysis were as described (25). In brief, RNA samples were isolated using the Qiagen's RNeasy RNA isolation kit. cDNA synthesis was performed using M-MLV reverse transcriptase from Promega (Madison, WI, USA). PCR primers were designed using MIT's Primer3 software. Real-time PCR analysis using B-R SYBR SuperMix (Quanta Biosciences, Gaithersburg, MD, USA) was performed with MyIQ PCR machine (Bio-Rad, Hercules, CA, USA). Melting curve analysis was performed to ensure a single amplicon for each gene. Data were analyzed by the 2ΔΔCT method using β2-microglobulin (b2M) as the reference gene. Hamster primer sequences and PCR product size in base pairs (bp) are: b2M-TCTCTTGGCTCACAGGGAGT (5′) and ATGTCTCGTTCCCAGGTGAC (3′), 108 bp; CD45- GGCGTACAGGCACCTACATT (5′) and ATGTACTGGGCCTCCACTTG (3′), 136 bp; M C P-1- TAGCATCCACGTGCTGTCTC (5′) and TGCTGCTGGTGATTCTCTTG (3′), 122 bp; IL-1a- ACGACTGGGTTTCAATCAGG (5′) and CTGCATGACTCGCCTTATCA (3′), 142 bp; IL-1b- CCAGGATGAGGACCTGAGAA (5′) CGAGGCATTTCTGTTGTTCA (3′), 149 bp; IL-4- CCACGGAGAAAGACCTCATCTG (5′) and GGGTCACCTCATGTTGGAAATAA (3′), 72 bp; IL-6-CTCCGCAAGAGACTTCCATC (5′) and ACCAAACCTCCGACTTGTTG (3′), 156 bp; NF-κB-GATCTCCCGAATGGACAAGA (5′) and GAACCGAACCTCAATGTCGT (3′), 100 bp; cyclin A-CCTGCAAACTGCAAAGTTGA (5′) and AAAGGCAGCTCCAGCAATAA (3′), 111 bp; cyclin B- CCAAATCCGAGAAATGGAGA (5′) and TTGGCTAGCGTGTGTTGTTC (3′), 132 bp; cyclin D-AGAACCTGCTGACCATCGAG (5′) and CCTCGCAGACCTCTAGCATC (3′), 123 bp; porcine 16s rRNA-ACATAACTTAACATGGACTAGCA (5′) and GGTTATTTTGACTGGTTTGTCTAG (3′). Oligonucleotides were synthesized by Midland Certified Reagent Company (Midland, TX, USA). Injected PMSCs were quantified by qRT-PCR using pig-specific 16S rRNA primers. A standard curve was created by generating a serial dilution curve plotting the threshold cycles of the 16s rRNA gene against known number of PMSCs.
Lipid peroxidation assay
Freshly isolated muscle was minced and homogenized in an ice-cold lysis solution (phosphate-buffered saline supplemented with 0.1% Triton X-100 and 2 mM EDTA). Tissue debris was removed by a 10-sec low speed spin. Crude extracts were recovered and protein concentrations were determined using the BCA protein assay method (Pierce, Rockford, IL, USA). Extracts were treated with 10% trichloroacetic acid (TCA) on ice for 30 min followed by centrifugation to remove precipitates. Lipid peroxidation assay kit was obtained from Biomedical Research Service (Buffalo, NY, USA). The TCA-treated muscle extracts were incubated with freshly prepared 6.5 mg/ml thiobarbituric acid at 95°C for 30 min. Reaction products were extracted with n-butanol and measured at 540 nm. MDA concentrations were calculated using a molar extinction coefficient of 1.56 × 105 cm-1M-1.
Blood cell and serum analysis
Hamster blood samples were collected by orbital bleeding. Blood leukocyte counts were analyzed by BioReliance (Rockville, MD, USA). Hamster sera were analyzed by Rules-Based Medicine, Inc. (Austin, TX, USA) using their rodent MAP analysis program.
Immunofluorescence analysis
Excised muscle was either quickly frozen in OCT medium in an isopentane/dry ice slurry to preserve DAPI fluorescence or fixed in freshly prepared 4% paraformaldehyde (PFA) overnight. Frozen tissue was cut to 10-μm sections, fixed in 2% PFA for 5 min at 4°C, and then washed with normal saline containing 0.1% Triton X-100 before histological analysis. Sections were stained with a myosin heavy chain antibody (28) or vWF antibody (#PC313; Calbiochem, San Diego, CA, USA) followed by TRITC- or FITC-conjugated secondary antibodies. Stained tissue sections were digitally imaged using an epifluorescence microscope equipped with a structured illumination system for confocal-like optical sectioning (Zeiss Axioimager Z1 with Apotome). 400× z-stack images were collected at 0.5-μ thick layers. Images were viewed using Axiovision LE Version 4.6 software.
Morphometric analysis
H&E staining of tissue sections was performed by the University at Buffalo histology core facility, and the sections were used for morphometric analysis using Axiovision software. 200× images were digitally collected using an epifluorescence microscope (Nikon Eclipse E600). For calculation of cross section fiber areas, pixel squares were converted to μm2 using a predetermined conversion factor for the microscope used. Muscle nuclear densities were obtained by dividing total counted muscle nuclei by total fiber areas in mm2. These analyses were performed in a blind-folded fashion.
Results
We have recently characterized bone marrow-derived MSCs (25, 26) and a δ-sarcoglycan-deficient dystrophic hamster model (BIO-TO2 strain) (29). Skeletal muscle degeneration in the TO2 strain hamsters is evident as early as one month of age, and the pathologic tissue remodeling process continues with age (30, 31). Using the dystrophic hamster model, we examined whether the immunomodulatory property of MSCs might promote skeletal muscle regeneration without the use of immunosuppression. Both human MSCs (HMSC) and porcine MSCs (PMSC) were tested here as the pig has long been considered the most desirable source species for organ transplantation due to similarities between human and porcine MHC antigens.
The TO2 hamsters also exhibit cardiac contractile dysfunction due to dilated cardiomyopathy, which can be conveniently monitored by echocardiograph (29). Since many muscular dystrophy patients also suffer from various forms of cardiomyopathy (32), our therapeutic regime was designed to target both the muscle and heart. We found that the trophic actions of MSCs elicited significantly improved cardiac contractile function even when the MSCs were delivered intramuscularly (manuscript in preparation). We thus took advantage of echocardiograph to determine the optimal injection interval and numbers of MSCs required for cardiac improvement. Initial cell dosage studies indicated 0.5-1 million MSCs per hamstring muscle to be sufficient to elicit initial cardiac improvement, and a boosting injection two weeks later to cause a greater cardiac improvement. This injection regime was adopted for both HMSCs and PMSCs. The injected muscle and blood were harvested for analysis at two weeks and one month after MSC treatment. In spite of the xenogeneic cell implantation, routine inspection revealed no adverse host tissue reaction. Blood cell analysis showed that total white blood cell counts and leukocyte distribution profiles were similar among the saline control, HMSC-, and PMSC-receiving hamsters (Fig. 1A). Immunoassays revealed that circulating levels of IgA along with those cytokines involved in inflammatory response such as interleukin (IL)-4 (Fig. 1B) as well as IL-1b, IL-10, lymphotactin, macrophage-derived chemokine (MDC), monocyte inhibitory protein (MIP)-2, and tumor necrosis factor (TNF)-α (data not shown) were not elevated by the MSC injections. Circulating levels of vascular cell adhesion molecule (VCAM)-1, an adhesion molecule highly expressed by inflamed endothelium, were also not elevated by MSCs (Fig. 1B). Notably, circulating levels of the neutrophil enzyme myeloperoxidase, which is another indicator of inflammation, were significantly reduced by MSCs (Fig. 1B).
Figure 1.

Analyses of blood cells and circulating markers show no sign of host immune response after intramuscular implantation of human and porcine MSCs in TO2 dystrophic hamsters. Panel A: Hamster blood samples were collected at two weeks and one month post injection by orbital bleeding. Similar results were obtained for both time points. Data shown were from the one-month time point. N=9 for TO2 control (saline injection). N=6 for HMSC- and PMSC-injected hamsters. No statistically significant difference was noted between groups. Panel B: Serum markers of inflammation were analyzed at one month. Data for IL-1b, IL-10, lymphotactin, MDC, MIP-2, and TNFα were not shown. N=3 for each group. The only statistically significant difference was noted for myeloperoxidase between control and PMSC (*P<0.05), showing reduced circulating myeloperoxidase after PMSC injection. Although HMSC also reduced myeloperoxidase, the difference did not reach statistical significance. Data shown were means ± SEM.
We further used the more sensitive real-time qRT-PCR method to quantify expression of several inflammatory cytokines in the injected muscle, and the assay showed increased IL-1b, CD45 (a leukocyte common antigen), and NF-κB expression in the TO2 muscle compared to the normal F1B muscle (Fig. 2A), reflecting the inflammatory nature of the disease. Decreased expression of IL-4, IL-6, CD45, and NF-κB were noted after MSC treatments (Fig. 2A). Significantly downregulated expression of NF-κB, a transcription factor involved in immune cytokine induction, is consistent with the observed down-regulation of myeloperoxidase after MSC administration (Fig. 1). Since myeloperoxidase is linked to the production of oxidized lipids, free radical-induced oxidative damages also represent a reliable indicator of tissue inflammation. We and other investigators have demonstrated that dystrophic muscles are exquisitely sensitive to the oxidative stress (29, 33). By measuring tissue levels of malondialdehyde (MDA), the end product of lipid peroxidation, we demonstrated that although the untreated TO2 hamster muscle contained significantly higher levels of MDA than the normal F1B hamster muscle, the MSC-treated TO2 muscle exhibited normalized levels of MDA (Fig. 2B), indicating significant attenuation of oxidative stress in the diseased muscle.
Figure 2.

qRT-PCR and lipid peroxidation analyses of injected muscle show no sign of host tissue inflammatory response after injection of xenogeneic MSCs. Panel A: qRT-PCR analysis comparing F1B, saline- and PMSC-injected TO2 hamstring muscles at one month post injection. Data were presented as fold change vs. the F1B group. N=4 for each group. Significantly reduced expression of IL-6 and NF-κB were noted after MSC treatment. *P<0.05 vs F1B; +P<0.05 vs TO2 control. Panel B: Lipid peroxidation assays measured muscle contents of malondialdehyde (MDA) in μM. Hamstring muscles from normal F1B, saline- and PMSC-injected TO2 hamsters were compared at one month. N=4 for each group. Data shown were means ± SEM. Statistically significant differences were indicated.
Previous studies showed that TO2 hamsters exhibit repeated cycles of skeletal muscle degeneration and regeneration marked by central nucleation in the newly regenerated fibers (30, 31). H&E staining shown in Figure 3A confirms the presence of central nuclei in muscle cells along with fibrosis in the TO2 but not F1B hamsters. Notably, the MSC-treated TO2 muscle exhibited further increased central nucleation with reduced fibrosis (Fig. 3A). Since central nucleation indicates skeletal muscle regeneration, qRT-PCR was used to analyze gene expression involved in cell cycle progression. Consistent with the MSC-mediated promotion of central nucleation, the MSC-injected muscle exhibited significantly increased expression of hamster cyclin D and c-myc genes, both of which are known to promote cell cycle progression (Fig. 3B). Morphometric analysis of the H&E-stained sections indicates that the control TO2 muscle contain ∼30% cells with central nuclei while the treated TO2 and normal F1B muscles contain ∼55% and ∼5% cells with central nuclei, respectively (Fig. 4A). Total nuclear densities are the highest in the MSC-treated TO2 muscle, and the lowest in the normal F1B muscle (Fig. 4B). Average fiber sizes were found to inversely correlate with the nuclear densities (Fig. 4C), reflecting the smaller fiber sizes of the newly regenerated muscle cells. Fiber size distribution profiles further illustrate the abundance of smaller muscle cells in the MSC-treated TO2 muscle and larger muscle cells in the normal F1B muscle (Fig. 4D).
Figure 3.
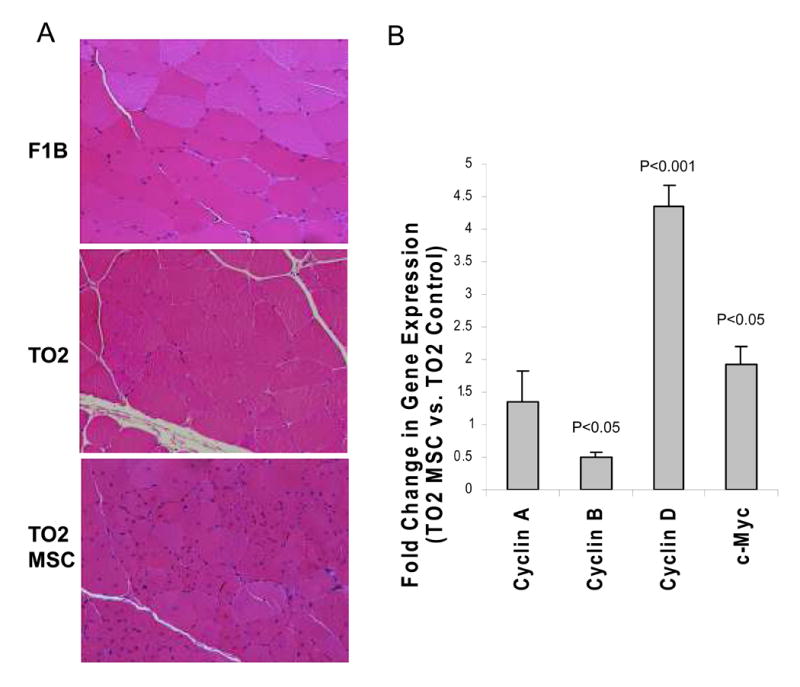
Histological and real time qRT-PCR analyses show active muscle regeneration after MSC treatment. Panel A: representative H&E staining of hamstring muscle sections from F1B, saline- and PMSC-injected TO2 hamsters at one month post treatment. Differences in fibrosis contents and central nucleation were clearly visible among groups. Panel B: Real time qRT-PCR analysis of saline- and PMSC-injected TO2 hamster muscles showing increased expression of cyclin D and c-myc three days post MSC injection. N=4 for each group. Data shown were means ± SEM. Statistically significant differences were indicated.
Figure 4.

Blind-folded morphometric analysis demonstrates MSC-mediated muscle regeneration. % fibers with central nucleation (panel A), muscle nuclear densities (panel B), average cross section areas (panel C), and fiber size distribution profiles (panel D) were analyzed in a blind-folded fashion. Numbers of hamstring fibers analyzed were N=157 for F1B hamsters, N=225 for saline-injected TO2 hamsters, and N=156 for PMSC-injected TO2 hamsters. Data shown were means ± SEM. Statistically significant differences were indicated.
We next determined whether the implanted xenogeneic MSCs might directly participate in myogenic and endothelial differentiation. We recently demonstrated that the membrane permeable nuclear affinity dye DAPI can be used to track MSCs implanted in small and large animals since the labeled MSCs emit prominent nuclear localized blue fluorescence, allowing for in vivo MSC tracking with little ambiguity (27). Immunostaining of the TO2 hamster muscle injected with DAPI-labeled MSCs was presented in Figure 5, illustrating anatomically distinct MSCs in four panels: (A) a string of peripheral MSC nuclei positioned along the length of preexisting fibers, (B) a centrally located MSC nucleus in a muscle fiber, (C) MSC nuclei embedded in a vWF-positive capillary, and (D) clusters of interstitial MSCs. The peripheral locations of the MSC nuclei identified in panel A suggest MSC contribution to preexisting muscle fibers likely through cell fusion. The central location of the MSC nucleus identified in panel B indicates its active participation in new fiber formation. The endothelial cell fate of implanted MSCs is identified in panel C, illustrating MSC participation in new capillary formation. qRT-PCR quantification of the injected PMSCs one month post implantation revealed little, if any, cell migration from the injected site to other tissues (Fig. 5E), indicating that the vast majority of the injected MSCs were trapped in the musculature.
Figure 5.
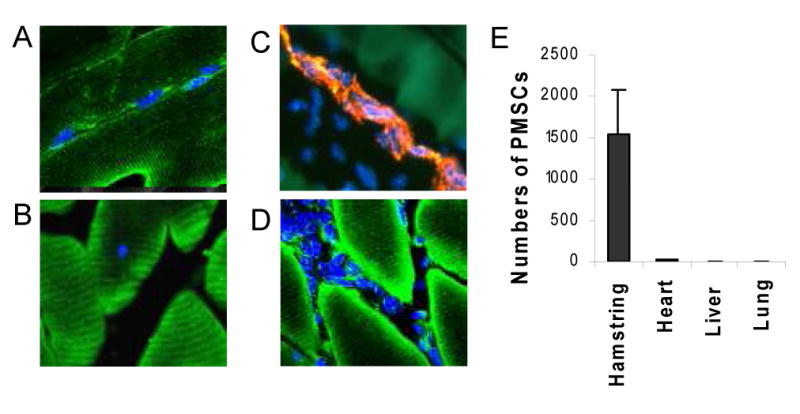
Cell tracking analysis illustrates injected MSCs contributing to preexisting and new fibers as well as capillaries. PMSC nuclei were labeled with DAPI (blue fluorescence) prior to injection in the TO2 hamstring muscle, and cryosections were prepared from the injected muscle two weeks after injection. FITC fluorescence (green) represents MHC immunostaining. TRITC fluorescence (red) represents vWF immunostaining. Panel A shows a string of peripheral MSC nuclei positioned along the length of preexisting fibers. Panel B shows a centrally located MSC nucleus. Panel C shows MSC nuclei embedded in a vWF-positive capillary. Panel D shows clusters of interstitial MSCs. Panel E: qRT-PCR quantification of retained PMSCs after 1 month using pig-specific 16S rRNA primers. Cell numbers were calculated based on each gram of tissue.
Discussion
Several types of adult stem cells have been used for skeletal muscle therapies including muscle satellite cells, side population cells, mesoangioblasts, hematopoietic stem cells, and MSCs (21, 22). Efforts were made in these cell transplantation studies to minimize immune rejection either through animal strain match, use of allogeneic stem cells, immunosuppression, or adoption of immunodeficient animals. Surprisingly, little data on potential host immune responses following stem cell administration are available in the literature. Given the recognition that host immune responses can compromise the efficacy of stem cell therapeutics (22), we focused our attention here on the hamster immune responses after the transplantation of human and porcine MSCs. Consistent with the general notion that MSCs are immune privileged cells (10), neither human nor porcine MSCs triggered adverse immune reactions based on our analyses of blood cells, serum inflammatory markers, tissue oxidative stress, and expression of immune cytokines, CD45, and NF-κB in muscle. Further, down-regulated immune cytokines, myeloperoxidase, and NF-κB, a transcription factor involved in immune cytokine induction, were observed after cell implantation, indicating a potent immunomodulatory property of MSCs. Since oxidative stress triggered by inflammation has been implicated in the pathogenesis of many diseases including muscular dystrophy, this immunomodulatory property of MSCs is ideally suited for treating degenerative and inflammatory diseases.
Our data suggest that the immunomodulatory, anti-oxidative, myogenic, and endothelial potentials of non-autologous MSCs can be harnessed to achieve muscle regeneration without the need for host immunosuppression. It remains unclear whether the immunosuppressive effect of intramuscularly injected MSCs can be sustained as the current study only involved a one-month time period. Further, studies demonstrating the induction of T cell anergy by MSCs were based on systemic infusion of MSCs (34, 35). Intramuscularly delivered MSCs may be less efficient in inducing T cell anergy as these cells were largely trapped in the musculature. Studies by Dezawa et al also reached the same conclusion that intramuscularly delivered MSCs were largely retained in the injected muscle bed (36). Another limitation of the current therapeutic approach is the rapid loss of the injected MSCs after implantation. Although the trophic actions of the retained MSCs appeared capable of activating the host satellite cells, new myofibers formed by the host satellite cells remain deficient in δ-sarcoglycan, and thus should remain injury prone. It will be of interest to determine whether the pro-survival trophic factors produced by MSCs may help maintain the viability of the newly formed δ-sarcoglycan-deficient myofibers.
We show that xenogeneic MSCs contributed to preexisting and new muscle fibers as well as capillaries one month after implantation. Analysis of hamster muscle cyclin and c-myc gene expression provided evidence that muscle satellite cells were also activated by MSCs, resulting in more than 50% fibers containing central nuclei in the injected dystrophic hamstring muscle. Importantly, we found that although MSCs were directly injected into the hamstring muscle, the quadriceps muscle also exhibited normalized lipid peroxidation and increased central nucleation (data not shown). We and others have shown that the δ-sarcoglyan deficient hamsters develop dilated cardiomyopathy leading to heart failure (29, 37, 38). Intramuscular injection of MSCs as reported here was further found to greatly improve cardiac function and promote cardiomyocyte regeneration in the TO2 hamster heart (manuscript in preparation). These beneficial effects of MSCs are most likely due to local production of humoral mediators capable of remote functioning. Indeed, the cardiovascular therapeutic effects of MSCs have been largely attributed to their trophic actions (39, 40), although the effective tropic factor(s) remains to be identified. Along this line, we and others have shown that MSCs are capable of producing a broad spectrum of trophic factors, and this capacity can be further boosted by genetic engineering of MSCs (25, 41, 42). Thus, MSC engineering aimed at promoting trophic factor outputs may represent an effective therapeutic module.
In conclusion, we show that non-autologous MSCs can achieve muscle regeneration in immunocompetent animals. Since skeletal muscle is the largest organ in the body, effective clinical therapy for dystrophic muscle is expected to require an enormous amount of MSCs, which can be fulfilled by the robust growth potential of MSCs in vitro. Although the long-term effect of non-autologous MSCs needs to be further examined, we envision that this approach can facilitate future stem cell clinical trials, which undoubtedly will require large quantities of safe, consistent, and potent batches of stem cells in a timely fashion.
Acknowledgments
The work is supported by NIH (HL84590) and New York State Stem Cell Science Program.
Abbreviations
- DAPI
4′,6′-diamidino-2-phenylindole
- MSC
mesenchymal stem cell
- qRT-PCR
quantitative reverse transcription-polymerase chain reaction
References
- 1.Keymel S, Kalka C, Rassaf T, Yeghiazarians Y, Kelm M, Heiss C. Impaired endothelial progenitor cell function predicts age-dependent carotid intimal thickening. Basic Res Cardiol. 2008;103(6):582. doi: 10.1007/s00395-008-0742-z. [DOI] [PubMed] [Google Scholar]
- 2.D'Ippolito G, Schiller PC, Ricordi C, Roos BA, Howard GA. Age-related osteogenic potential of mesenchymal stromal stem cells from human vertebral bone marrow. J Bone Miner Res. 1999;14(7):1115. doi: 10.1359/jbmr.1999.14.7.1115. [DOI] [PubMed] [Google Scholar]
- 3.Lehrke S, Mazhari R, Durand DJ, et al. Aging impairs the beneficial effect of granulocyte colony-stimulating factor and stem cell factor on post-myocardial infarction remodeling. Circ Res. 2006;99(5):553. doi: 10.1161/01.RES.0000238375.88582.d8. [DOI] [PubMed] [Google Scholar]
- 4.Satoh M, Ishikawa Y, Takahashi Y, Itoh T, Minami Y, Nakamura M. Association between oxidative DNA damage and telomere shortening in circulating endothelial progenitor cells obtained from metabolic syndrome patients with coronary artery disease. Atherosclerosis. 2008;198(2):347. doi: 10.1016/j.atherosclerosis.2007.09.040. [DOI] [PubMed] [Google Scholar]
- 5.Kissel CK, Lehmann R, Assmus B, et al. Selective functional exhaustion of hematopoietic progenitor cells in the bone marrow of patients with postinfarction heart failure. J Am Coll Cardiol. 2007;49(24):2341. doi: 10.1016/j.jacc.2007.01.095. [DOI] [PubMed] [Google Scholar]
- 6.Fadini GP, Sartore S, Schiavon M, et al. Diabetes impairs progenitor cell mobilisation after hindlimb ischaemia-reperfusion injury in rats. Diabetologia. 2006;49(12):3075. doi: 10.1007/s00125-006-0401-6. [DOI] [PubMed] [Google Scholar]
- 7.Gallagher KA, Liu ZJ, Xiao M, et al. Diabetic impairments in NO-mediated endothelial progenitor cell mobilization and homing are reversed by hyperoxia and SDF-1 alpha. J Clin Invest. 2007;117(5):1249. doi: 10.1172/JCI29710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Wallace SR, Oken MM, Lunetta KL, Panoskaltsis-Mortari A, Masellis AM. Abnormalities of bone marrow mesenchymal cells in multiple myeloma patients. Cancer. 2001;91(7):1219. doi: 10.1002/1097-0142(20010401)91:7<1219::aid-cncr1122>3.0.co;2-1. [DOI] [PubMed] [Google Scholar]
- 9.Li Y, Zhang C, Xiong F, et al. Comparative study of mesenchymal stem cells from C57BL/10 and mdx mice. BMC Cell Biol. 2008;9:24. doi: 10.1186/1471-2121-9-24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tyndall A, Walker UA, Cope A, et al. Immunomodulatory properties of mesenchymal stem cells: a review based on an interdisciplinary meeting held at the Kennedy Institute of Rheumatology Division, London, UK, 31 October 2005. Arthritis Res Ther. 2007;9(1):301. doi: 10.1186/ar2103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Gotherstrom C. Immunomodulation by multipotent mesenchymal stromal cells. Transplantation. 2007;84(1 Suppl):S35. doi: 10.1097/01.tp.0000269200.67707.c8. [DOI] [PubMed] [Google Scholar]
- 12.Tse WT, Pendleton JD, Beyer WM, Egalka MC, Guinan EC. Suppression of allogeneic T-cell proliferation by human marrow stromal cells: implications in transplantation. Transplantation. 2003;75(3):389. doi: 10.1097/01.TP.0000045055.63901.A9. [DOI] [PubMed] [Google Scholar]
- 13.Fan TX, Hisha H, Jin TN, et al. Successful allogeneic bone marrow transplantation (BMT) by injection of bone marrow cells via portal vein: stromal cells as BMT-facilitating cells. Stem Cells. 2001;19(2):144. doi: 10.1634/stemcells.19-2-144. [DOI] [PubMed] [Google Scholar]
- 14.Le Blanc K, Tammik C, Rosendahl K, Zetterberg E, Ringden O. HLA expression and immunologic properties of differentiated and undifferentiated mesenchymal stem cells. Exp Hematol. 2003;31(10):890. doi: 10.1016/s0301-472x(03)00110-3. [DOI] [PubMed] [Google Scholar]
- 15.Dean RM, Bishop MR. Graft-versus-host disease: emerging concepts in prevention and therapy. Curr Hematol Rep. 2003;2(4):287. [PubMed] [Google Scholar]
- 16.Aggarwal S, Pittenger MF. Human mesenchymal stem cells modulate allogeneic immune cell responses. Blood. 2005;105(4):1815. doi: 10.1182/blood-2004-04-1559. [DOI] [PubMed] [Google Scholar]
- 17.Poncelet AJ, Vercruysse J, Saliez A, Gianello P. Although pig allogeneic mesenchymal stem cells are not immunogenic in vitro, intracardiac injection elicits an immune response in vivo. Transplantation. 2007;83(6):783. doi: 10.1097/01.tp.0000258649.23081.a3. [DOI] [PubMed] [Google Scholar]
- 18.McNally EM, Pytel P. Muscle diseases: the muscular dystrophies. Annu Rev Pathol. 2007;2:87. doi: 10.1146/annurev.pathol.2.010506.091936. [DOI] [PubMed] [Google Scholar]
- 19.Rando TA. The dystrophin-glycoprotein complex, cellular signaling, and the regulation of cell survival in the muscular dystrophies. Muscle Nerve. 2001;24(12):1575. doi: 10.1002/mus.1192. [DOI] [PubMed] [Google Scholar]
- 20.Ozawa E, Noguchi S, Mizuno Y, Hagiwara Y, Yoshida M. From dystrophinopathy to sarcoglycanopathy: evolution of a concept of muscular dystrophy. Muscle Nerve. 1998;21(4):421. doi: 10.1002/(sici)1097-4598(199804)21:4<421::aid-mus1>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]
- 21.Blau HM. Cell therapies for muscular dystrophy. N Engl J Med. 2008;359(13):1403. doi: 10.1056/NEJMcibr0805708. [DOI] [PubMed] [Google Scholar]
- 22.Peault B, Rudnicki M, Torrente Y, et al. Stem and progenitor cells in skeletal muscle development, maintenance, and therapy. Mol Ther. 2007;15(5):867. doi: 10.1038/mt.sj.6300145. [DOI] [PubMed] [Google Scholar]
- 23.Yuasa K, Sakamoto M, Miyagoe-Suzuki Y, et al. Adeno-associated virus vector-mediated gene transfer into dystrophin-deficient skeletal muscles evokes enhanced immune response against the transgene product. Gene Ther. 2002;9(23):1576. doi: 10.1038/sj.gt.3301829. [DOI] [PubMed] [Google Scholar]
- 24.Herzog RW, Arruda VR. Substantial immune suppression required in gene therapy for muscular dystrophy? Neuromuscul Disord. 2008;18(1):83. doi: 10.1016/j.nmd.2007.08.007. [DOI] [PubMed] [Google Scholar]
- 25.Lin H, Shabbir A, Molnar M, et al. Adenoviral expression of vascular endothelial growth factor splice variants differentially regulate bone marrow-derived mesenchymal stem cells. J Cell Physiol. 2008;216:458. doi: 10.1002/jcp.21414. [DOI] [PubMed] [Google Scholar]
- 26.Vacanti V, Kong E, Suzuki G, Sato K, Canty JM, Jr, Lee T. Phenotypic changes of adult porcine mesenchymal stem cells induced by prolonged passaging in culture. J Cell Physiol. 2005;205:194. doi: 10.1002/jcp.20376. [DOI] [PubMed] [Google Scholar]
- 27.Leiker M, Suzuki G, Iyer VS, Canty JMJ, Lee T. Assessment of a Nuclear Affinity Labeling Method for Tracking Implanted Mesenchymal Stem Cells. Cell Transplantation. 2008;17:911. doi: 10.3727/096368908786576444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kositprapa C, Zhang B, Berger S, Canty JMJ, Lee TC. Calpain-mediated proteolytic cleavage of troponin I induced by hypoxia or metabolic inhibition in cultured neonatal cardiomyocytes. Mol Cell Biochem. 2000;214:47. doi: 10.1023/a:1007160702275. [DOI] [PubMed] [Google Scholar]
- 29.Missihoun C, Zisa D, Shabbir A, Lin H, Lee T. Myocardial oxidative stress, osteogenic phenotype, and energy metabolism are differentially involved in the initiation and early progression of delta-sarcoglycan-null cardiomyopathy. Mol Cell Biochem. 2008 doi: 10.1007/s11010-008-9908-0. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kato Y, Iwase M, Takagi K, et al. Differential myolysis of myocardium and skeletal muscle in hamsters with dilated cardiomyopathy. Circ J. 2006;70:1497. doi: 10.1253/circj.70.1497. [DOI] [PubMed] [Google Scholar]
- 31.Straub V, Duclos F, Venzke DP, et al. Molecular pathogenesis of muscle degeneration in the delta-sarcoglycan-deficient hamster. Am J Pathol. 1998;153(5):1623. doi: 10.1016/s0002-9440(10)65751-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hoogerwaard EM, Bakker E, Ippel PF, et al. Signs and symptoms of Duchenne muscular dystrophy and Becker muscular dystrophy among carriers in The Netherlands: a cohort study. Lancet. 1999;353:2116. doi: 10.1016/s0140-6736(98)10028-4. [DOI] [PubMed] [Google Scholar]
- 33.Disatnik MH, Chamberlain JS, Rando TA. Dystrophin mutations predict cellular susceptibility to oxidative stress. Muscle Nerve. 2000;23(5):784. doi: 10.1002/(sici)1097-4598(200005)23:5<784::aid-mus17>3.0.co;2-y. [DOI] [PubMed] [Google Scholar]
- 34.Zappia E, Casazza S, Pedemonte E, et al. Mesenchymal stem cells ameliorate experimental autoimmune encephalomyelitis inducing T-cell anergy. Blood. 2005;106(5):1755. doi: 10.1182/blood-2005-04-1496. [DOI] [PubMed] [Google Scholar]
- 35.Glennie S, Soeiro I, Dyson PJ, Lam EW, Dazzi F. Bone marrow mesenchymal stem cells induce division arrest anergy of activated T cells. Blood. 2005;105(7):2821. doi: 10.1182/blood-2004-09-3696. [DOI] [PubMed] [Google Scholar]
- 36.Dezawa M, Ishikawa H, Itokazu Y, et al. Bone marrow stromal cells generate muscle cells and repair muscle degeneration. Science. 2005;309(5732):314. doi: 10.1126/science.1110364. [DOI] [PubMed] [Google Scholar]
- 37.Fiaccavento R, Carotenuto F, Minieri M, et al. Stem cell activation sustains hereditary hypertrophy in hamster cardiomyopathy. J Pathol. 2005;205(3):397. doi: 10.1002/path.1717. [DOI] [PubMed] [Google Scholar]
- 38.Ryoke T, Gu Y, Ikeda Y, et al. Apoptosis and oncosis in the early progression of left ventricular dysfunction in the cardiomyopathic hamster. Basic Res Cardiol. 2002;97(1):65. doi: 10.1007/s395-002-8389-4. [DOI] [PubMed] [Google Scholar]
- 39.Gnecchi M, He H, Liang OD, et al. Paracrine action accounts for marked protection of ischemic heart by Akt-modified mesenchymal stem cells. Nat Med. 2005;11(4):367. doi: 10.1038/nm0405-367. [DOI] [PubMed] [Google Scholar]
- 40.Tang YL, Zhao Q, Qin X, et al. Paracrine action enhances the effects of autologous mesenchymal stem cell transplantation on vascular regeneration in rat model of myocardial infarction. Ann Thorac Surg. 2005;80:229. doi: 10.1016/j.athoracsur.2005.02.072. [DOI] [PubMed] [Google Scholar]
- 41.Kinnaird T, Stabile E, Burnett MS, et al. Marrow-derived stromal cells express genes encoding a broad spectrum of arteriogenic cytokines and promote in vitro and in vivo arteriogenesis through paracrine mechanisms. Circ Res. 2004;94(5):678. doi: 10.1161/01.RES.0000118601.37875.AC. [DOI] [PubMed] [Google Scholar]
- 42.Wieczorek G, Steinhoff C, Schulz R, et al. Gene expression profile of mouse bone marrow stromal cells determined by cDNA microarray analysis. Cell Tissue Res. 2003;311(2):227. doi: 10.1007/s00441-002-0671-3. [DOI] [PubMed] [Google Scholar]