

Abstract
Nicotinic acetylcholine receptors (nAChRs) regulate critical aspects of brain maturation during the prenatal, early postnatal, and adolescent periods. During these developmental windows, nAChRs are often transiently upregulated or change subunit composition in those neural structures that are undergoing major phases of differentiation and synaptogenesis, and are sensitive to environmental stimuli. Nicotine exposure, most often via tobacco smoke, but increasingly via nicotine replacement therapy, has been shown to have unique effects on the developing human brain. Consistent with a dynamic developmental role for acetylcholine, exogenous nicotine produces effects that are unique to the period of exposure and that impact the developing structures regulated by acetylcholine at that time. Here we present a review of the evidence, available from both the clinical literature and preclinical animal models, which suggests that the diverse effects of nicotine exposure are best evaluated in the context of regional and temporal expression patterns of nAChRs during sensitive maturational periods, and disruption of the normal developmental influences of acetylcholine. We present evidence that nicotine interferes with catecholamine and brainstem autonomic nuclei development during the prenatal period of the rodent (equivalent to first and second trimester of the human), alters the neocortex, hippocampus, and cerebellum during the early postnatal period (third trimester of the human), and influences limbic system and late monoamine maturation during adolescence.
Keywords: acetylcholine, adolescent, catecholamine, neonatal, nicotinic receptor, prenatal, smoking, tobacco
1. Introduction
Tobacco is one of the most heavily abused drugs in the world, with an estimated 21% of adults in the US identifying themselves as regular smokers (CDC, 2005). Smoking is highly addicting, with a nearly 97% relapse rate for unaided quit attempts (Hughes et al., 1992), and may cause one out of every five deaths each year (CDC, Health effects of smoking, 2008). Tobacco exposure is not only a health concern for adults, however. It has also been shown to exert deleterious effects on the health of the fetus, newborn, child, and adolescent (Rogers, 2008; Prokhorov et al., 2006; Kum-Nji et al., 2006; Mathers et al., 2006). Indeed, the long-term effects of tobacco exposure may be more profound at these younger ages, since maturation of neural circuitry is not yet complete and the inherent plasticity of the developing brain makes it particularly vulnerable to drug-induced alteration.
Whereas tobacco smoke contains over four thousand chemicals (US Dept. of Health and Human Services, 1989), there is substantial evidence that nicotine, the main psychoactive ingredient, exerts neurotoxic effects on developing brain. Nicotine can both activate and desensitize neuronal nicotinic acetylcholine receptors (nAChRs), which are ligand gated ion channels that mediate the physiological effects of the neurotransmitter, acetylcholine (ACh) (Dani, 2001). nAChRs are expressed by the first trimester in human brain and exhibit a complex pattern of developmental expression that is both region-specific and temporally regulated. In many brain areas there is a transient appearance of nAChRs during critical phases of development. Such findings suggest that acetylcholine, acting through nAChRs, may have an important functional role in modulating brain development, particularly during critical periods when brain maturation is most sensitive to perturbation.
The present review will discuss what is known of the complex roles of nAChRs in regulating brain development and will provide a conceptual framework for understanding the developmental consequences of clinical nicotine exposure, through smoking, secondhand smoke, or nicotine replacement therapy. Substantial emphasis will be placed on rodent models that provide valuable insight into the mechanisms underlying nicotine-induced pathophysiology. Although the rate of maturation of human and rodent nervous systems are vastly different, the brain contains core elements and developmental patterns that are universally shared across mammalian species, giving rise to strong parallels in their morphological development. Anatomical studies have correlated periods of rodent neural development to corresponding phases of human development, and have validated use of animal models to study human brain maturation (Bayer et al., 1993; Quinn, 2005). As is described below, there is substantial comparability in patterns of nAChR expression in developing human and rodent brain, and increasing evidence of convergence in the findings of the effects of nicotine on developmental outcomes in the clinical and animal literature. This body of work shows that the effects of nicotine are highly dependent on the timing of exposure, with specific neural vulnerabilities at each developmental phase. Furthermore, there is ample evidence that prenatal, early postnatal, and adolescent brain maturation is physiologically regulated by ACh via activation of nAChRs, and that exogenous nicotine produces marked and unique long-term deficits in developing structures by interfering with these cholinergic regulatory processes.
2. Pharmacology of nAChRs
nAChRs are pentameric ligand-gated cation channels that are widely distributed throughout human and rodent brain during all phases of development (Zoli et al., 1995; Broide and Leslie, 1999; Hellstrom-Lindahl and Court, 2000; Pentel et al., 2006). Depending on their subunit composition, nAChRs can gate both Na+ and Ca++, and exist in one of three conformational states: open, closed at rest, and desensitized, in which ligand binding cannot induce channel opening (Dani and Bertrand, 2007). There are twelve distinct genes that encode the subunit proteins, yielding seven α (α2-α10) and three β subunits (β2-β4) (McGehee, 1999). These combine in both homomeric (α7-α10) and heteromeric (α2-α6, β2-β4) configurations, which determine the pharmacological specificity, ion selectivity, and desensitization characteristics of the nAChR (Gotti et al., 2006).
The most common neuronal nAChR is the heteromeric α4β2, formed in 2α:3β stoichiometry that produces two ligand binding pockets at the α/β interfaces. α4β2 nAChRs bind nicotine with high affinity, in addition to epibatidine and cytisine (Dani and Bertrand, 2007), although epibatidine also binds to other heteromeric subunit combinations (e.g. α3β2 and α3β4) (Perry et al., 2002). These receptors primarily gate Na+ and have a range of effects at the neuronal level depending on their location. When located postsynaptically, they can mediate direct depolarization (Roerig et al., 1997; Alkondon et al., 1998). However, they are more predominant in presynaptic and preterminal regions, where they enhance overall cell excitability, determine the efficacy and spread of converging inputs, and indirectly modulate neurotransmitter release by triggering voltage-gated Ca++ channels (Tredway et al., 1999). They are slow to desensitize, but do so to concentrations of agonist much lower than that required for activation (Fenster et al., 1999). Thus, at concentrations of nicotine found in the brains of smokers, most of these receptors are in the desensitized state (Brody et al., 2006). Desensitization is an important concept in study interpretation where acute exposure to nicotine may potentiate a normal cholinergic response via activation, but chronic exposure may inhibit the normal actions of ACh via desensitization.
The other abundant nAChR in the brain is the α7 homomer, which contains five identical ligand binding pockets that are antagonized by α-bungarotoxin (Orr-Urtreger et al., 1997, Gotti et al., 2006; Mansvelder and Role, 2006). In contrast to α4β2, α7 nAChRs have a low affinity for nicotine, but can also be activated by endogenous choline (Dani and Bertrand, 2007). These receptors desensitize rapidly to high concentrations of agonist and have distinct ion selectivity, gating both Na+ and a large amount of Ca++ (1Na+:10Ca++) (Dani and Bertrand, 2007). When located presynaptically, α7 nAChRs may play critical modulatory roles by directly inducing Ca++-dependent neurotransmitter release. α7 nAChRs gate as much Ca++ as the NMDA glutamate receptor, but are not restricted by the depolarization-dependent Mg++ block and, thus, are implicated in synaptic plasticity at more hyperpolarized membrane potentials (McGehee and Role, 1995). This property may be particularly important during early cortical development, when depolarizing input from sensory experience is not yet available (see below).
Whereas α4β2 and α7 nAChRs are widely expressed in many brain regions, other subunit combinations are found in more spatially limited patterns, consistent with more distinct functional roles. Expression of the α3β4 nAChR, which is common in the periphery, is largely restricted to sensory structures and preganglionic autonomic nuclei in the brainstem (Winzer-Serhan and Leslie, 1997; Perry et al., 2002). These nAChRs have low agonist affinity, slow desensitization kinetics, and can induce burst firing (Luetje and Patrick, 1991; Papke and Heinemann, 1991). Cholinergic regulation of burst firing may be particularly important in generating the spontaneous waves of activity that lay the foundations of developing neural and sensory circuitry (see below).
Other nAChR subunits have a more limited distribution but also play important physiological roles. Whereas α2 subunit mRNA expression is largely restricted to cortex and hippocampus (Son and Winzer-Serhan, 2006), it has a critical role in learning and memory processes. α2-containing nAChRs gate synaptic plasticity and information flow in the hippocampus, and may mediate nicotine’s cognitive-enhancing effects in adults (Nakauchi et al., 2007). Although α5, α6, and β3 subunits only form functional cation channels when combined with both α and β subunits (Ramirez-Latorre et al., 1996; Wang et al., 1996), and also have a more restricted spatial pattern of expression than α4β2 and α7 nAChRs, these subunits have a profound impact on cholinergic signaling by altering Ca++ conductance, agonist sensitivity, and desensitization kinetics of nAChRs (Girod et al., 1999; Grady et al., 2007). For example, addition of the α5 subunit to α4 and β2 retains the high affinity binding site for nicotine, but induces a six fold increase in Ca++ gating (Tapia et al., 2007). Thus, the α4α5β2 nAChR retains the high agonist sensitivity of the α4β2, but has a Ca++ conductance that is comparable to that of the NMDA glutamate receptor or α7 nAChR (Girod et al., 1999; Tapia et al., 2007). Furthermore, in contrast to the traditional α4β2 nAChR, the α4α5β2 nAChR is rapidly desensitized by even brief agonist exposure (Ramierez-Latorre et al., 1996) and is resistant to upregulation by chronic nicotine (Mao et al., 2007). Given the critical roles of Ca++ in synaptic plasticity and development, and the suggestion that α5 is implicated in predisposition to early and heavy tobacco use (Schlaepfer et al., 2008; Berrettini et al., 2008), the regional and temporal expression profile of this subunit is of particular interest. The expression of α6 and β3 subunit mRNAs is most abundant in the catecholaminergic nuclei of the substantia nigra (SN), ventral tegmental area (VTA), locus coeruleus (LC) and nucleus tractus solitarius (Le Novere et al., 1996; Klink et al., 2001; Azam et al., 2007; O’Leary et al., 2008). These subunits are also expressed on dopaminergic terminals in the striatum and nucleus accumbens in suggested combinations of α6α4β2β3 and α6β2β3 (Gotti et al., 2006; Zoli et al., 2002; Exley et al., 2008).
All of these subunits and their resulting nAChRs display transient, region-specific spikes of expression that correspond temporally to critical developmental events. The cholinergic system drives maturational processes from the earliest stages of brain formation through adolescence, and in many cases the functional roles of these transiently-expressed nAChRs have been illustrated. Elucidating the pharmacological properties of complex nAChRs and characterizing their change in expression during the prenatal, postnatal, and adolescent stages of brain development is critical to understanding the mechanisms of nicotine’s effects on brain and behavior at each age.
3. Prenataldevelopment
3.1 Prenatal morphological development
Rats and mice are altricial animals that are born at a stage of brain development that is considerably more immature than that of humans. The gestational period of the rat spans only 22 days, which is roughly equivalent to the first 2 trimesters of human brain development. During this prenatal period, in both humans and rodents, most neuronal cell groups are born and early synaptogenesis occurs (Bayer et al., 1993). In the rat, the central nervous system begins to form around gestational day (G) 11 as the neuroepithelium, composed of a ventricle surrounded by actively proliferating cells. This generates waves of neurons and glia that ultimately form the cell populations of the more mature central nervous system. The caudal portion of the neural tube gives rise to the medulla, pons and spinal cord, with early functional maturation of processes that are critical for independent life. The rostral part of the neuroepithelium gives rise to the forebrain and a portion of these cells migrates to form the densely packed cortical plate. The development of the cortex is organized by an “inside-out” pattern in which the first neurons born occupy the deepest layers of cortex while the subsequently generated cells actively migrate to settle in more superficial cortical layers.
After neurons migrate, they begin to form synaptic connections and chemical transmission soon follows. Dopamine and other monoamine neurons are an exception in that the cell bodies and axons can express neurotransmitter before their target neurons are fully differentiated (Pendleton et al., 1998). In the adult, the monoamine systems regulate the neural circuitries underlying motivation, attention, cognition, and mood (Brennan and Arnsten, 2008; Nutt, 2008; Nestler et al., 2006; Palmiter, 2008). However, these systems are among the earliest to appear in the brain and may have distinct developmental roles in the prenatal period. Serotonergic cells are among the first generated in the brain at G11 in rat, and these cells project to the forebrain and aid in induction of differentiation of neuronal progenitor cells (Herlenius and Lagercrantz, 2001). Noradrenergic neurons of the LC appear at G12, and their cell bodies express norepinephrine soon after they are born (Altman and Bayer, 1980). Axons of these neurons are the first to penetrate and form synapses in the cortical plate during gestation and are thought to mediate neuronal migration and laminar formation (Herlenius and Langercrantz, 2001). Dopamine neurons of the SN and VTA originate around G13, and their fibers containing dopamine innervate the neocortex as early as G15 (Voorn et al., 1988; Kalsbeek et al., 1988).
Both synthetic (choline acetyltransferase) and degradative enzymes (acetylcholinesterase) for ACh are found in the fetus as early as the gastrulation phase (Mansvelder and Role, 2006), where ACh is suggested to regulate morphogenic cell movements (Lauder and Schambra, 1999). Theses enzymes also appear early in human fetal brain development, by 8–12 gestational weeks (Candy et al., 1985; Perry et al., 1986). In the rat, cholinergic projection neurons are first generated in the spinal cord and brainstem from G11-14, and forebrain cells are generated from G14-16 (Semba and Fibiger, 1988). Whereas target innervation by forebrain cholinergic afferents occurs largely after birth, transient populations of cholinergic interneurons appear in the cortex and hippocampus during the prenatal period (Lauder and Schambra, 1999). Thus, ACh is available to serve key regulatory roles throughout ontogenesis.
3.2. Prenatal expression of nAChRs
In humans, mRNA for most of the nAChR subunits and their corresponding binding sites are detected in the first trimester and appear in a caudal to rostral gradient (Hellstrom-Lindahl et al., 1998). α4β2 nAChRs are detected earliest in the spinal cord, pons, and medulla at six weeks, followed shortly thereafter by the cortex, subcortical forebrain, and cerebellum at eight weeks (Hellstrom-Lindahl et al., 1998), then spreading to the thalamus, hippocampal formation, and basal ganglia by twenty five weeks (Agulhon et al., 1998). In contrast, α7 nAChRs are found first in the midbrain and pons at five weeks and do not appear in spinal cord, subcortical forebrain, or cortex until nine weeks (Falk et al., 2002; Hellstrom-Lindahl and Court, 2000; Hellstrom-Lindahl et al., 1998).
Similar patterns of nAChR ontogeny have been observed when studying the development of the rodent brain. As in humans, α4 and β2 mRNAs are detected earliest in the rat spinal cord (G11), then more rostrally by G12, extending to the neocortex by G17-19 (Zoli et al., 1995). High affinity nicotine binding also appears in a caudal to rostral pattern, and is detectable in the neocortex by G20 (Naeff et al., 1992). α7 nAChR transcripts and binding sites are also detectable in rat brain during early development, first appearing in cortical and thalamic neuroepithelium between G13 and 15 (Broide et al., 1995). By G16, there is discrete expression of α7 nAChRs throughout the brain and spinal cord (Tribollet et al., 2004). Both α3 and β4 transcripts are widely distributed in rat brain throughout mid-gestation, but decline at later developmental stages (Winzer-Serhan and Leslie, 1997). In contrast to the postnatal period, there are many areas of the embryonic brain where expression of these transcripts is non-overlapping, suggesting that they are not always incorporated into α3β4 nAChRs. The α5 subunit, which has been shown to be transiently incorporated into α4β2 nAChRs during embryonic development (Wang et al., 1996), is also expressed during late gestation in embryonic cortex and hippocampus, as well as in catecholamine neurons (Winzer-Serhan and Leslie, 2005; Leslie et al., 2006; Azam et al., 2007; O’Leary et al., 2008). In contrast, α6 and β3 subunit mRNAs, which are strongly expressed in adult catecholamine neurons, are barely detectable during the prenatal period (Leslie et al., 2006; Azam et al., 2007; O’Leary et al., 2008).
3.3. Prenatal functional roles of nAChRs
nAChRs are vital to proper brain organization during the prenatal period by virtue of their roles in cell survival and targeting, the formation of rudimentary neural and sensory circuits via generation of spontaneous activity, and the development of catecholamine systems by regulation of neurotransmitter release. ACh plays a general regulatory role in neuronal pathfinding and survival during these early periods, mediated by Ca++ entry induced directly and indirectly by α7 nAChRs. Stimulation of α7 by ACh induces neurite retraction (Pugh and Berg, 1994; Small et al., 1995), while antagonism induces extension (Lipton et al. 1988). Developing cells may spontaneously release ACh with resulting asymmetrical gradients determining the direction of neurite growth (Zheng et al., 1994; Erskine and McCaig, 1995). α7 nAChRs also regulate developmental apotosis (Renshaw et al., 1993; Hory-Lee and Frank, 1995), together controlling cell number and the projection path of surviving neurons. Even at their earliest detection point, G11 in the spinal cord, nAChRs serve a role in the formation of essential neural circuitry. ACh released from developing motoneurons generates spontaneous bursts of activity that can propagate the entire length of the spinal cord via α4β2 nAChR activation in concert with inhibitory glycinergic input, or can produce bursting confined to local circuits via non-α4β2 nAChRs coupled to GABA excitation (Hanson and Landmesser, 2003). This coordinated activity is essential for accurate pathfinding of motoneurons to their targets (Myers et al., 2005; Hanson et al., 2008).
Cholinergic modulation of spontaneous activity is also critical for the initial creation of sensory circuitry (Galli and Maffei, 1988; Feller, 2002). In developing sensory structures, α3 and β4 subunit mRNAs show robust codistribution in the early prenatal period, most markedly in the retina, whisker barrels, and taste buds of the tongue (Zoli et al., 1995; Winzer-Serhan and Leslie, 1997). As functionally expressed α3β4 channels can induce burst firing patterns (Luetje and Patrick, 1991; Papke and Heinemann, 1991), their timing, location, and physiological properties are ideal for creating and propagating waves of activity. This probably occurs in the retina, where ACh released from starburst amacrine cells stimulates burst firing in ganglion cells via α3β4 and α3β2 nAChRs (Bansal et al., 2000). Cholinergic regulation of this process is transient, extending from G16 through the early postnatal period, at which point eye opening occurs and glutamateric signaling from bipolar cells becomes responsible for wave formation (Bansal et al., 2000).
Developing catecholamine neurons are also regulated by nAChRs during prenatal development. Both dopamine cell bodies in the SN/VTA and developing efferent terminals express α4β2 nAChRs throughout maturation. Expression of α4 and β2 mRNAs is first detected at G15, just days after these cells are born (Azam et al., 2007). α4 mRNA and high affinity nicotine binding are nearly twice as high in the SN compared to the VTA at G15, but decline to VTA levels by G17 (Azam et al., 2007). This is precisely the period when dopaminergic neurons begin to innervate their targets, and they are thus poised for cholinergic regulation. Given the higher expression of α4β2 nAChRs in the SN, the cholinergic system potentially has greater influence on SN neurons projecting to the striatum than those in the VTA projecting to limbic and cortical targets. ACh may influence both development of dopamine neurons and that of their target structures since striatal D1 and D2 dopamine receptors require afferent dopaminergic stimulation to be translated into functional proteins (Jung and Bennett, 1996). α4β2 nAChRs on dopamine terminals are functional during the prenatal period (Azam et al., 2007) and, thus, may coordinate the development of dopamine receptor systems by controlling the timing of neurotransmitter release. Studies using genetically engineered mice have also supported a critical, but complex, role for the α4β2 nAChR in dopamine system development. Mutants with a hypersensitive α4 subunit show initially intact dopamine neurons at G14, but these cells fail to mature and are rapidly lost by G16-18 (Labarca et al., 2001). Mice that lack α4 or β2 subunits also show maturational deficiencies, with loss of α4 producing unpruned terminal arbors, and loss of either subunit causing distorted presynaptic signaling and transmitter release (Parish et al., 2005; Picciotto et al., 1998). Adrenergic neurons of the LC and brainstem also express high levels of nAChRs early in their development (Leslie et al., 2006; O’Leary et al., 2008), with evidence of nAChR regulation of embryonic transmitter release (Gallardo and Leslie, 1998; O’Leary and Leslie, 2006). Such findings suggest that nAChRs may additionally regulate the early maturation of these neuronal pathways which play pivotal roles in regulating arousal, maternal bonding, and autonomic function during the perinatal period (Nelson and Panksepp, 1998; Itoi, 2008).
nAChRs also serve critical physiological functions in regulating the brainstem networks that mediate cardiorespiratory responses. Parasympathetic cardioinhibitory neurons in the nucleus ambiguous are excited both directly and indirectly by nAChRs, and play an essential role in controlling heart rate (Neff et al., 1998). These neurons can also be inhibited via nAChRs, with α4β2-like induction of local GABA release (Wang et al., 2003). Brainstem nuclei exhibit a transient increase of both α4 and α5 mRNA just prior to birth, suggesting that the perinatal period is one that is highly dependent on nAChR regulation (O’Leary et al., 2008). Given the important roles of endogenous ACh on the development of catecholamine and brainstem autonomic nuclei, it is not surprising that these are the circuits that are impacted by nicotine exposure during the prenatal period.
3.4. Effects of prenatal nicotine exposure
3.4.1. Methodological issues
Rodents are born at an earlier stage of brain maturation than humans (Bayer et al., 1993; Quinn, 2005). As a result, prenatal nicotine exposure in rats or mice only models tobacco exposure during the first two trimesters of human gestation and not the full term. Several different approaches to prenatal nicotine treatment have been used, each with its advantages and potential confounds, and none of which fully model prenatal tobacco smoke exposure (Slotkin, 1998). Most groups use an osmotic minipump to deliver nicotine to the pregnant dam, which produces constant blood levels of drug that decline gradually as pregnancy progresses and the dam gains weight. Initial infusion of 2–3 mg nicotine base/kg/day produces maternal blood levels of nicotine analogous to that resulting from moderate to heavy smoking (Benowitz and Jacob, 1984). Although this delivery method does not fully model the pharmacokinetics of nicotine in smokers, it minimizes stress to the dam from repeated drug treatments and prevents hypoxia that results from spikes in blood nicotine concentration. Since this approach is a form of controlled drug exposure, which neither models the daily variations in nicotine levels nor the effects of the many other constituents of tobacco, caution is needed in comparing resulting experimental effects with those of maternal smoking. This methodological approach is, however, much more analogous to nicotine delivery via patch. Many physicians, and the UK National Institute for Clinical Excellence guidelines, have endorsed the nicotine patch as a substitute for maternal smoking (National Institute for Clinical Excellence, 2002; Coleman, 2008), despite continued debate regarding its safety and clinical efficacy in pregnant women (Barron et al., 2007; Pauly and Slotkin, 2008).
Notwithstanding these limitations, there is strong evidence from the animal literature for widespread deleterious effects of prenatal exposure to nicotine, consistent with an important developmental role for nAChRs. There is also substantial convergence between the findings of animal studies and those of the clinical literature, suggesting that nicotine may be an important neuroteratogen within tobacco smoke. Thus, the results of these animal studies provide an important conceptual framework for understanding the mechanisms underlying clinical disorders in the offspring of women who smoke during pregnancy.
3.4.2. General effects
Prenatal nicotine exposure has substantial effects on the developing brain at doses that do not retard general growth (Slotkin, 1998). High affinity nicotine binding is increased in fetal and neonatal brain following chronic prenatal nicotine delivery (Slotkin et al., 1987a; Navarro et al., 1989; Pentel et al., 2006), providing critical evidence that nicotine reaches fetal brain, and upregulates nAChRs as it does in adult rats (Nguyen et al., 2003). Regions exhibiting persistent nAChR upregulation are more restricted by postnatal day (P) 18, however, being confined to somatosensory and visual cortices (Tizabi and Perry, 2000). This upregulation disappears entirely by adolescence and may in fact reverse in some regions, with less high affinity binding in the SN/VTA, nucleus accumbens, and prefrontal cortex in rats treated with prenatal nicotine compared to controls (Chen et al., 2005).
Gestational nicotine treatment has also been shown to decrease total numbers of cells in whole brain and in certain subregions, during the fetal and early neonatal period, consistent with a role of nAChRs in apotosis (Slotkin et al., 1987b; Onal et al., 2004). The activity of ornithine decarboxylase in neonatal rat brain is also increased by prenatal nicotine exposure, suggesting an abnormal switch from cellular proliferation to differentiation (Slotkin et al., 1987b; Navarro et al., 1989). Endocrine systems in males are impacted by this treatment, with nicotine increasing corticosterone levels at G18 through a direct action on the adrenal, which then inhibits testosterone production during the perinatal hormonal surge (von Ziegler et al., 1991; Sarasin et al., 2003). Nicotine may also have direct effects on neural sexual differentiation by inhibiting aromatase activity in the male fetal brain (Barbieri et al., 1986). The acute endocrine effects of nicotine during gestation appear to have long-term consequences in male rats, resulting in the abolition of sex differences in saccharin preference (Lichtensteiger and Schlumpf, 1985). In humans, prenatal tobacco exposure also alters the onset of puberty in males, further suggesting functional relevance for early endocrine disruption (Fried et al., 2001).
3.4.3. Catecholamine systems
Both in vivo and in vitro studies have shown that acute nicotine treatment stimulates dopamine release in fetal rat forebrain (Ribary and Lichtensteiger, 1988; Azam et al., 2007). Chronic nicotine infusion also alters brain levels of dopamine, norepinephrine and their metabolites during the late prenatal period (Ribary and Lichtensteiger, 1989; Onal et al., 2004). In the early postnatal period, both dopamine and norepinephrine levels are elevated in the forebrain of rats treated with nicotine during gestation, although they return to control levels by adulthood (Ribary and Lichtensteiger, 1989). Total tissue content, however, does not reflect changes at the synaptic level, and functional analyses reveal profound alterations in cholinergic regulation of catecholamine signaling that, in many cases, persist through adulthood. Prenatal nicotine treatment markedly increases nicotine-induced norepinephrine release from in vitro slice preparations of parietal cortex after birth, with increased efficacy during early postnatal development, adolescence, and adulthood (Leslie et al., 2006). Conversely, in vivo nicotine-induced dopamine release is substantially reduced in the nucleus accumbens of adolescent rats treated with prenatal nicotine (Kane et al., 2003).
Behavioral analysis of prenatal nicotine-treated rats reveals phenotypes consistent with alterations in the dopamine system, with changes in both motor function and reward. Gestational nicotine-induced alterations in both spontaneous and drug-induced motor activity have been reported (Fung, 1988; Le Sage et al., 2006; Paz et al., 2007). Males, but not females, exposed to prenatal nicotine have enhanced locomotor responses to nicotine challenge on P14 (Shacka et al., 1997). In contrast, cocaine-induced stereotypy in adolescent males and females is severely blunted, apparently as a result of loss of functional D2 receptors (Franke et al., 2008; Franke et al., submitted for publication). There are also complex persisting effects of prenatal nicotine exposure on nicotine, cocaine, and natural reward during adolescence and adulthood. Whereas prenatally exposed adolescent females initially self-administer nicotine to the same degree as controls, they take significantly more drug following a withdrawal period (Levin et al., 2006). Additionally, rats treated with prenatal nicotine show less initial motivation for food reward (Franke et al., 2008), but increased motivation for and total intake of cocaine (Paz et al., 2007; Franke et al., 2008). Cocaine-induced locomotor sensitization, an adaptive neural process which has been linked to the processes underlying drug abuse (Robinson and Berridge, 2001), is also increased by prenatal nicotine exposure (Franke et al., 2007). Together these data confirm that nicotine exposure during the prenatal period, a critical time for the initial development of catecholamine systems, has lasting effects on behaviors that are regulated by dopamine systems (see Figure 1).
Figure 1.
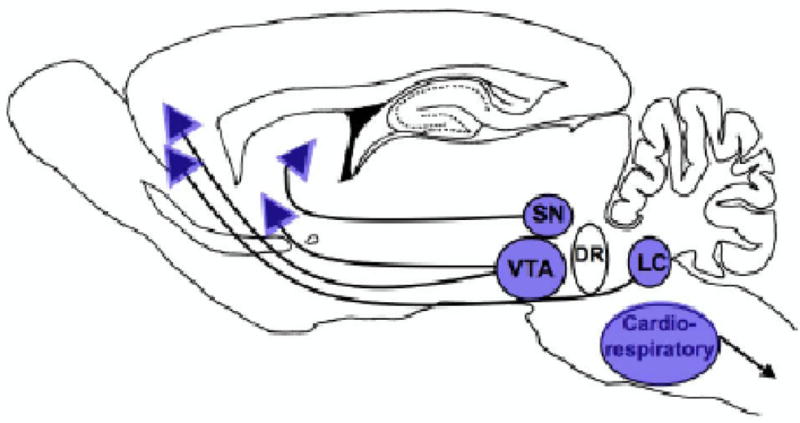
Brain regions impacted by nicotine exposure during the prenatal period. The dopamine-producing cell bodies of the SN/VTA and the noradrenergic cell bodies of the LC are vulnerable to exogenous nicotine-induced deficits during the prenatal period. nAChRs are located on both the cell bodies and terminals of these projections and are able to modulate neurotransmitter release in fetal brain. Additionally, the brainstem nuclei controlling cardiorespiratory and autonomic responses are also sensitive to nicotine exposure during this period. Prenatal nicotine-induced defects in these structures may underlie the increase in dopamine-mediated disorders such as attention deficit hyperactivity disorder and substance abuse, as well as the increased risk for sudden infant death syndrome reported in the clinical literature (see text for detail).
Changes in dopamine release and signaling during adolescence have important implications for understanding the mechanisms underlying some neurobehavioral disorders that have been associated with maternal smoking. Clinical studies have shown that smoking during pregnancy can lead to long-standing neurobehavioral deficits in the offspring that may result from catecholaminergic dysfunction, including attention deficit hyperactivity disorder, conduct disorder, and cognitive deficits (Thapar et al., 2003; Weissman et al., 1999). A significant gene × environment interaction has also been found, with hyperactivity-impulsivity and oppositional behavior being significantly associated with polymorphisms of the dopamine transporter in the children of pregnant smokers (Kahn et al., 2003; Neuman et al., 2007). Recent evidence suggests this may be an extremely potent interaction, since maternal exposure to second hand smoke has been found to be significantly associated with vulnerability to externalizing psychopathology in the offspring (Gatzke-Kopp and Beauchaine, 2007). Maternal smoking has also been significantly associated with increased risks of addiction to both tobacco and other drugs of abuse during adolescence, which is likely mediated by alteration of catecholaminergic reward circuitry (Weissman et al., 1999; Buka et al., 2003; Porath and Fried, 2005). Adolescent smokers whose mothers used tobacco during pregnancy also experience more severe memory deficits during nicotine withdrawal, potentially making quitting more difficult for adolescents who were exposed to tobacco during pregnancy (Jacobsen et al., 2006). Taken together, studies using rodent models of prenatal nicotine exposure have produced results remarkably consistent with that seen in the clinical literature. Rodent studies have confirmed the presence of functional nAChRs in developing catecholaminergic circuitry during the prenatal period, which may be aberrantly activated, or more likely, desensitized by nicotine exposure at this time. Nicotine-induced alterations in these circuits that regulate motivation, cognition, and hyperactivity may not manifest until their full maturation during childhood and adolescence, which is in fact when deficits in both humans and lab animals emerge.
3.4.4. Cardiorespiratory responses
Fetal exposure to tobacco via maternal smoking is a well-established risk factor for sudden infant death syndrome (SIDS), as cigarette smoking during pregnancy dose dependently increases the odds ratio of developing SIDS in the offspring (Fleming and Blair, 2007). While the full etiology of SIDS is unknown, it is thought to involve dysregulation of neural control of cardiorespiratory processes and aberrant responses to hypoxia (Moon et al., 2007). Rats treated prenatally with nicotine exhibit behavioral phenotypes consistent with SIDS in the early postnatal period, showing increased spontaneous apnea in the first days of life (Huang et al., 2004) and increased vulnerability to death from hypoxic challenge (Slotkin et al., 1995; Fewell et al., 2001).
Recent electrophysiological studies have elegantly shown that prenatal nicotine alters the activity of cardiorespiratory neurons in the brainstem during both normal breathing patterns and episodes of hypoxia via persisting alteration of cholinergic regulation (Huang et al., 2004; Kamendi et al., 2006; Huang et al., 2007). During normal inspiration, rats treated prenatally with nicotine show cholinergic enhancement of cardioinhibitory vagal neuron disinhibition, resulting in increased heart rates in these animals (Neff et al., 2003). More importantly, these neonates also have impaired responses to hypoxic challenge. Cardioinhibitory neurons, which are normally silent during hypoxic episodes, are excited during hypoxia in rats exposed to prenatal nicotine. This aberrant excitement occurs via cholinergic enhancement of glutamate release onto these cells, producing decreased heart rate during the hypoxic episode, potentially enhancing the likelihood of mortality (Evans et al., 2005; Huang et al., 2007). Changes in cholinergic signaling may be mediated in part by alteration in the subunit composition of nAChRs expressed on glutamatergic terminals. Prenatal nicotine treatment induces a change from α3β2 and α6-containing nAChRs to α3β4 nAChRs, which may mediate the irregular signaling during hypoxia (Kamendi et al., 2006).
Changes in central and peripheral adrenergic signaling may also contribute to the aberrant response to hypoxia following gestational nicotine exposure (Slotkin et al., 1995). Adrenergic systems are critically involved in regulating neonatal arousal and ventilatory response (Bamford and Hawkins, 1990; Karlsson et al., 2004; Viemari, 2008), which are dysregulated in SIDS (Leiter and Bohm, 2007). Prenatal nicotine exposure decreases the baseline activity of noradrenergic neurons, but greatly sensitizes them to hypoxia-induced activation (Slotkin et al., 1995). This excessive release of central norepinephrine during hypoxia may inhibit the ventilatory response via activation of brainstem α2-adrenergic receptors. Conversely, hypoxia-induced release of norepinephrine from adrenal chromaffin cells is greatly diminished by prenatal nicotine exposure (Slotkin et al., 1995; Buttigieg et al., 2008). Since massive release of adrenal catecholamines, and the resulting redistribution of blood flow, is required for neonates to survive hypoxia (Hedner et al., 1980; Thompson et al., 1997), this may contribute significantly to the increased incidence of SIDS seen in the offspring of smoking mothers (see Figure 1).
4. Early postnatal development
4.1 Early postnatal morphological development
The first twelve days of life in the rat are approximately comparable to the third trimester of human gestation (Dobbing, 1971; Quinn, 2005). This period is characterized by rapid brain growth including dendritic arborization, axonal growth, peak synaptogenesis, gliogenesis, and maturation of neurotransmission (Dobbing, 1971; Dobbing and Sands, 1979). During this time, neurogenesis of the olfactory bulb, hippocampus and cerebellum is just beginning (Bayer et al., 1993). For example, the hippocampus undergoes an increase in size and a change in excitatory neurotransmission during this period to allow for adult-like synaptic plasticity by the end of the second postnatal week (Dumas, 2005). This transiently heightened level of brain plasticity is shaped by environmental factors and stimuli such as stress, malnutrition, and drugs, which have profound effects on this brain growth spurt (Goodlett et al., 1989; Burns, 1990).
Some neurotransmitters change their functional role in the brain during this developmental period. For example, GABAergic signaling is excitatory because of the reversed chloride gradient during prenatal and early postnatal maturation (Rivera et al., 1999). GABA-induced excitation allows for neuronal proliferation, migration and targeting. A switch from depolarizing to hyperpolarizing GABAergic signaling occurs during the first two postnatal weeks, and this change is triggered by a shift in the balance of chloride transporter expression from those that increase (i.e. NKCC1) intracellular chloride to those that decrease (i.e. KCC2). The maturation of this change in signaling follows a sex-specific pattern, in which females demonstrate an earlier switch than males in the hippocampus and substantia nigra (Galanopoulou et al., 2003; Galanopoulou, 2008). Modulation of GABAergic signaling during this time window, such as exposure to alcohol, can alter cell proliferation and migration of cortical neurons (Belhage et al., 1998).
In both humans and rodents, there are sensitive periods during early postnatal life during which sensory input shapes the maturation of sensory circuits (Berardi et al., 2000; Lewis and Maurer, 2005). In rats, the second postnatal week is a critical period for sensory cortical development as the animal begins to hear (Jewett and Romano, 1972), starts whisking (Welker, 1964), and opens its eyes (Morishita and Hensch, 2008). During this time, synaptic connections are established between sensory thalamic afferents and their cortical targets (for review, Metherate, 2004), and disruptions of this process can have serious and persisting consequences. Basal forebrain cholinergic innervation of the cortex coincides with the development of cortical synapse formation (Hohmann and Berger-Sweeney, 1998). Thalamic synaptic contacts are closely aligned with acetylcholinesterase positive fibers throughout sensory cortical maturation, which suggests that ACh acts as a regulating factor for cortical plasticity (Hohmann, 2003).
4.2. Early postnatal expression and function of nAChRs
4.2.1 Neocortex
In the human, the peaks of both α4β2 and α7 nAChR receptor binding in the neocortex and hippocampus occur during the third trimester of development (Court et al., 1997; Court et al., 2000). In the rodent, high affinity nicotine binding in the cortex increases during the first postnatal week, and then moderately declines to adult levels by P28 (Zhang et al., 1998). α7 mRNA, while evident in the cortex by G13, peaks during the second postnatal week (Broide et al., 1995; Mansvelder and Role, 2006). Particularly striking is the transition to the heterogeneous distribution of cortical α7 nAChRs seen during the postnatal peak. Sensory and limbic cortices show preferential upregulation of α7 mRNA and α-bungarotoxin binding (Fuchs, 1989; Bina et al., 1995; Broide et al., 1995), and these areas are mirrored by increased α7 levels in corresponding thalamic nuclei that send their afferents to innervate the cortex precisely at this time (Broide et al., 1996). Similar to prenatal development, α7 nAChRs may regulate developmental programmed cell death during the early postnatal period, as gain of function mutations in α7 cause massive cell loss in the somatosensory regions of the neonate cortex (Orr-Urtreger et al., 2000). α7 nAChRs also regulate thalamocortical development at the synaptic level by enhancing glutamate release from presynaptic terminals onto layer IV pyramidal cells. During the early postnatal period, this enhanced release appears to convert NMDA-only, or silent, synapses to mature synapses via depolarization-dependent AMPA insertion (Metherate and Hsieh, 2003). Cortical expression of α7 nAChRs is also regulated by thalamic input and by the onset of sensory signaling (Broide et al., 1996; Bina et al., 1998; Aztiria et al., 2004), suggesting an important role for these receptors in experience-dependent plasticity.
Cortical expression patterns of some heteromeric nAChRs also undergo dynamic changes during the early postnatal period. α3 and β4 subunit transcripts are transiently expressed within layer IV neurons of the visual cortex at this time (Winzer-Serhan and Leslie, 1997). The developmental distribution of α5 also suggests that this subunit may play an important role in cortical development during the early postnatal period. α5 mRNA appears by G18 in layer VI of cortex, followed by expression in interneuron-rich layers II/III and V at birth. Cortical α5 mRNA expression rises to peak in the first two postnatal weeks and then declines to low levels in the adult (Winzer-Serhan and Leslie, 2005). It has recently been shown that corticothalamic pyramidal neurons in layer VI of the prefrontal cortex generate excitatory currents via α4α5β2 stimulation that peak during the first postnatal month (Kassam et al., 2008), suggesting an important developmental function for this receptor type. Loss of the β2 subunit on these neurons during these first two postnatal weeks, but not later in life, impairs the learning of passive avoidance tasks in adulthood (King et al., 2003). Thus, corticothalamic neurons, that are important for attentional processes and emotional learning, appear to be programmed by cholinergic input via activation of α4α5β2 nAChRs during the early postnatal period.
4.2.2. Hippocampus
Like the neocortex, the hippocampus also develops primarily during the early postnatal period (Bayer and Altman, 1974), and shows similar transient increases of α7 and α5 subunit expression. In CA1 and CA3 pyramidal cell layers, α7 and α5 mRNA levels rise to peak in the first two postnatal weeks and decline to adult levels thereafter (Zhang et al., 1998; Adams et al., 2002; Winzer-Serhan and Leslie, 2005). Hippocampal interneurons also express α5 mRNA after birth, increasing to reach high adult levels by the third postnatal week (Winzer-Serhan and Leslie, 2005). During this time, there is significant colocalization of α5 and α7 mRNAs in developing pyramidal neurons, but only slight colocalization in interneurons, suggesting alternate nAChR subunit combinations involving α5 in this population (Winzer-Serhan and Leslie, 2005). α3 and β4 mRNAs are also codistributed transiently in the pyramidal layer of the CA3 during the first postnatal weeks, which may contribute to more complex subunit combinations (Winzer-Serhan and Leslie, 1997).
As with the retina and the spinal cord, early hippocampal development is driven by spontaneous oscillatory activity (Feller, 1999). These activity waves are driven by excitatory GABA, but are indirectly controlled by ACh by virtue of its influence over the GABA excitatory to inhibitory switch. During the first postnatal week, Ca++ influx via α7 nAChRs induces expression of the adult chloride transporter (KCC2), which decreases intracellular chloride to establish the inhibitory GABA gradient (Liu et al., 2006). During their period of peak expression, α7 nAChRs also mediate silent synapse activation, as in the cortex, to lay the foundation for adult hippocampal circuitry (Maggi et al., 2003).
4.2.3. Cerebellum
Another structure that develops largely postnatally in rodent is the cerebellum (Altman, 1972; Bayer et al., 1993). Neurogenesis of cerebellar granule cells occurs entirely following birth within the external germinal layer, which expresses high levels of α3 and β4 mRNAs during the perinatal period (Winzer-Serhan and Leslie, 1997). Furthermore, during early postnatal development this region expresses heteromeric nicotinic receptor binding sites that have pharmacological properties consistent with α3β4 nAChRs (Opanashuk et al., 2001).
There is a large ACh input to the developing cerebellum, with Purkinje cells transiently expressing a cholinergic phenotype during the early postnatal period (Gould and Butcher, 1987). Substantial evidence indicates that ACh regulates early cerebellar development via activation of nAChRs both on developing cerebellar neurons and their afferents. In vitro studies have shown that nAChR activation promotes neurogenesis and survival of cerebellar granule cells, and reduces their motility (Opanashuk et al., 2001; Fucile et al., 2004). Furthermore, heteromeric nAChRs enhance synaptic glutamate and GABA transmission onto Purkinje cells, as well as cerebellar norepinephrine release, effects that are prominent during the perinatal period but decrease substantially as the cerebellum matures (Kawa et al., 2002; O’Leary and Leslie, 2003).
4.2.4. Catecholamine neurons
Some earlier maturing structures also show changes in nAChR distribution during the early postnatal period. The noradrenergic LC exhibits a substantial postnatal decrease in α4 mRNA levels, which is paralleled by a decline in high affinity nicotine binding (Leslie et al., 2006). There is a corresponding postnatal increase in the expression of α6 and β3 mRNAs, which contribute to the heterogeneous nAChR types seen in mature LC (Léna et al., 1999). The dopaminergic cell bodies of the SN and VTA show a second sharp increase in high affinity nicotine binding at birth, followed by a slow decline to adult levels by P20 (Azam et al., 2007). Paralleling the increase in nicotine binding in the dopamine synthesizing cell bodies, nicotine binding and α4/β2 subunit mRNA expression in striatal terminal fields also peaks around birth, but remains sustained through the first two postnatal weeks (Zhang et al., 1998; Zoli et al., 1995; Naeff et al., 1992). As with the LC, there is a postnatal increase in the expression of α6 and β3 mRNAs in the SN/VTA, which contribute to the heterogeneous nAChR types seen in the mature dopamine system (Charpantier et al., 1998; Zoli et al., 2002).
Cholinergic regulation of the catecholamine system continues to mature during the early postnatal period, and nAChR regulation of transmitter release in terminal fields is highly region specific. nAChR efficacy in stimulating norepinephrine release is low in the hippocampus at birth and increases over the first ten days of life (Leslie et al., 2002). At all stages of postnatal development, however, there are two components of nAChR-mediated norepinephrine release; a direct effect on hippocampal LC terminals and an indirect effect mediated by stimulation of an excitatory GABA input. In contrast, in other LC terminal fields such as the cerebellum, neocortex, and hypothalamus, nAChRs directly stimulate norepinephrine release and are more efficacious during the early postnatal period (O’Leary and Leslie, 2003, 2006; Leslie et al., 2006). In these terminal regions, there is a clear change in the pharmacological properties of the nAChRs that regulate norepinephrine release as maturation progresses, suggesting that they subserve differing functional roles at different developmental stages.
Similarly, the pharmacological properties of nAChRs that stimulate dopamine release also continue to change during the early postnatal period. After birth, nicotine efficacy at nAChRs that stimulate striatal dopamine release decreases (Azam et al., 2007). Significant sex differences are also observed during this period. After P7, however, nicotine efficacy and potency steadily increases until the third postnatal week. These complex changes in acute responses to nicotine highlight the complexity of cholinergic signaling, and the functional importance of changing nAChR subunit profiles, during early postnatal development.
4.3. Effects of early postnatal nicotine exposure
During early postnatal development, nAChRs begin to shape the formation of maturing cortical and limbic circuitry, and their regulation of gene expression becomes essential for proper synaptic formation and neuronal signaling. ACh plays important roles in all phases of brain development, but its effect at each age is dependent on the timing of critical periods for different neural structures. Thus, while disruption of cholinergic signaling during prenatal development has long term effects on the brainstem catecholamine systems that are maturing at this time, nicotine administration in the early postnatal period alters later developing structures like the cortex and hippocampus (see Figure 2).
Figure 2.
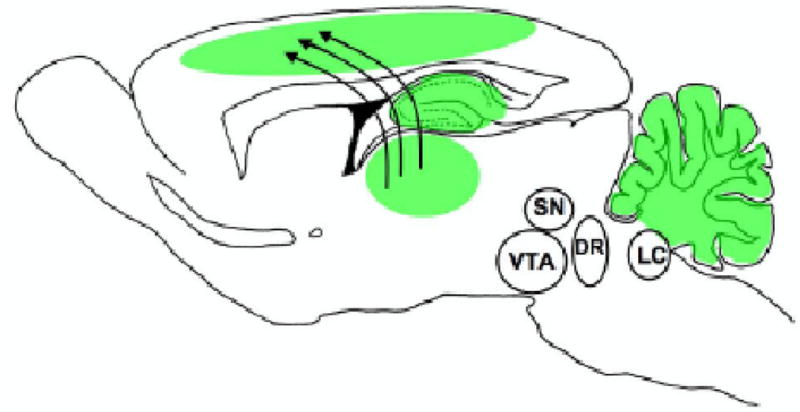
Brain regions impacted by nicotine exposure during the early postnatal period. In the early postnatal period, the cortex, hippocampus, and cerebellum are all in sensitive periods of development and are actively regulated by transient populations of nAChRs. During this time exogenous nicotine may disrupt normal thalmocortical and hippocampal development. Offspring of children whose mothers smoked during pregnancy are at increased risk for cognitive and auditory processing deficits, which are likely related to cortical dysfunction induced by early nicotine exposure (see text for detail).
Nicotine exposure during the early postnatal period induces both acute and persisting changes in high affinity nicotine binding sites. Nicotine treatment from P1-P7 results in increased high affinity binding in the hippocampus, cortex, and thalamus, but not cerebellum (Huang and Winzer-Serhan, 2006; Huang et al., 2007a). These increases in binding are not accompanied by changes in mRNA, indicating that the upregulation occurs via post-translational mechanisms. Treatment of rat pups during the second postnatal week induces upregulation of high affinity sites in the cortex, hippocampus, and striatum that persist through adulthood, whereas nicotine given from P1 through P21 induces only transient upregulation (Miao et al, 1998). Functional changes in heteromeric nAChRs are also found after early postnatal exposure. Either prenatal or early postnatal exposure to nicotine eliminates the peak of nicotine-stimulated Rb+ efflux in synaptosomes from the cortex, hippocampus, striatum, and thalamus that normally occurs at P35. Early postnatal exposure, however, causes an earlier decrease of Rb+ efflux from the cortex compared to control at P28, an effect not seen with prenatal exposure alone (Britton et al., 2007). This effect confirms that the cortex is preferentially impacted by early postnatal exposure to nicotine.
Nicotine exposure during the second postnatal week induces long lasting effects on sensory cortical function. Rats treated with nicotine at this time show enhanced corticothalamic excitability, evidenced by longer duration and more erratic excitatory postsynaptic potentials in the auditory cortex (Aramakis et al., 2000). These effects result from changes in the NMDA current, whereas AMPA currents are unaffected, and persist for at least 10 days following treatment (Aramakis et al., 2000). NMDA receptor subunits are altered by early postnatal nicotine treatment, showing increased NR2A expression levels up to two weeks following chronic neonatal nicotine (Hsieh et al., 2002). NR2A is typically upregulated by neuronal activity, which may be mediated by nAChR-induced stimulation of glutamate release (Carmignoto and Vicini, 1992; Metherate and Hsieh, 2003). While the immediate effects of neonatal nicotine exposure on thalamocortical excitability and NMDA subunit expression subside by the adolescent period, the organization and sensitivity of these circuits is permanently altered. Adult rats that were exposed to nicotine during the second postnatal week fail to show nicotine-induced potentiation of neural responses to auditory stimuli, and exhibit deficits in cognitive processing requiring auditory cues (Liang et al., 2006). Thus, nicotine exposure in the rodent during the early postnatal period produces permanent changes in cortical circuitry that result in deficiencies in sensory and cognitive processing. Such findings may clarify the mechanisms underlying the well established defects in human auditory cognitive processing that result from maternal smoking (Sexton et al., 1990; McCartney et al., 1994).
The hippocampus also undergoes substantial maturation during the early postnatal period, and there is some evidence that neonatal nicotine exposure may impair hippocampal function later in life. The combination of prenatal and postnatal nicotine exposure has been reported to impair spatial reference learning in adult females, but not males, in the Morris water maze task (Eppolito and Smith, 2006). Others, however, examining only early neonatal exposure, have not shown deficiencies in hippocampal mediated spatial learning, but have found increased anxiety-like behaviors in both males and females (Huang et al., 2007b). This phenotype may result from nicotine-induced changes in ventral hippocampus, as recent findings suggest a role for this structure in anxiolytic behavior (Engin and Treit, 2007).
5. Adolescent Development
5.1 Adolescent morphological development
Adolescence, which is conservatively defined as approximately 12 to 20 years in humans and P28 to 42 in rats, is the final developmental epoch leading to adulthood (Spear, 2000). This maturational period is defined by characteristic behaviors such as risk-taking, novelty-seeking, high play and increased social interactions, features that are conserved across species (Spear, 2000). Another hallmark feature of this period is the development of executive functions such as decision-making and impulse control (Leon-Carrion et al., 2004; Toga et al., 2006). Underlying these behavioral changes are dynamic alterations in the brain.
During adolescence, there is an increase in myelination and synaptic pruning to allow more efficient neural signaling. Human magnetic resonance studies have shown a linear increase in white matter (Paus et al., 1999; Giedd, 2004) and an inverted U-shaped change in gray matter volume during the adolescent period (Giedd et al., 1999; Giedd, 2004). Correlated with gray matter changes, synaptic connections are overproduced in early puberty and rapidly pruned back in late adolescence (Giedd, 2004). Specifically, synaptic or receptor pruning occurs in the amygdala (Zehr et al., 2006), striatum (Teicher et al., 1995) and prefrontal cortex (Andersen et al., 2000). Despite synaptic pruning, fiber density of amygdalocortical (Cunningham et al., 2002) and corticoaccumbens connections (Brenhouse et al., 2008) continues to increase into early adulthood. This process allows for experience-dependent strengthening of connections that are frequently activated in response to the environment, and elimination of those that are less often used (Rakic et al., 1994). Prefrontal cortex control of executive function requires maturation of connectivity to inhibit impulsive limbic signals. Thus, such remodeling is necessary for development of adult-like behavior.
In addition to these gross structural changes, the adolescent brain shows dramatic alterations in neurochemical transmission. In particular, there is marked maturation of the mesocorticolimbic dopamine system and its targets, which are ultimately responsible for the integration of the external environment with internal drives to produce motivated behavior (Benes et al., 2000; Chambers et al., 2003). Dopaminergic innervation of the prefrontal cortex increases in density (Benes et al., 2000), and its control of prefrontal interneurons and pyramidal firing states matures during adolescence (Tseng and O’Donnell, 2005; 2007). There is also a transient increase in dopamine D1 receptor expression on maturing cortical-accumbens afferents (Brenhouse et al., 2008). Both animal and human studies reveal that dopamine D1, D2, and D4 receptors are overproduced during adolescence and undergo rapid elimination in striatal regions (Seeman et al., 1987; Teicher et al., 1995; Tarazi et al., 1999), whereas dopamine D3 receptors steadily rise to reach peak levels in adulthood (Stanwood et al., 1997;Gurevich et al., 1999). Age differences are also evident in the functional consequences of striatal dopamine receptor activation (Bolanos et al., 1998; Andersen, 2002). The dopaminergic system is dynamically changing during adolescence and stimulation by nicotine, possibly via nAChRs on dopamine neurons and terminals, may alter maturation of the mesocorticolimbic system.
5.2. Adolescent expression and function of nAChRs
In rodents, levels of α4β2 and α7 mRNA and binding in most brain regions have been reported to be stable from adolescence through adulthood by some (Zhang et al., 1998), although others have described increased nAChR binding in adolescents relative to adults (Doura et al., 2008). Despite these discrepancies, α4β2 nAChRs show functional changes in the cortex, hippocampus, striatum, and thalamus during adolescence, with increased nicotine-induced Rb+ efflux compared to adults (Kota et al., 2007). These changes may be mediated by increased receptor number in these regions, or by insertion of subunits such as α5 that alter gating properties.
The dopamine system also exhibits marked functional changes in its regulation by nAChRs during adolescence (Azam et al., 2007). However, the mechanisms underlying these complex changes are not entirely clear. Whereas high affinity nicotine binding has reached adult levels in the SN/VTA region by adolescence, α4β2-like binding in the striatum remains elevated relative to adult (Zhang et al., 1998; Doura et al., 2008). Although expression of most nAChR subunit mRNAs within SN/VTA has reached adult levels by adolescence, levels of α5 mRNA, which peak just prior to adolescence at P20, remain somewhat elevated, particularly in the SN (Azam et al., 2007). In the adult, α5 is expressed on dopaminergic terminals in the striatum and prefrontal cortex in nAChRs with a α4α5β2 combination (Zoli et al., 2002; Gotti et al., 2006). However, it is not yet known whether expression of α5 on dopaminergic terminals also changes with age. Given the elevated expression of α4β2 nAChRs, however, adolescent-specific changes of α4α5β2 are likely. The α6 subunit may also play an important role in adolescence, as its mRNA expression peaks in the SN and VTA at P21 (Azam et al., 2007). The SN contains more α6 mRNA than the VTA throughout adolescence, but levels decline to equal expression by adulthood. α6-containing nAChRs presynaptically regulate dopamine release in the nucleus accumbens, but not the striatum, in adulthood (Exley et al., 2008). As yet, however, age-specific changes in α6 protein levels in these regions have not been determined. The biochemical and behavioral effects of nicotine administration during adolescence, however, suggest changes in subunit expression and nAChR function. Further characterization of nAChR subunit profiles, particularly on the terminals of catecholamine neurons in the striatum and cortex are needed.
Whereas the functional role of nAChRs in adolescent maturational processes has not been fully explored, the neurochemical and behavioral studies outlined below suggest that they may regulate limbic system circuitry that is undergoing critical experience-dependent reshaping during this period (see Figure 3). Thus, adolescence may be a particularly vulnerable period during which nicotine exposure may produce long-term changes in limbic function.
Figure 3.
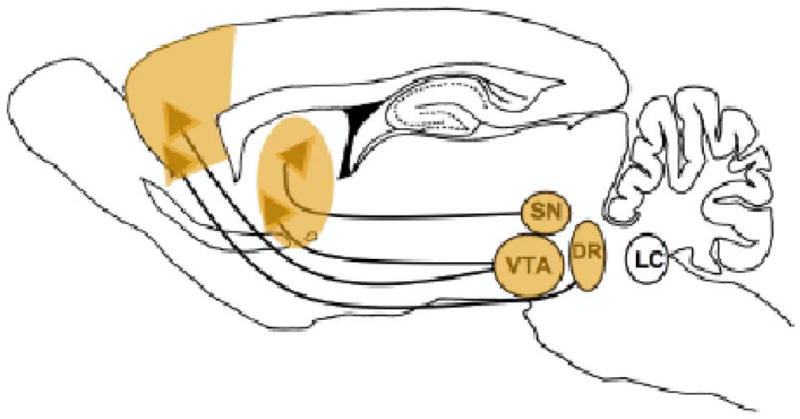
Brain regions impacted by nicotine exposure during the adolescent period. During adolescence, there is substantial plasticity in limbic circuitry including the amygdala, prefrontal cortex, and nucleus accumbens. The dopamine systems of the SN/VTA also undergo their last major phase of development during this period. These late maturing catecholaminergic projections possess functional nAChRs on the cell bodies and terminals that allow nicotine to alter their excitation and release. nAChRs have also been shown to alter the dopamine and serotonin transporter function in prefrontal cortex and nucleus accumbens. Thus, nicotine exposure during this time produces changes within the limbic and dopaminergic circuitry that underlies motivated behaviors, potentially enhancing the vulnerability to nicotine addiction during this time period. The serotonin system, originating in the dorsal raphe (DR), also appears to be affected by adolescent nicotine exposure. Nicotine-induced alterations in serotonin function may contribute to the increased incidence of mood disorders seen after adolescent smoking (see text for more detail).
5.3. Effects of adolescent nicotine exposure
5.3.1. Acute effects
Nicotine administered during adolescence induces unique neurochemical responses in limbic circuitry. A recent in vivo microdialysis study has shown nicotine to induce greater increases in extracellular levels of dopamine and its metabolite, 3,4-dihydroxyphenylacetic acid, in the nucleus accumbens of the adolescent as compared to the adult rat (Shearman et al., 2008). A more striking difference, however, was the large nicotine-induced increase in extracellular serotonin levels in the nucleus accumbens of the adolescent, but not adult, rat. This finding suggests that the serotonin system may mediate some acute nicotine effects in adolescence. In contrast, in prefrontal cortex, nicotine stimulates increases in extracellular levels of dopamine, serotonin, and norepinephrine in adults, but not adolescents, indicating a late maturation of nAChR regulation of these systems.
Adolescents also exhibit some unique patterns of neuronal activation in response to nicotine, as evidenced by expression of immediate early genes such as c-fos and arc. Adolescent animals have greater nicotine-induced c-fos and/or arc mRNA activation in reward-related regions, such as the nucleus accumbens, the extended amygdala and the prefrontal cortex (Schochet et al., 2005; Shram et al., 2007). Recent evidence suggests a critical developmental role for α7 nAChRs in mediating these unique neuronal responses in the adolescent prefrontal cortex (Thomsen et al., 2008). Nicotine also selectively stimulates c-fos mRNA expression in the adolescent paraventricular nucleus of the thalamus (Cao et al., 2007), which is highly interconnected with limbic regions and an important inhibitory regulator of the stress response (Jaferi and Bhatnagar, 2006). Consistent with this finding, adolescents are less sensitive to nicotine-induced activation of the paraventricular nucleus of the hypothalamus and subsequent corticosterone release (Cao et al., 2007), despite the fact that the hypothalamic-pituitary-adrenal axis response to other stressors reaches adult levels by this time (Goldman et al, 1973; Vazquez and Akil, 1993; Choi and Kellogg, 1996).
These unique responses of limbic circuitry to nicotine administration in adolescents are paralleled by age differences in nicotine-induced behaviors. Recent animal studies have provided evidence consistent with the clinical literature (Chen and Millar, 1998; Everett et al., 1999), that adolescents are more sensitive to the rewarding properties of nicotine, potentially enhancing their vulnerability to addiction. Adolescent rats have been shown to acquire intravenous nicotine self-administration more readily, and to take higher amounts of drug, than adults (Levin et al., 2003, 2007; Chen et al., 2007). Although these studies were conducted in older adolescents, other reports in which different behavioral approaches are used have also shown an enhanced sensitivity to nicotine reward during early adolescence (P28-35). Mice of this age have a greater preference than adults for drinking sucrose solution containing nicotine (Adriani et al., 2002). In conditioned place preference tests, adolescent rats have been shown to spend more time in the nicotine-paired chamber at lower doses than found in adults (Belluzzi et al., 2004; Kota et al., 2007; Shram et al., 2006; Torres et al., 2008; Vastola et al., 2002). Young adolescent rats also form these associations more readily than the adult, in that a single conditioning trial can elicit a conditioned place preference response (Belluzzi et al., 2004; Brielmaier et al., 2007).
Not only are the rewarding effects of nicotine enhanced during adolescence, but the aversive effects are also reduced. High doses of nicotine are perceived as aversive in adult animals, but not in adolescents, as demonstrated with conditioned place or taste aversion procedures (Shram et al., 2006; Torres et al., 2008; Wilmouth and Spear, 2004). Nicotine withdrawal is also experienced less in adolescents than adults. Adolescents display fewer physical signs under both spontaneous and precipitated withdrawal after continuous nicotine infusion (Kota et al., 2007; O’Dell et al., 2004, 2006; Shram et al., 2008). Furthermore, in nicotine-dependent adults, withdrawal causes a decrease in brain reward function as indicated by an elevation of intracranial self-stimulation thresholds (Kenny and Markou, 2001; Panagis et al., 2000), whereas this effect is absent in adolescents (O’Dell et al., 2006, 2008). These findings of decreased sensitivity to the aversive effects of nicotine in adolescent animals are in contrast to the clinical literature, in which teenagers have been shown to exhibit symptoms of withdrawal shortly after the initiation of smoking (DiFranza et al., 2000). This discrepancy may reflect the synergistic interactions of nicotine with other components of tobacco, which have been shown to influence the behavioral response of adolescents (Belluzzi et al., 2005).
Nicotine exposure during adolescence can also alter behaviors associated with mood. Despite discrepancies in the adult literature, acute nicotine treatment consistently reduces anxiety in adolescent male rats (Cheeta et al., 2001; Elliott et al., 2004). This property of nicotine may provide a psychological incentive to initiate tobacco use in young smokers, but likely produces long-term changes in neural circuitry making smokers more vulnerable to future mood disorders (see below).
5.3.2. Chronic effects
Chronic nicotine in adolescence has been shown to cause greater and longer-lasting alterations in cholinergic signaling than in adults. Daily adolescent nicotine treatment from P34-43 results in persisting increased mRNA expression of α5, α6, and β2 nAChR subunits in the ventral midbrain that is not seen in rats treated as adults (Adriani et al., 2003). Furthermore, greater upregulation of α4β2* and α7-nAChR binding (Trauth et al., 1999; Slotkin et al., 2004) and downregulation of striatal α6* nAChR binding are seen in adolescents as compared to adults (Doura et al., 2008). Presynaptic markers of cholinergic function, such as choline acetyltransferase activity and hemicholinium binding, are also altered many months after adolescent nicotine treatment, with decrements in neocortex, and increases in the hippocampus and midbrain (Slotkin et al., 2007). These lasting changes in cholinergic activity may impact a variety of neurotransmitter systems that are modulated by ACh.
Given that nAChRs are found on monoamine cell bodies and terminals that continue to mature during adolescence, it is not surprising that adolescent nicotine treatment has been found to result in long-term changes in these neurotransmitter systems. Striatal dopamine transporter density is upregulated after a week of daily nicotine injections in adolescent, but not adult, rats (Collins et al., 2004a). Similarly, eight-day continuous infusion of nicotine results in persistent increases in striatal dopamine content (Slotkin and Seidler, 2007). Functional studies have also shown increased electrically-evoked release of dopamine from prefrontal cortex slices of nicotine-treated adolescents but not adults (Counotte et al, 2008). Transient decreases in striatal serotonin turnover and increases in serotonergic receptors have been observed after adolescent nicotine exposure (Slotkin et al., 2006; Slotkin and Seidler, 2007). Despite these increases in receptor density, cessation of continuous nicotine induces an overall suppression of presynaptic serotonergic activity (Xu et al., 2002). Furthermore, months after chronic adolescent nicotine treatment, persistent increases in serotonergic transporter and receptors are observed in cortex of male, but not female, rats (Slotkin et al., 2007). These long-term alterations in serotonergic signaling may be responsible for the prevalence of mood disorders, such as depression in teenage smokers (Wu and Anthony, 1999; Goodman and Capitman, 2000).
Behavioral studies have similarly implicated a unique role for adolescent nicotine exposure in the development of mood disorders. Several groups have shown that continuous infusion of nicotine during adolescence causes an anxiogenic phenotype in adulthood (Slawecki et al., 2003, 2005; Smith et al., 2006). Adolescent nicotine exposure also enhances adult acquisition of fear conditioning and a prolonged extinction of freezing behavior (Smith et al., 2006), as well as performance on a passive avoidance task, another measure of emotional learning and memory (Trauth et al., 2000). These findings suggest that adolescent nicotine exposure may induce maladaptive learning in emotional contexts, which may lead to life-long mood disorders that are associated with smoking (Gehricke et al., 2007).
Other studies have also revealed persisting deficits in cognition after continuous infusion of nicotine during adolescence. Adolescent, but not adult, nicotine treatment reduces accuracy of correct stimulus detection in a visuospatial attentional task, with an increase in premature and time-out responding that suggests a lack of impulsive control which is part of normal adolescent maturation (Counotte et al., 2008). Similar nicotine-induced deficits have been found in a serial pattern learning paradigm (Fountain et al., 2008). Such alterations in executive functioning resulting from early adolescent nicotine exposure have recently been suggested as a basis for the development of dependence in smokers (deBry and Tiffany, 2008). These studies also shed light on questions in the human literature of whether adolescent nicotine treatment induces enhanced impulsivity or whether adolescents who are more impulsive are simply more prone to smoke (Tercyak et al., 2002; DeBry and Tiffany, 2008). These animal studies suggest that nicotine may play a causal role in the persistence of impulsivity during and beyond adolescence.
Epidemiological studies have also suggested that tobacco is gateway drug that increases the likelihood of subsequent use of other addictive substances (Kandel et al., 1992; Lai et al., 2000). Support for this concept has come from animal studies in which brief exposure to nicotine during adolescence has been shown to enhance the reinforcing value of other abused drugs. Nicotine treatment for seven days produces cross-sensitization to the locomotor effects of cocaine and amphetamine in adolescent, but not adult, male rats (Collins and Izenwasser, 2004; Collins et al., 2004b). A similar enhancement of self-administration behavior is also observed. Intravenous administration of a low nicotine dose, equivalent to approximately 4 cigarettes per day, for four days during adolescence also enhances subsequent acquisition of cocaine self-administration (McQuown et al., 2007). This effect is long-lasting and specific to adolescents, and is associated with enhanced maturation of limbic circuitry (McQuown et al., unpublished observations).
6. Conclusion
nAChRs are present in the brain from the earliest phases of neural development through childhood and adolescence, and into adulthood. However, their patterns of expression are regionally and temporally heterogeneous and, in many cases, unique to the developmental period. The multitude of nAChR subunits, and the resulting range of pharmacological and physiological properties of the nAChR, allows the cholinergic system immense flexibility to regulate many aspects of brain development. The transient increases in nAChR expression within a given brain structure often coincide with the most crucial phases of its development. Thus, nAChRs critically regulate catecholamine and autonomic development in the prenatal period (see Figure 1), cortical, hippocampal, and cerebellar development during the early postnatal period (see Figure 2), and limbic and postnatal catecholamine development during the adolescent period (see Figure 3).
This exquisite regulation of nAChR expression during development predicts that exogenous nicotine exposure may produce a diverse array of functional consequences that depend critically on the timing of exposure. This prediction has been supported not only by studies in laboratory animals, but also by clinical observations. Prenatal nicotine exposure produces autonomic deficits, which may underlie the increased incidence of SIDS seen in the human literature. Nicotine exposure during this time also appears to alter developing catecholamine systems, with particular vulnerability of the dopamine system. Prenatal nicotine-induced deficits reflective of altered dopaminergic processing appear later in life as these circuits undergo postnatal maturation, with children of mothers who smoked exhibiting increased incidence of ADHD and substance abuse during childhood and adolescence. Early postnatal nicotine exposure in rodents, or third trimester exposure in humans, appears to preferentially interfere with cortical development, with human newborns and children exhibiting long-lasting defects in auditory cognitive processing. Finally, exposure to nicotine during adolescence may preferentially interfere with limbic circuitry, producing enhanced vulnerability to nicotine addiction, increased impulsivity, and mood disorders.
Nicotine has dynamic effects on the developing brain, and continued exploration of the developmental patterns of nAChR expression and the impact of nicotine exposure is needed. Completing the characterization of the regional ontogeny of nAChRs, differentiating the effects of nicotine through activation versus desensitization, and better understanding the acute and long-term effects of nicotine at each age will allow better predictive power in the clinical setting and novel therapeutic approaches to nicotine-induced pathologies.
Acknowledgments
This work was supported by U.S. Public Health Service grants DA 10612, DA 19138 and GM 08620 and TRDRP fellowship 16DT-0189.
Abbreviations
- ACh
acetylcholine
- AMPA
alpha-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid glutamate receptor
- G
gestational day
- GABA
Gamma-aminobutyric acid
- KCC2
adult chloride transporter
- LC
locus coeruleus
- nAChRs
nicotinic acetylcholine receptors
- NKCC1
prenatal chloride transporter
- NMDA
N- methyl D-aspartate glutamate receptor
- P
postnatal day
- SIDS
sudden infant death syndrome
- SN
substantia nigra
- VTA
ventral tegmental area
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Adams CE, Broide RS, Chen Y, Winzer-Serhan UH, Henderson TA, Leslie FM, Freedman R. Development of the alpha7 nicotinic cholinergic receptor in rat hippocampal formation. Brain Res Dev Brain Res. 2002;139(2):175–87. doi: 10.1016/s0165-3806(02)00547-3. [DOI] [PubMed] [Google Scholar]
- Adriani W, Macrì S, Pacifici R, Laviola G. Peculiar vulnerability to nicotine oral self-administration in mice during early adolescence. Neuropsychopharmacology. 2002;27(2):212–24. doi: 10.1016/S0893-133X(02)00295-6. [DOI] [PubMed] [Google Scholar]
- Adriani W, Spijker S, Deroche-Gamonet V, Laviola G, Le Moal M, Smit AB, Piazza PV. Evidence for enhanced neurobehavioral vulnerability to nicotine during periadolescence in rats. J Neurosci. 2003;23(11):4712–6. doi: 10.1523/JNEUROSCI.23-11-04712.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agulhon C, Charnay Y, Vallet P, Abitbol M, Kobetz A, Bertrand D, Malafosse A. Distribution of mRNA for the alpha4 subunit of the nicotinic acetylcholine receptor in the human fetal brain. Brain Res Mol Brain Res. 1998;58(1–2):123–31. doi: 10.1016/s0169-328x(98)00113-2. [DOI] [PubMed] [Google Scholar]
- Alkondon M, Pereira EF, Albuquerque EX. alpha-bungarotoxin- and methyllycaconitine-sensitive nicotinic receptors mediate fast synaptic transmission in interneurons of rat hippocampal slices. Brain Res. 1998;810(1–2):257–63. doi: 10.1016/s0006-8993(98)00880-4. [DOI] [PubMed] [Google Scholar]
- Altman J, Bayer SA. Development of the brain stem in the rat. IV. Thymidine- radiographic study of the time of origin of neurons in the pontine region. J Comp Neurol. 1980;194(4):905–29. doi: 10.1002/cne.901940411. [DOI] [PubMed] [Google Scholar]
- Altman J. Postnatal development of the cerebellar cortex in the rat. I. The external germinal layer and the transitional molecular layer. J Comp Neurol. 1972;145(3):353–97. doi: 10.1002/cne.901450305. [DOI] [PubMed] [Google Scholar]
- Andersen SL, Thompson AT, Rutstein M, Hostetter JC, Teicher MH. Dopamine receptor pruning in prefrontal cortex during the periadolescent period in rats. Synapse. 2000;37(2):167–9. doi: 10.1002/1098-2396(200008)37:2<167::AID-SYN11>3.0.CO;2-B. [DOI] [PubMed] [Google Scholar]
- Andersen SL. Changes in the second messenger cyclic AMP during development may underlie motoric symptoms in attention deficit/hyperactivity disorder (ADHD) Behav Brain Res. 2002;130(1–2):197–201. doi: 10.1016/s0166-4328(01)00417-x. [DOI] [PubMed] [Google Scholar]
- Aramakis VB, Hsieh CY, Leslie FM, Metherate R. A critical period for nicotine- induced disruption of synaptic development in rat auditory cortex. J Neurosci. 2000;20(16):6106–16. doi: 10.1523/JNEUROSCI.20-16-06106.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azam L, Chen Y, Leslie FM. Developmental regulation of nicotinic acetylcholine receptors within midbrain dopamine neurons. Neuroscience. 2007;144(4):1347–60. doi: 10.1016/j.neuroscience.2006.11.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aztiria E, Gotti C, Domenici L. Alpha7 but not alpha4 AChR subunit expression is regulated by light in developing primary visual cortex. J Comp Neurol. 2004;480(4):378–91. doi: 10.1002/cne.20358. [DOI] [PubMed] [Google Scholar]
- Bamford O, Hawkins RL. Central effects of an alpha 2-adrenergic antagonist on fetal lambs: a possible mechanism for hypoxic apnea. J Dev Physiol. 1990;13(6):353–8. [PubMed] [Google Scholar]
- Bansal A, Singer JH, Hwang BJ, Xu W, Beaudet A, Feller MB. Mice lacking specific nicotinic acetylcholine receptor subunits exhibit dramatically altered spontaneous activity patterns and reveal a limited role for retinal waves in forming ON and OFF circuits in the inner retina. J Neurosci. 2000;20(20):7672–81. doi: 10.1523/JNEUROSCI.20-20-07672.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbieri RL, Gochberg J, Ryan KJ. Nicotine, cotinine, and anabasine inhibit aromatase in human trophoblast in vitro. J Clin Invest. 1986;77(6):1727–33. doi: 10.1172/JCI112494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barron J, Petrilli F, Strath L, McCaffrey R. Successful interventions for smoking cessation in pregnancy. MCN Am J Matern Child Nurs. 2007;32(1):42–7. doi: 10.1097/00005721-200701000-00009. [DOI] [PubMed] [Google Scholar]
- Bayer SA, Altman J. Hippocampal development in the rat: cytogenesis and morphogenesis examined with autoradiography and low-level X-irradiation. J Comp Neurol. 1974;158(1):55–79. doi: 10.1002/cne.901580105. [DOI] [PubMed] [Google Scholar]
- Bayer SA, Altman J, Russo RJ, Zhang X. Timetables of neurogenesis in the human brain based on experimentally determined patterns in the rat. Neurotoxicology. 1993;14(1):83–144. [PubMed] [Google Scholar]
- Belhage B, Hansen GH, Elster L, Schousboe A. Effects of gamma-aminobutyric acid (GABA) on synaptogenesis and synaptic function. Perspect Dev Neurobiol. 1998;5(2–3):235–46. [PubMed] [Google Scholar]
- Belluzzi JD, Lee AG, Oliff HS, Leslie FM. Age-dependent effects of nicotine on locomotor activity and conditioned place preference in rats. Psychopharmacology (Berl) 2004;174(3):389–95. doi: 10.1007/s00213-003-1758-6. [DOI] [PubMed] [Google Scholar]
- Benes FM, Taylor JB, Cunningham MC. Convergence and plasticity of monoaminergic systems in the medial prefrontal cortex during the postnatal period: implications for the development of psychopathology. Cereb Cortex. 2000;10(10):1014–27. doi: 10.1093/cercor/10.10.1014. [DOI] [PubMed] [Google Scholar]
- Benowitz NL, Jacob P., 3rd Daily intake of nicotine during cigarette smoking. Clin Pharmacol Ther. 1984;35(4):499–504. doi: 10.1038/clpt.1984.67. [DOI] [PubMed] [Google Scholar]
- Berardi N, Pizzorusso T, Maffei L. Critical periods during sensory development. Curr Opin Neurobiol. 2000;10(1):138–45. doi: 10.1016/s0959-4388(99)00047-1. [DOI] [PubMed] [Google Scholar]
- Berrettini W, Yuan X, Tozzi F, Song K, Francks C, Chilcoat H, Waterworth D, Muglia P, Mooser V. Alpha-5/alpha-3 nicotinic receptor subunit alleles increase risk for heavy smoking. Mol Psychiatry. 2008;13(4):368–73. doi: 10.1038/sj.mp.4002154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bina KG, Guzman P, Broide RS, Leslie FM, Smith MA, O’Dowd DK. Localization of alpha 7 nicotinic receptor subunit mRNA and alpha-bungarotoxin binding sites in developing mouse somatosensory thalamocortical system. J Comp Neurol. 1995;363(2):321–332. doi: 10.1002/cne.903630212. [DOI] [PubMed] [Google Scholar]
- Bina KG, Park M, O’Dowd DK. Regulation of alpha7 nicotinic acetylcholine receptors in mouse somatosensory cortex following whisker removal at birth. J Comp Neurol. 1998;397(1):1–9. [PubMed] [Google Scholar]
- Bolanos CA, Glatt SJ, Jackson D. Subsensitivity to dopaminergic drugs in periadolescent rats: a behavioral and neurochemical analysis. Brain Res Dev Brain Res. 1998;111(1):25–33. doi: 10.1016/s0165-3806(98)00116-3. [DOI] [PubMed] [Google Scholar]
- Brenhouse HC, Sonntag KC, Andersen SL. Transient D1 dopamine receptor expression on prefrontal cortex projection neurons: relationship to enhanced motivational salience of drug cues in adolescence. J Neurosci. 2008;28(10):2375–82. doi: 10.1523/JNEUROSCI.5064-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brennan AR, Arnsten AF. Neuronal mechanisms underlying attention deficit hyperactivity disorder: the influence of arousal on prefrontal cortical function. Ann N Y Acad Sci. 2008;1129:236–45. doi: 10.1196/annals.1417.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brielmaier JM, McDonald CG, Smith RF. Immediate and long-term behavioral effects of a single nicotine injection in adolescent and adult rats. Neurotoxicol Teratol. 2007;29(1):74–80. doi: 10.1016/j.ntt.2006.09.023. [DOI] [PubMed] [Google Scholar]
- Britton AF, Vann RE, Robinson SE. Perinatal nicotine exposure eliminates peak in nicotinic acetylcholine receptor response in adolescent rats. J Pharmacol Exp Ther. 2007;320(2):871–6. doi: 10.1124/jpet.106.112730. [DOI] [PubMed] [Google Scholar]
- Brody AL, Mandelkern MA, London ED, Olmstead RE, Farahi J, Scheibal D, Jou J, Allen V, Tiongson E, Chefer SI, Koren AO, Mukhin AG. Cigarette smoking saturates brain alpha 4 beta 2 nicotinic acetylcholine receptors. Arch Gen Psychiatry. 2006;63(8):907–15. doi: 10.1001/archpsyc.63.8.907. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Broide RS, Leslie FM. The alpha7 nicotinic acetylcholine receptor in neuronal plasticity. Mol Neurobiol. 1999;20(1):1–16. doi: 10.1007/BF02741361. [DOI] [PubMed] [Google Scholar]
- Broide RS, O’Connor LT, Smith MA, Smith JA, Leslie FM. Developmental expression of alpha 7 neuronal nicotinic receptor messenger RNA in rat sensory cortex and thalamus. Neuroscience. 1995;67(1):83–94. doi: 10.1016/0306-4522(94)00623-d. [DOI] [PubMed] [Google Scholar]
- Broide RS, Robertson RT, Leslie FM. Regulation of alpha7 nicotinic acetylcholine receptors in the developing rat somatosensory cortex by thalamocortical afferents. J Neurosci. 1996;16(9):2956–71. doi: 10.1523/JNEUROSCI.16-09-02956.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buka SL, Shenassa ED, Niaura R. Elevated risk of tobacco dependence among offspring of mothers who smoked during pregnancy: a 30-year prospective study. Am J Psychiatry. 2003;160(11):1978–84. doi: 10.1176/appi.ajp.160.11.1978. [DOI] [PubMed] [Google Scholar]
- Burns EM. The effects of stress during the brain growth spurt. Annu Rev Nurs Res. 1990;8:57–82. [PubMed] [Google Scholar]
- Buttigieg J, Brown S, Zhang M, Lowe M, Holloway AC, Nurse CA. Chronic nicotine in utero selectively suppresses hypoxic sensitivity in neonatal rat adrenal chromaffin cells. FASEB J. 2008;22(5):1317–26. doi: 10.1096/fj.07-9194com. [DOI] [PubMed] [Google Scholar]
- Candy JM, Perry EK, Perry RH, Bloxham CA, Thompson J, Johnson M, Oakley AE, Edwardson JA. Evidence for the early prenatal development of cortical cholinergic afferents from the nucleus of Meynert in the human foetus. Neurosci Lett. 1985;61(1–2):91–5. doi: 10.1016/0304-3940(85)90406-9. [DOI] [PubMed] [Google Scholar]
- Cao J, Belluzzi JD, Loughlin SE, Keyler DE, Pentel PR, Leslie FM. Acetaldehyde, a major constituent of tobacco smoke, enhances behavioral, endocrine, and neuronal responses to nicotine in adolescent and adult rats. Neuropsychopharmacology. 2007;32(9):2025–35. doi: 10.1038/sj.npp.1301327. [DOI] [PubMed] [Google Scholar]
- Carmignoto G, Vicini S. Activity-dependent decrease in NMDA receptor responses during development of the visual cortex. Science. 1992;258(5084):1007–11. doi: 10.1126/science.1279803. [DOI] [PubMed] [Google Scholar]
- Centers for Disease Control and Prevention (CDC) Fact Sheet: Health Effects of Cigarette Smoking. Smoking and Tobacco Use 2008 [Google Scholar]
- Centers for Disease Control and Prevention (CDC) Tobacco use among adults-- United States, 2005. MMWR Morb Mortal Wkly Rep. 2006;55(42):1145–8. [PubMed] [Google Scholar]
- Chambers RA, Taylor JR, Potenza MN. Developmental neurocircuitry of motivation in adolescence: a critical period of addiction vulnerability. AmJ Psychiatry. 2003;160:1041–1052. doi: 10.1176/appi.ajp.160.6.1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charpantier E, Barnéoud P, Moser P, Besnard F, Sgard F. Nicotinic acetylcholine subunit mRNA expression in dopaminergic neurons of the rat substantia nigra and ventral tegmental area. Neuroreport. 1998;9(13):3097–101. doi: 10.1097/00001756-199809140-00033. [DOI] [PubMed] [Google Scholar]
- Cheeta S, Irvine EE, Tucci S, Sandhu J, File SE. In adolescence, female rats are more sensitive to the anxiolytic effect of nicotine than are male rats. Neuropsychopharmacology. 2001;25(4):601–7. doi: 10.1016/S0893-133X(01)00258-5. [DOI] [PubMed] [Google Scholar]
- Chen H, Matta SG, Sharp BM. Acquisition of nicotine self-administration in adolescent rats given prolonged access to the drug. Neuropsychopharmacology. 2007;32(3):700–9. doi: 10.1038/sj.npp.1301135. [DOI] [PubMed] [Google Scholar]
- Chen J, Millar WJ. Age of smoking initiation: implications for quitting. Health Rep. 1998;9(4):39–46. [PubMed] [Google Scholar]
- Chen H, Parker SL, Matta SG, Sharp BM. Gestational nicotine exposure reduces nicotinic cholinergic receptor (nAChR) expression in dopaminergic brain regions of adolescent rats. Eur J Neurosci. 2005;22(2):380–8. doi: 10.1111/j.1460-9568.2005.04229.x. [DOI] [PubMed] [Google Scholar]
- Choi S, Kellogg CK. Adolescent development influences functional responsiveness of noradrenergic projections to the hypothalamus in male rats. Brain Res Dev Brain Res. 1996;94(2):144–51. doi: 10.1016/s0165-3806(96)80005-8. [DOI] [PubMed] [Google Scholar]
- Coleman T. Reducing harm from tobacco smoke exposure during pregnancy. Birth Defects Res C Embryo Today. 2008;84(1):73–9. doi: 10.1002/bdrc.20115. [DOI] [PubMed] [Google Scholar]
- Collins SL, Izenwasser S. Chronic nicotine differentially alters cocaine-induced locomotor activity in adolescent vs. adult male and female rats. Neuropharmacology. 2004;46(3):349–62. doi: 10.1016/j.neuropharm.2003.09.024. [DOI] [PubMed] [Google Scholar]
- Collins SL, Wade D, Ledon J, Izenwasser S. Neurochemical alterations produced by daily nicotine exposure in periadolescent vs. adult male rats. Eur J Pharmacol. 2004a;502(1–2):75–85. doi: 10.1016/j.ejphar.2004.08.039. [DOI] [PubMed] [Google Scholar]
- Collins SL, Montano R, Izenwasser S. Nicotine treatment produces persistent increases in amphetamine-stimulated locomotor activity in periadolescent male but not female or adult male rats. Brain Res Dev Brain Res. 2004b;153(2):175–87. doi: 10.1016/j.devbrainres.2004.08.004. [DOI] [PubMed] [Google Scholar]
- Counotte DS, Spijker S, Van de Burgwal LH, Hogenboom F, Schoffelmeer AN, De Vries TJ, Smit AB, Pattij T. Long-Lasting Cognitive Deficits Resulting from Adolescent Nicotine Exposure in Rats. Neuropsychopharmacology. 2008 doi: 10.1038/npp.2008.96. Epub on Jun 25. [DOI] [PubMed] [Google Scholar]
- Court JA, Lloyd S, Johnson M, Griffiths M, Birdsall NJ, Piggott MA, Oakley AE, Ince PG, Perry EK, Perry RH. Nicotinic and muscarinic cholinergic receptor binding in the human hippocampal formation during development and aging. Brain Res Dev Brain Res. 1997;101(1–2):93–105. doi: 10.1016/s0165-3806(97)00052-7. [DOI] [PubMed] [Google Scholar]
- Court JA, Martin-Ruiz C, Graham A, Perry E. Nicotinic receptors in human brain: topography and pathology. J Chem Neuroanat. 2000;20(3–4):281–98. doi: 10.1016/s0891-0618(00)00110-1. [DOI] [PubMed] [Google Scholar]
- Cunningham MG, Bhattacharyya S, Benes FM. Amygdalo-cortical sprouting continues into early adulthood: implications for the development of normal and abnormal function during adolescence. J Comp Neurol. 2002;453(2):116–30. doi: 10.1002/cne.10376. [DOI] [PubMed] [Google Scholar]
- Dani JA, Bertrand D. Nicotinic acetylcholine receptors and nicotinic cholinergic mechanisms of the central nervous system. Annu Rev Pharmacol Toxicol. 2007;47:699–729. doi: 10.1146/annurev.pharmtox.47.120505.105214. [DOI] [PubMed] [Google Scholar]
- Dani JA. Overview of nicotinic receptors and their roles in the central nervous system. Biol Psychiatry. 2001;49(3):166–74. doi: 10.1016/s0006-3223(00)01011-8. [DOI] [PubMed] [Google Scholar]
- DeBry SC, Tiffany ST. Tobacco-induced neurotoxicity of adolescent cognitive development (TINACD): a proposed model for the development of impulsivity in nicotine dependence. Nicotine Tob Res. 2008;10(1):11–25. doi: 10.1080/14622200701767811. [DOI] [PubMed] [Google Scholar]
- DiFranza JR, Rigotti NA, McNeill AD, Ockene JK, Savageau JA, St Cyr D, Coleman M. Initial symptoms of nicotine dependence in adolescents. Tob Control. 2000;9(3):313–9. doi: 10.1136/tc.9.3.313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobbing J, Sands J. Comparative aspects of the brain growth spurt. Early Hum Dev. 1979;3(1):79–83. doi: 10.1016/0378-3782(79)90022-7. [DOI] [PubMed] [Google Scholar]
- Dobbing J. Undernutrition and the developing brain: the use of animal models to elucidate the human problem. Psychiatr Neurol Neurochir. 1971;74(6):433–42. [PubMed] [Google Scholar]
- Doura MB, Gold AB, Keller AB, Perry DC. Adult and periadolescent rats differ in expression of nicotinic cholinergic receptor subtypes and in the response of these subtypes to chronic nicotine exposure. Brain Res. 2008;1215:40–52. doi: 10.1016/j.brainres.2008.03.056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumas TC. Late postnatal maturation of excitatory synaptic transmission permits adult-like expression of hippocampal-dependent behaviors. Hippocampus. 2005;15(5):562–78. doi: 10.1002/hipo.20077. [DOI] [PubMed] [Google Scholar]
- Elliott BM, Faraday MM, Phillips JM, Grunberg NE. Effects of nicotine on elevated plus maze and locomotor activity in male and female adolescent and adult rats. Pharmacol Biochem Behav. 2004;77(1):21–8. doi: 10.1016/j.pbb.2003.09.016. [DOI] [PubMed] [Google Scholar]
- Engin E, Treit D. The role of hippocampus in anxiety: intracerebral infusion studies. Behav Pharmacol. 2007;18(5–6):365–74. doi: 10.1097/FBP.0b013e3282de7929. [DOI] [PubMed] [Google Scholar]
- Eppolito AK, Smith RF. Long-term behavioral and developmental consequences of pre- and perinatal nicotine. Pharmacol Biochem Behav. 2006;85(4):835–41. doi: 10.1016/j.pbb.2006.11.020. [DOI] [PubMed] [Google Scholar]
- Erskine L, McCaig CD. Growth cone neurotransmitter receptor activation modulates electric field-guided nerve growth. Dev Biol. 1995;171(2):330–9. doi: 10.1006/dbio.1995.1285. [DOI] [PubMed] [Google Scholar]
- Evans C, Wang J, Neff R, Mendelowitz D. Hypoxia recruits a respiratory-related excitatory pathway to brainstem premotor cardiac vagal neurons in animals exposed to prenatal nicotine. Neuroscience. 2005;133(4):1073–9. doi: 10.1016/j.neuroscience.2005.03.053. [DOI] [PubMed] [Google Scholar]
- Everett SA, Warren CW, Sharp D, Kann L, Husten CG, Crossett LS. Initiation of cigarette smoking and subsequent smoking behavior among U.S. high school students. Prev Med. 1999;29(5):327–33. doi: 10.1006/pmed.1999.0560. [DOI] [PubMed] [Google Scholar]
- Exley R, Clements MA, Hartung H, McIntosh JM, Cragg SJ. Alpha6-containing nicotinic acetylcholine receptors dominate the nicotine control of dopamine neurotransmission in nucleus accumbens. Neuropsychopharmacology. 2008;33(9):2158–66. doi: 10.1038/sj.npp.1301617. [DOI] [PubMed] [Google Scholar]
- Falk L, Nordberg A, Seiger A, Kjaeldgaard A, Hellström-Lindahl E. The alpha7 nicotinic receptors in human fetal brain and spinal cord. J Neurochem. 2002;80(3):457–65. doi: 10.1046/j.0022-3042.2001.00714.x. [DOI] [PubMed] [Google Scholar]
- Feller MB. Spontaneous correlated activity in developing neural circuits. Neuron. 1999;22(4):653–6. doi: 10.1016/s0896-6273(00)80724-2. [DOI] [PubMed] [Google Scholar]
- Feller MB. The role of nAChR-mediated spontaneous retinal activity in visual system development. J Neurobiol. 2002;53(4):556–67. doi: 10.1002/neu.10140. [DOI] [PubMed] [Google Scholar]
- Fenster CP, Hicks JH, Beckman ML, Covernton PJ, Quick MW, Lester RA. Desensitization of nicotinic receptors in the central nervous system. Ann N Y Acad Sci. 1999;868:620–3. doi: 10.1111/j.1749-6632.1999.tb11335.x. [DOI] [PubMed] [Google Scholar]
- Fewell JE, Smith FG, Ng VK. Prenatal exposure to nicotine impairs protective responses of rat pups to hypoxia in an age-dependent manner. Respir Physiol. 2001;127(1):61–73. doi: 10.1016/s0034-5687(01)00232-8. [DOI] [PubMed] [Google Scholar]
- Fleming P, Blair PS. Sudden Infant Death Syndrome and parental smoking. Early Hum Dev. 2007;83(11):721–5. doi: 10.1016/j.earlhumdev.2007.07.011. [DOI] [PubMed] [Google Scholar]
- Fountain SB, Rowan JD, Kelley BM, Willey AR, Nolley EP. Adolescent exposure to nicotine impairs adult serial pattern learning in rats. Exp Brain Res. 2008;187(4):651–6. doi: 10.1007/s00221-008-1346-4. [DOI] [PubMed] [Google Scholar]
- Franke RM, Belluzzi JD, Leslie FM. Gestational exposure to nicotine and monoamine oxidase inhibitors influences cocaine-induced locomotion in adolescent rats. Psychopharmacology (Berl) 2007;195(1):117–24. doi: 10.1007/s00213-007-0876-y. [DOI] [PubMed] [Google Scholar]
- Franke RM, Park M, Belluzzi JD, Leslie FM. Prenatal nicotine exposure changes natural and drug-induced reinforcement in adolescent male rats. Eur J Neurosci. 2008;27(11):2952–61. doi: 10.1111/j.1460-9568.2008.06253.x. [DOI] [PubMed] [Google Scholar]
- Fried PA, James DS, Watkinson B. Growth and pubertal milestones during adolescence in offspring prenatally exposed to cigarettes and marihuana. Neurotoxicol Teratol. 2001;23(5):431–6. doi: 10.1016/s0892-0362(01)00161-1. [DOI] [PubMed] [Google Scholar]
- Fuchs JL. [125I]alpha-bungarotoxin binding marks primary sensory area developing rat neocortex. Brain Res. 1989;501(2):223–234. doi: 10.1016/0006-8993(89)90640-9. [DOI] [PubMed] [Google Scholar]
- Fucile S, Renzi M, Lauro C, Limatola C, Ciotti T, Eusebi F. Nicotinic cholinergic stimulation promotes survival and reduces motility of cultured rat cerebellar granule cells. Neuroscience. 2004;127(1):53–61. doi: 10.1016/j.neuroscience.2004.04.017. [DOI] [PubMed] [Google Scholar]
- Fung YK. Postnatal behavioural effects of maternal nicotine exposure in rats. J Pharm Pharmacol. 1988;40(12):870–2. doi: 10.1111/j.2042-7158.1988.tb06290.x. [DOI] [PubMed] [Google Scholar]
- Galanopoulou AS, Kyrozis A, Claudio OI, Stanton PK, Moshé SL. Sex-specific KCC2 expression and GABA(A) receptor function in rat substantia nigra. Exp Neurol. 2003;183(2):628–37. doi: 10.1016/s0014-4886(03)00213-9. [DOI] [PubMed] [Google Scholar]
- Galanopoulou AS. Sexually dimorphic expression of KCC2 and GABA function. Epilepsy Res. 2008;80(2–3):99–113. doi: 10.1016/j.eplepsyres.2008.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallardo KA, Leslie FM. Nicotine-stimulated release of [3H]norepinephrine from fetal rat locus coeruleus cells in culture. J Neurochem. 1998;70(2):663–70. doi: 10.1046/j.1471-4159.1998.70020663.x. [DOI] [PubMed] [Google Scholar]
- Galli L, Maffei L. Spontaneous impulse activity of rat retinal ganglion cells in prenatal life. Science. 1988 Oct 7;242(4875):90–1. doi: 10.1126/science.3175637. [DOI] [PubMed] [Google Scholar]
- Gatzke-Kopp LM, Beauchaine TP. Direct and passive prenatal nicotine exposure and the development of externalizing psychopathology. Child Psychiatry Hum Dev. 2007;38(4):255–69. doi: 10.1007/s10578-007-0059-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehricke JG, Loughlin SE, Whalen CK, Potkin SG, Fallon JH, Jamner LD, Belluzzi JD, Leslie FM. Smoking to self-medicate attentional and emotional dysfunctions. Nicotine Tob Res. 2007;9(Suppl 4):S523–36. doi: 10.1080/14622200701685039. [DOI] [PubMed] [Google Scholar]
- Giedd JN, Blumenthal J, Jeffries NO, Castellanos FX, Liu H, Zijdenbos A, Paus T, Evans AC, Rapoport JL. Brain development during childhood and adolescence: a longitudinal MRI study. Nat Neurosci. 1999;2(10):861–3. doi: 10.1038/13158. [DOI] [PubMed] [Google Scholar]
- Giedd JN. Structural magnetic resonance imaging of the adolescent brain. Ann N Y Acad Sci. 2004;1021:77–85. doi: 10.1196/annals.1308.009. [DOI] [PubMed] [Google Scholar]
- Girod R, Crabtree G, Ernstrom G, Ramirez-Latorre J, McGehee D, Turner J, Role L. Heteromeric complexes of alpha 5 and/or alpha 7 subunits. Effects of calcium and potential role in nicotine-induced presynaptic facilitation. Ann N Y Acad Sci. 1999;868:578–90. doi: 10.1111/j.1749-6632.1999.tb11331.x. [DOI] [PubMed] [Google Scholar]
- Goldman L, Winget C, Hollingshead GW, Levine S. Postweaning development of negative feedback in the pituitary-adrenal system of the rat. Neuroendocrinology. 1973;12(3):199–211. doi: 10.1159/000122169. [DOI] [PubMed] [Google Scholar]
- Goodlett CR, Mahoney JC, West JR. Brain growth deficits following a single day of alcohol exposure in the neonatal rat. Alcohol. 1989;6(2):121–6. doi: 10.1016/0741-8329(89)90036-0. [DOI] [PubMed] [Google Scholar]
- Goodman E, Capitman J. Depressive symptoms and cigarette smoking among teens. Pediatrics. 2000;106(4):748–55. doi: 10.1542/peds.106.4.748. [DOI] [PubMed] [Google Scholar]
- Gotti C, Zoli M, Clementi F. Brain nicotinic acetylcholine receptors: native subtypes and their relevance. Trends Pharmacol Sci. 2006;27(9):482–91. doi: 10.1016/j.tips.2006.07.004. [DOI] [PubMed] [Google Scholar]
- Gould E, Butcher LL. Transient expression of choline acetyltransferase-like immunoreactivity in Purkinje cells of the developing rat cerebellum. Brain Res. 1987;431(2):303–6. doi: 10.1016/0165-3806(87)90218-5. [DOI] [PubMed] [Google Scholar]
- Grady SR, Salminen O, Laverty DC, Whiteaker P, McIntosh JM, Collins AC, Marks MJ. The subtypes of nicotinic acetylcholine receptors on dopaminergic terminals of mouse striatum. Biochem Pharmacol. 2007;74(8):1235–46. doi: 10.1016/j.bcp.2007.07.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gurevich EV, Himes JW, Joyce JN. Developmental regulation of expression of the D3 dopamine receptor in rat nucleus accumbens and islands of Calleja. J Pharmacol Exp Ther. 1999;289(1):587–98. [PubMed] [Google Scholar]
- Hanson MG, Landmesser LT. Characterization of the circuits that generate spontaneous episodes of activity in the early embryonic mouse spinal cord. J Neurosci. 2003;23(2):587–600. doi: 10.1523/JNEUROSCI.23-02-00587.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanson MG, Milner LD, Landmesser LT. Spontaneous rhythmic activity in early chick spinal cord influences distinct motor axon pathfinding decisions. Brain Res Rev. 2008;57(1):77–85. doi: 10.1016/j.brainresrev.2007.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hedner T, Bergman B, Holmgren M. Adrenal catecholamines during and following hypoxia in neonatal rats. Med Biol. 1980;58(4):228–31. [PubMed] [Google Scholar]
- Hellström-Lindahl E, Court JA. Nicotinic acetylcholine receptors during prenatal development and brain pathology in human aging. Behav Brain Res. 2000;113(1–2):159–68. doi: 10.1016/s0166-4328(00)00210-2. [DOI] [PubMed] [Google Scholar]
- Hellström-Lindahl E, Gorbounova O, Seiger A, Mousavi M, Nordberg A. Regional distribution of nicotinic receptors during prenatal development of human brain and spinal cord. Brain Res Dev Brain Res. 1998;108(1–2):147–60. doi: 10.1016/s0165-3806(98)00046-7. [DOI] [PubMed] [Google Scholar]
- Herlenius E, Lagercrantz H. Neurotransmitters and neuromodulators during early human development. Early Hum Dev. 2001;65(1):21–37. doi: 10.1016/s0378-3782(01)00189-x. [DOI] [PubMed] [Google Scholar]
- Hohmann CF, Berger-Sweeney J. Cholinergic regulation of cortical development and plasticity. New twists to an old story. Perspect Dev Neurobiol. 1998;5(4):401–25. [PubMed] [Google Scholar]
- Hohmann CF. A morphogenetic role for acetylcholine in mouse cerebral neocortex. Neurosci Biobehav Rev. 2003;27(4):351–63. doi: 10.1016/s0149-7634(03)00066-6. [DOI] [PubMed] [Google Scholar]
- Hory-Lee F, Frank E. The nicotinic blocking agents d-tubocurare and alpha- bungarotoxin save motoneurons from naturally occurring death in the absence of neuromuscular blockade. J Neurosci. 1995;15(10):6453–60. doi: 10.1523/JNEUROSCI.15-10-06453.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsieh CY, Leslie FM, Metherate R. Nicotine exposure during a postnatal critical period alters NR2A and NR2B mRNA expression in rat auditory forebrain. Brain Res Dev Brain Res. 2002;133(1):19–25. doi: 10.1016/s0165-3806(01)00314-5. [DOI] [PubMed] [Google Scholar]
- Huang ZG, Griffioen KJ, Wang X, Dergacheva O, Kamendi H, Gorini C, Mendelowitz Huang ZG, Wang X, Evans C, Gold A, Bouairi E, Mendelowitz D. Prenatal nicotine exposure alters the types of nicotinic receptors that facilitate excitatory inputs to cardiac vagal neurons. J Neurophysiol. 2004;92(4):2548–54. doi: 10.1152/jn.00500.2004. [DOI] [PubMed] [Google Scholar]
- Huang ZG, Griffioen KJ, Wang X, Dergacheva O, Kamendi H, Gorini C, Mendelowitz D. Nicotinic receptor activation occludes purinergic control of central cardiorespiratory network responses to hypoxia/hypercapnia. J Neurophysiol. 2007;98(4):2429–38. doi: 10.1152/jn.00448.2007. [DOI] [PubMed] [Google Scholar]
- Huang LZ, Abbott LC, Winzer-Serhan UH. Effects of chronic neonatal nicotine exposure on nicotinic acetylcholine receptor binding, cell death and morphology in hippocampus and cerebellum. Neuroscience. 2007a;146(4):1854–68. doi: 10.1016/j.neuroscience.2007.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang LZ, Liu X, Griffith WH, Winzer-Serhan UH. Chronic neonatal nicotine increases anxiety but does not impair cognition in adult rats. Behav Neurosci. 2007b;121(6):1342–52. doi: 10.1037/0735-7044.121.6.1342. [DOI] [PubMed] [Google Scholar]
- Huang LZ, Winzer-Serhan UH. Chronic neonatal nicotine upregulates heteromeric nicotinic acetylcholine receptor binding without change in subunit mRNA expression. Brain Res. 2006;1113(1):94–109. doi: 10.1016/j.brainres.2006.06.084. [DOI] [PubMed] [Google Scholar]
- Hughes JR, Gulliver SB, Fenwick JW, Valliere WA, Cruser K, Pepper S, Shea P, Solomon LJ, Flynn BS. Smoking cessation among self-quitters. Health Psychol. 1992;11(5):331–4. doi: 10.1037//0278-6133.11.5.331. [DOI] [PubMed] [Google Scholar]
- Itoi K. Ablation of the central noradrenergic neurons for unraveling their roles in stress and anxiety. Ann N Y Acad Sci. 2008;1129:47–54. doi: 10.1196/annals.1417.012. [DOI] [PubMed] [Google Scholar]
- Jacobsen LK, Slotkin TA, Westerveld M, Mencl WE, Pugh KR. Visuospatial memory deficits emerging during nicotine withdrawal in adolescents with prenatal exposure to active maternal smoking. Neuropsychopharmacology. 2006;31(7):1550–61. doi: 10.1038/sj.npp.1300981. [DOI] [PubMed] [Google Scholar]
- Jaferi A, Bhatnagar S. Corticosterone can act at the posterior paraventricular thalamus to inhibit hypothalamic-pituitary-adrenal activity in animals that habituate to repeated stress. Endocrinology. 2006;147(10):4917–30. doi: 10.1210/en.2005-1393. [DOI] [PubMed] [Google Scholar]
- Jewett DL, Romano MN. Neonatal development of auditory system potentials averaged from the scalp of rat and cat. Brain Res. 1972;36(1):101–15. doi: 10.1016/0006-8993(72)90769-x. [DOI] [PubMed] [Google Scholar]
- Jung AB, Bennett JP., Jr Development of striatal dopaminergic function. I. Pre- and postnatal development of mRNAs and binding sites for striatal D1 (D1a) and D2 (D2a) receptors. Brain Res Dev Brain Res. 1996;94(2):109–20. doi: 10.1016/0165-3806(96)00033-8. [DOI] [PubMed] [Google Scholar]
- Kahn RS, Khoury J, Nichols WC, Lanphear BP. Role of dopamine transporter genotype and maternal prenatal smoking in childhood hyperactive-impulsive, inattentive, and oppositional behaviors. J Pediatr. 2003;143(1):104–10. doi: 10.1016/S0022-3476(03)00208-7. [DOI] [PubMed] [Google Scholar]
- Kalsbeek A, Voorn P, Buijs RM, Pool CW, Uylings HB. Development of the dopaminergic innervation in the prefrontal cortex of the rat. J Comp Neurol. 1988;269(1):58–72. doi: 10.1002/cne.902690105. [DOI] [PubMed] [Google Scholar]
- Kamendi H, Stephens C, Dergacheva O, Wang X, Huang ZG, Bouairi E, Gorini C, Kane VB, Fu Y, Matta SG, Sharp BM. Gestational nicotine exposure attenuates nicotine-stimulated dopamine release in the nucleus accumbens shell of adolescent Lewis rats. J Pharmacol Exp Ther. 2004;308(2):521–8. doi: 10.1124/jpet.103.059899. [DOI] [PubMed] [Google Scholar]
- Kandel DB, Yamaguchi K, Chen K. Stages of progression in drug involvement from adolescence to adulthood: further evidence for the gateway theory. J Stud Alcohol. 1992;53:447–457. doi: 10.15288/jsa.1992.53.447. [DOI] [PubMed] [Google Scholar]
- Karlsson KA, Kreider JC, Blumberg MS. Hypothalamic contribution to sleep- wake cycle development. Neuroscience. 2004;123(2):575–82. doi: 10.1016/j.neuroscience.2003.09.025. [DOI] [PubMed] [Google Scholar]
- Kassam SM, Herman PM, Goodfellow NM, Alves NC, Lambe EK. Developmental excitation of corticothalamic neurons by nicotinic acetylcholine receptors. J Neurosci. 2008;28(35):8756–64. doi: 10.1523/JNEUROSCI.2645-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawa K. Acute synaptic modulation by nicotinic agonists in developing cerebellar Purkinje cells of the rat. J Physiol. 2002;538(Pt 1):87–102. doi: 10.1113/jphysiol.2001.012885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kenny PJ, Markou A. Neurobiology of the nicotine withdrawal syndrome. Pharmacol Biochem Behav. 2001;70(4):531–49. doi: 10.1016/s0091-3057(01)00651-7. [DOI] [PubMed] [Google Scholar]
- King SL, Marks MJ, Grady SR, Caldarone BJ, Koren AO, Mukhin AG, Collins AC, Picciotto MR. Conditional expression in corticothalamic efferents reveals a developmental role for nicotinic acetylcholine receptors in modulation of passive avoidance behavior. J Neurosci. 2003;23(9):3837–43. doi: 10.1523/JNEUROSCI.23-09-03837.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klink R, de Kerchove d’Exaerde A, Zoli M, Changeux JP. Molecular and physiological diversity of nicotinic acetylcholine receptors in the midbrain dopaminergic nuclei. J Neurosci. 2001;21(5):1452–63. doi: 10.1523/JNEUROSCI.21-05-01452.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kota D, Martin BR, Robinson SE, Damaj MI. Nicotine dependence and reward differ between adolescent and adult male mice. J Pharmacol Exp Ther. 2007;322(1):399–407. doi: 10.1124/jpet.107.121616. [DOI] [PubMed] [Google Scholar]
- Kum-Nji P, Meloy L, Herrod HG. Environmental tobacco smoke exposure: prevalence and mechanisms of causation of infections in children. Pediatrics. 2006;117(5):1745–54. doi: 10.1542/peds.2005-1886. [DOI] [PubMed] [Google Scholar]
- Labarca C, Schwarz J, Deshpande P, Schwarz S, Nowak MW, Fonck C, Nashmi R, Kofuji P, Dang H, Shi W, Fidan M, Khakh BS, Chen Z, Bowers BJ, Boulter J, Wehner JM, Lester HA. Point mutant mice with hypersensitive alpha 4 nicotinic receptors show dopaminergic deficits and increased anxiety. Proc Natl Acad Sci U S A. 2001;98(5):2786–91. doi: 10.1073/pnas.041582598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai S, Lai H, Page JB, McCoy CB. The association between cigarette smoking and drug abuse in the United States. J Addict Dis. 2000;19:11–24. doi: 10.1300/J069v19n04_02. [DOI] [PubMed] [Google Scholar]
- Lauder JM, Schambra UB. Morphogenetic roles of acetylcholine. Environ Health Perspect. 1999;107(Suppl 1):65–9. doi: 10.1289/ehp.99107s165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leiter JC, Böhm I. Mechanisms of pathogenesis in the Sudden Infant Death Syndrome. Respir Physiol Neurobiol. 2007;159(2):127–38. doi: 10.1016/j.resp.2007.05.014. [DOI] [PubMed] [Google Scholar]
- Le Novère N, Zoli M, Changeux JP. Neuronal nicotinic receptor alpha 6 subunit mRNA is selectively concentrated in catecholaminergic nuclei of the rat brain. Eur J Neurosci. 1996;8(11):2428–39. doi: 10.1111/j.1460-9568.1996.tb01206.x. [DOI] [PubMed] [Google Scholar]
- Léna C, de Kerchove D’Exaerde A, Cordero-Erausquin M, Le Novère N, del Mar Arroyo-Jimenez M, Changeux JP. Diversity and distribution of nicotinic acetylcholine receptors in the locus ceruleus neurons. Proc Natl Acad Sci U S A. 1999;96(21):12126–31. doi: 10.1073/pnas.96.21.12126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leon-Carrion J, García-Orza J, Pérez-Santamaría FJ. Development of the inhibitory component of the executive functions in children and adolescents. Int J Neurosci. 2004;114(10):1291–311. doi: 10.1080/00207450490476066. [DOI] [PubMed] [Google Scholar]
- LeSage MG, Gustaf E, Dufek MB, Pentel PR. Effects of maternal intravenous nicotine administration on locomotor behavior in pre-weanling rats. Pharmacol Biochem Behav. 2006;85(3):575–83. doi: 10.1016/j.pbb.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leslie FM, Azam L, Gallardo K, O’Leary K, Franke R, Lotfipour S. Nicotinic Receptor Regulation of Developing Catecholamine Systems. In: Miller Michael W., editor. Brain Development: Normal Processes and the Effects of Alcohol and Nicotine. New York, NY: Oxford University Press; 2006. pp. 381–398. [Google Scholar]
- Leslie FM, Gallardo KA, Park MK. Nicotinic acetylcholine receptor-mediated release of [3H]norepinephrine from developing and adult rat hippocampus: direct and indirect mechanisms. Neuropharmacology. 2002;42(5):653–61. doi: 10.1016/s0028-3908(02)00019-9. [DOI] [PubMed] [Google Scholar]
- Levin ED, Lawrence S, Petro A, Horton K, Seidler FJ, Slotkin TA. Increased nicotine self-administration following prenatal exposure in female rats. Pharmacol Biochem Behav. 2006;85(3):669–74. doi: 10.1016/j.pbb.2006.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin ED, Lawrence SS, Petro A, Horton K, Rezvani AH, Seidler FJ, Slotkin TA. Adolescent vs. adult-onset nicotine self-administration in male rats: duration of effect and differential nicotinic receptor correlates. Neurotoxicol Teratol. 2007;29(4):458–65. doi: 10.1016/j.ntt.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levin ED, Rezvani AH, Montoya D, Rose JE, Swartzwelder HS. Adolescent- onset nicotine self-administration modeled in female rats. Psychopharmacology (Berl) 2003;169(2):141–9. doi: 10.1007/s00213-003-1486-y. [DOI] [PubMed] [Google Scholar]
- Lewis TL, Maurer D. Multiple sensitive periods in human visual development: evidence from visually deprived children. Dev Psychobiol. 2005;46(3):163–83. doi: 10.1002/dev.20055. [DOI] [PubMed] [Google Scholar]
- Liang K, Poytress BS, Chen Y, Leslie FM, Weinberger NM, Metherate R. Neonatal nicotine exposure impairs nicotinic enhancement of central auditory processing and auditory learning in adult rats. Eur J Neurosci. 2006;24(3):857–66. doi: 10.1111/j.1460-9568.2006.04945.x. [DOI] [PubMed] [Google Scholar]
- Lichtensteiger W, Schlumpf M. Prenatal nicotine affects fetal testosterone and sexual dimorphism of saccharin preference. Pharmacol Biochem Behav. 1985;23(3):439–44. doi: 10.1016/0091-3057(85)90018-8. [DOI] [PubMed] [Google Scholar]
- Lipton SA, Frosch MP, Phillips MD, Tauck DL, Aizenman E. Nicotinic antagonists enhance process outgrowth by rat retinal ganglion cells in culture. Science. 1988;239(4845):1293–6. doi: 10.1126/science.3344435. [DOI] [PubMed] [Google Scholar]
- Liu A, Neff RA, Berg DK. Sequential Interplay of Nicotinic and GABAergic Signaling Guides Neuronal Development. Science. 2006;314:1610–1613. doi: 10.1126/science.1134246. [DOI] [PubMed] [Google Scholar]
- Luetje CW, Patrick J. Both alpha- and beta-subunits contribute to the agonist sensitivity of neuronal nicotinic acetylcholine receptors. J Neurosci. 1991;11(3):837–45. doi: 10.1523/JNEUROSCI.11-03-00837.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maggi L, Le Magueresse C, Changeux JP, Cherubini E. Nicotine activates immature “silent” connections in the developing hippocampus. Proc Natl Acad Sci U S A. 2003;100(4):2059–64. doi: 10.1073/pnas.0437947100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansvelder HD, Role LW. Neuronal Receptors for Nicotine: Functional Diversity and Developmental Changes. In: Miller Michael W., editor. Brain Development: Normal Processes and the Effects of Alcohol and Nicotine. New York, NY: Oxford University Press; 2006. pp. 341–362. [Google Scholar]
- Mao D, Perry DC, Yasuda RP, Wolfe BB, Kellar KJ. The alpha4beta2alpha5 nicotinic cholinergic receptor in rat brain is resistant to up-regulation by nicotine in vivo. J Neurochem. 2008;104(2):446–56. doi: 10.1111/j.1471-4159.2007.05011.x. [DOI] [PubMed] [Google Scholar]
- Mathers M, Toumbourou JW, Catalano RF, Williams J, Patton GC. Consequences of youth tobacco use: a review of prospective behavioural studies. Addiction. 2006;101(7):948–58. doi: 10.1111/j.1360-0443.2006.01438.x. [DOI] [PubMed] [Google Scholar]
- McCartney JS, Fried PA, Watkinson B. Central auditory processing in school-age children prenatally exposed to cigarette smoke. Neurotoxicol Teratol. 1994;16(3):269– 76. doi: 10.1016/0892-0362(94)90048-5. [DOI] [PubMed] [Google Scholar]
- McGehee DS, Role LW. Physiological diversity of nicotinic acetylcholine receptors expressed by vertebrate neurons. Annu Rev Physiol. 1995;57:521–46. doi: 10.1146/annurev.ph.57.030195.002513. [DOI] [PubMed] [Google Scholar]
- McGehee DS. Molecular diversity of neuronal nicotinic acetylcholine receptors. Ann N Y Acad Sci. 1999;868:565–77. doi: 10.1111/j.1749-6632.1999.tb11330.x. [DOI] [PubMed] [Google Scholar]
- McIntosh JM, Mendelowitz D. Prenatal nicotine exposure alters the nicotinic receptor subtypes that modulate excitation of parasympathetic cardiac neurons in the nucleus ambiguus from primarily alpha3beta2 and/or alpha6betaX to alpha3beta4. Neuropharmacology. 2006;51(1):60–6. doi: 10.1016/j.neuropharm.2006.03.001. [DOI] [PubMed] [Google Scholar]
- McQuown SC, Belluzzi JD, Leslie FM. Low dose nicotine treatment during early adolescence increases subsequent cocaine reward. Neurotoxicol Teratol. 2007;29(1):66– 73. doi: 10.1016/j.ntt.2006.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metherate R, Hsieh CY. Regulation of glutamate synapses by nicotinic acetylcholine receptors in auditory cortex. Neurobiol Learn Mem. 2003;80(3):285–90. doi: 10.1016/s1074-7427(03)00062-5. [DOI] [PubMed] [Google Scholar]
- Metherate R, Hsieh CY. Regulation of glutamate synapses by nicotinic acetylcholine receptors in auditory cortex. Neurobiol Learn Mem. 2003;80(3):285–90. doi: 10.1016/s1074-7427(03)00062-5. [DOI] [PubMed] [Google Scholar]
- Metherate R. Nicotinic acetylcholine receptors in sensory cortex. Learn Mem. 2004;11(1):50–9. doi: 10.1101/lm.69904. [DOI] [PubMed] [Google Scholar]
- Miao H, Liu C, Bishop K, Gong ZH, Nordberg A, Zhang X. Nicotine exposure during a critical period of development leads to persistent changes in nicotinic acetylcholine receptors of adult rat brain. J Neurochem. 1998;70(2):752–62. doi: 10.1046/j.1471-4159.1998.70020752.x. [DOI] [PubMed] [Google Scholar]
- Moon RY, Horne RS, Hauck FR. Sudden infant death syndrome. Lancet. 2007;370(9598):1578–87. doi: 10.1016/S0140-6736(07)61662-6. [DOI] [PubMed] [Google Scholar]
- Morishita H, Hensch TK. Critical period revisited: impact on vision. Curr Opin Neurobiol. 2008;18(1):101–7. doi: 10.1016/j.conb.2008.05.009. [DOI] [PubMed] [Google Scholar]
- Myers CP, Lewcock JW, Hanson MG, Gosgnach S, Aimone JB, Gage FH, Lee KF, Landmesser LT, Pfaff SL. Cholinergic input is required during embryonic development to mediate proper assembly of spinal locomotor circuits. Neuron. 2005;46(1):37–49. doi: 10.1016/j.neuron.2005.02.022. [DOI] [PubMed] [Google Scholar]
- Naeff B, Schlumpf M, Lichtensteiger W. Pre- and postnatal development of high- affinity [3H]nicotine binding sites in rat brain regions: an autoradiographic study. Brain Res Dev Brain Res. 1992;68(2):163–74. doi: 10.1016/0165-3806(92)90058-5. [DOI] [PubMed] [Google Scholar]
- Nakauchi S, Brennan RJ, Boulter J, Sumikawa K. Nicotine gates long-term potentiation in the hippocampal CA1 region via the activation of alpha2* nicotinic ACh receptors. Eur J Neurosci. 2007;25(9):2666–81. doi: 10.1111/j.1460-9568.2007.05513.x. [DOI] [PubMed] [Google Scholar]
- National Institute for Clinical Excellence. Technology Appraisal Guidance No 39. London: National Institute for Clinical Excellence; 2002. Guidance on the use of nicotine replacement therapy (NRT) and bupropion for smoking cessation. [Google Scholar]
- Navarro HA, Seidler FJ, Schwartz RD, Baker FE, Dobbins SS, Slotkin TA. Prenatal exposure to nicotine impairs nervous system development at a dose which does not affect viability or growth. Brain Res Bull. 1989;23(3):187–92. doi: 10.1016/0361-9230(89)90146-9. [DOI] [PubMed] [Google Scholar]
- Neff RA, Humphrey J, Mihalevich M, Mendelowitz D. Nicotine enhances presynaptic and postsynaptic glutamatergic neurotransmission to activate cardiac parasympathetic neurons. Circ Res. 1998;83(12):1241–7. doi: 10.1161/01.res.83.12.1241. [DOI] [PubMed] [Google Scholar]
- Neff RA, Wang J, Baxi S, Evans C, Mendelowitz D. Respiratory sinus arrhythmia: endogenous activation of nicotinic receptors mediates respiratory modulation of brainstem cardioinhibitory parasympathetic neurons. Circ Res. 2003;93(6):565–72. doi: 10.1161/01.RES.0000090361.45027.5B. [DOI] [PubMed] [Google Scholar]
- Nelson EE, Panksepp J. Brain substrates of infant-mother attachment: contributions of opioids, oxytocin, and norepinephrine. Neurosci Biobehav Rev. 1998;22(3):437–52. doi: 10.1016/s0149-7634(97)00052-3. [DOI] [PubMed] [Google Scholar]
- Nestler EJ, Carlezon WA., Jr The mesolimbic dopamine reward circuit in depression. Biol Psychiatry. 2006;59(12):1151–9. doi: 10.1016/j.biopsych.2005.09.018. [DOI] [PubMed] [Google Scholar]
- Neuman RJ, Lobos E, Reich W, Henderson CA, Sun LW, Todd RD. Prenatal smoking exposure and dopaminergic genotypes interact to cause a severe ADHD subtype. Biol Psychiatry. 2007;61(12):1320–8. doi: 10.1016/j.biopsych.2006.08.049. [DOI] [PubMed] [Google Scholar]
- Nguyen HN, Rasmussen BA, Perry DC. Subtype-selective up-regulation by chronic nicotine of high-affinity nicotinic receptors in rat brain demonstrated by receptor autoradiography. J Pharmacol Exp Ther. 2003;307:1090–1097. doi: 10.1124/jpet.103.056408. [DOI] [PubMed] [Google Scholar]
- Nutt DJ. Relationship of neurotransmitters to the symptoms of major depressive disorder. J Clin Psychiatry. 2008;69(Suppl E1):4–7. [PubMed] [Google Scholar]
- O’Dell LE, Bruijnzeel AW, Ghozland S, Markou A, Koob GF. Nicotine withdrawal in adolescent and adult rats. Ann N Y Acad Sci. 2004;1021:167–74. doi: 10.1196/annals.1308.022. [DOI] [PubMed] [Google Scholar]
- O’Dell LE, Bruijnzeel AW, Smith RT, Parsons LH, Merves ML, Goldberger BA, Richardson HN, Koob GF, Markou A. Diminished nicotine withdrawal in adolescent rats: implications for vulnerability to addiction. Psychopharmacology (Berl) 2006;186(4):612–9. doi: 10.1007/s00213-006-0383-6. [DOI] [PubMed] [Google Scholar]
- O’Dell LE. A psychobiological framework of the substrates that mediate nicotine use during adolescence. Neuropharmacology. 2008 doi: 10.1016/j.neuropharm.2008.07.039. Epub on Aug 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Leary KT, Leslie FM. Developmental regulation of nicotinic acetylcholine receptor-mediated [3H]norepinephrine release from rat cerebellum. J Neurochem. 2003;84(5):952–9. doi: 10.1046/j.1471-4159.2003.01575.x. [DOI] [PubMed] [Google Scholar]
- O’Leary KT, Leslie FM. Enhanced nicotinic acetylcholine receptor-mediated [3H]norepinephrine release from neonatal rat hypothalamus. Neuropharmacology. 2006;50(1):81–8. doi: 10.1016/j.neuropharm.2005.08.013. [DOI] [PubMed] [Google Scholar]
- O’Leary KT, Loughlin SE, Chen Y, Leslie FM. Nicotinic acetylcholine receptor subunit mRNA expression in adult and developing rat medullary catecholamine neurons. J Comp Neurol. 2008;510(6):655–72. doi: 10.1002/cne.21833. [DOI] [PubMed] [Google Scholar]
- Onal A, Uysal A, Ulker S, Delen Y, Yurtseven ME, Evinç A. Alterations of brain tissue in fetal rats exposed to nicotine in utero: possible involvement of nitric oxide and catecholamines. Neurotoxicol Teratol. 2004;26(1):103–12. doi: 10.1016/j.ntt.2003.08.005. [DOI] [PubMed] [Google Scholar]
- Opanashuk LA, Pauly JR, Hauser KF. Effect of nicotine on cerebellar granule neuron development. Eur J Neurosci. 2001;13(1):48–56. [PMC free article] [PubMed] [Google Scholar]
- Orr-Urtreger A, Broide RS, Kasten MR, Dang H, Dani JA, Beaudet AL, Patrick JW. Mice homozygous for the L250T mutation in the alpha7 nicotinic acetylcholine receptor show increased neuronal apoptosis and die within 1 day of birth. J Neurochem. 2000;74(5):2154–66. doi: 10.1046/j.1471-4159.2000.0742154.x. [DOI] [PubMed] [Google Scholar]
- Orr-Urtreger A, Göldner FM, Saeki M, Lorenzo I, Goldberg L, De Biasi M, Dani JA, Patrick JW, Beaudet AL. Mice deficient in the alpha7 neuronal nicotinic acetylcholine receptor lack alpha-bungarotoxin binding sites and hippocampal fast nicotinic currents. J Neurosci. 1997;17(23):9165–71. doi: 10.1523/JNEUROSCI.17-23-09165.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palmiter RD. Dopamine signaling in the dorsal striatum is essential for motivated behaviors: lessons from dopamine-deficient mice. Ann N Y Acad Sci. 2008;1129:35–46. doi: 10.1196/annals.1417.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Panagis G, Kastellakis A, Spyraki C, Nomikos G. Effects of methyllycaconitine (MLA), an alpha 7 nicotinic receptor antagonist, on nicotine- and cocaine-induced potentiation of brain stimulation reward. Psychopharmacology (Berl) 2000 May;149(4):388–96. doi: 10.1007/s002130000384. [DOI] [PubMed] [Google Scholar]
- Papke RL, Heinemann SF. The role of the beta 4-subunit in determining the kinetic properties of rat neuronal nicotinic acetylcholine alpha 3-receptors. J Physiol. 1991;440:95–112. doi: 10.1113/jphysiol.1991.sp018698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parish CL, Nunan J, Finkelstein DI, McNamara FN, Wong JY, Waddington JL, Brown RM, Lawrence AJ, Horne MK, Drago J. Mice lacking the alpha4 nicotinic receptor subunit fail to modulate dopaminergic neuronal arbors and possess impaired dopamine transporter function. Mol Pharmacol. 2005;68(5):1376–86. doi: 10.1124/mol.104.004820. [DOI] [PubMed] [Google Scholar]
- Pauly JR, Slotkin TA. Maternal tobacco smoking, nicotine replacement and neurobehavioural development. Acta Paediatr. 2008;97(10):1331–7. doi: 10.1111/j.1651-2227.2008.00852.x. [DOI] [PubMed] [Google Scholar]
- Paus T, Zijdenbos A, Worsley K, Collins DL, Blumenthal J, Giedd JN, Rapoport JL, Evans AC. Structural maturation of neural pathways in children and adolescents: in vivo study. Science. 1999;283(5409):1908–11. doi: 10.1126/science.283.5409.1908. [DOI] [PubMed] [Google Scholar]
- Paz R, Barsness B, Martenson T, Tanner D, Allan AM. Behavioral teratogenicity induced by nonforced maternal nicotine consumption. Neuropsychopharmacology. 2007;32(3):693–9. doi: 10.1038/sj.npp.1301066. [DOI] [PubMed] [Google Scholar]
- Pendleton RG, Rasheed A, Roychowdhury R, Hillman R. A new role for catecholamines: ontogenesis. Trends Pharmacol Sci. 1998;19(7):248–51. doi: 10.1016/s0165-6147(98)01218-8. [DOI] [PubMed] [Google Scholar]
- Pentel PR, Keyler DE, Chen Y, LeSage MG, Dufek MB, Le C, Leslie FM. Vaccination against nicotine does not prevent nicotine-induced changes in fetal nicotinic receptor binding and c-fos mRNA expression in rats. Neurotoxicol Teratol. 2006;28(5):589–96. doi: 10.1016/j.ntt.2006.08.001. [DOI] [PubMed] [Google Scholar]
- Perry DC, Xiao Y, Nguyen HN, Musachio JL, Dávila-García MI, Kellar KJ. Measuring nicotinic receptors with characteristics of alpha4beta2, alpha3beta2 and alpha3beta4 subtypes in rat tissues by autoradiography. J Neurochem. 2002;82(3):468–81. doi: 10.1046/j.1471-4159.2002.00951.x. [DOI] [PubMed] [Google Scholar]
- Perry EK, Smith CJ, Atack JR, Candy JM, Johnson M, Perry RH. Neocortical cholinergic enzyme and receptor activities in the human fetal brain. J Neurochem. 1986;47(4):1262–9. doi: 10.1111/j.1471-4159.1986.tb00749.x. [DOI] [PubMed] [Google Scholar]
- Picciotto MR, Zoli M, Rimondini R, Léna C, Marubio LM, Pich EM, Fuxe K, Changeux JP. Acetylcholine receptors containing the beta2 subunit are involved in the reinforcing properties of nicotine. Nature. 1998;391(6663):173–7. doi: 10.1038/34413. [DOI] [PubMed] [Google Scholar]
- Porath AJ, Fried PA. Effects of prenatal cigarette and marijuana exposure on drug use among offspring. Neurotoxicol Teratol. 2005;27(2):267–77. doi: 10.1016/j.ntt.2004.12.003. [DOI] [PubMed] [Google Scholar]
- Prokhorov AV, Winickoff JP, Ahluwalia JS, Ossip-Klein D, Tanski S, Lando HA, Moolchan ET, Muramoto M, Klein JD, Weitzman M, Ford KH Tobacco Consortium; American Academy of Pediatrics Center for Child Health Research. Youth tobacco use: a global perspective for child health care clinicians. Pediatrics. 2006;118(3):e890–903. doi: 10.1542/peds.2005-0810. [DOI] [PubMed] [Google Scholar]
- Pugh PC, Berg DK. Neuronal acetylcholine receptors that bind alpha- bungarotoxin mediate neurite retraction in a calcium-dependent manner. J Neurosci. 1994;14(2):889–96. doi: 10.1523/JNEUROSCI.14-02-00889.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinn R. Comparing rat’s to human’s age: how old is my rat in people years? Nutrition. 2005;21(6):775–7. doi: 10.1016/j.nut.2005.04.002. [DOI] [PubMed] [Google Scholar]
- Rakic P, Bourgeois JP, Goldman-Rakic PS. Synaptic development of the cerebral cortex: implications for learning, memory, and mental illness. Prog Brain Res. 1994;102:227–43. doi: 10.1016/S0079-6123(08)60543-9. [DOI] [PubMed] [Google Scholar]
- Ramirez-Latorre J, Yu CR, Qu X, Perin F, Karlin A, Role L. Functional contributions of alpha5 subunit to neuronal acetylcholine receptor channels. Nature. 1996;380(6572):347–51. doi: 10.1038/380347a0. [DOI] [PubMed] [Google Scholar]
- Renshaw G, Rigby P, Self G, Lamb A, Goldie R. Exogenously administered alpha- bungarotoxin binds to embryonic chick spinal cord: implications for the toxin- induced arrest of naturally occurring motoneuron death. Neuroscience. 1993;53(4):1163–72. doi: 10.1016/0306-4522(93)90498-5. [DOI] [PubMed] [Google Scholar]
- Ribary U, Lichtensteiger W. Effects of acute and chronic prenatal nicotine treatment on central catecholamine systems of male and female rat fetuses and offspring. J Pharmacol Exp Ther. 1989;248(2):786–92. [PubMed] [Google Scholar]
- Rivera C, Voipio J, Payne JA, Ruusuvuori E, Lahtinen H, Lamsa K, Pirvola U, Saarma M, Kaila K. The K+/Cl− co-transporter KCC2 renders GABA hyperpolarizing during neuronal maturation. Nature. 1999;397(6716):251–5. doi: 10.1038/16697. [DOI] [PubMed] [Google Scholar]
- Robinson TE, Berridge KC. Incentive-sensitization and addiction. Addiction. 2001;96(1):103–14. doi: 10.1046/j.1360-0443.2001.9611038.x. [DOI] [PubMed] [Google Scholar]
- Roerig B, Nelson DA, Katz LC. Fast synaptic signaling by nicotinic acetylcholine and serotonin 5-HT3 receptors in developing visual cortex. J Neurosci. 1997;17(21):8353–62. doi: 10.1523/JNEUROSCI.17-21-08353.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rogers JM. Tobacco and pregnancy: overview of exposures and effects. Birth Defects Res C Embryo Today. 2008;84(1):1–15. doi: 10.1002/bdrc.20119. [DOI] [PubMed] [Google Scholar]
- Sarasin A, Schlumpf M, Müller M, Fleischmann I, Lauber ME, Lichtensteiger W. Adrenal-mediated rather than direct effects of nicotine as a basis of altered sex steroid synthesis in fetal and neonatal rat. Reprod Toxicol. 2003;17(2):153–62. doi: 10.1016/s0890-6238(02)00119-3. [DOI] [PubMed] [Google Scholar]
- Schlaepfer IR, Hoft NR, Collins AC, Corley RP, Hewitt JK, Hopfer CJ, Lessem JM, McQueen MB, Rhee SH, Ehringer MA. The CHRNA5/A3/B4 gene cluster variability as an important determinant of early alcohol and tobacco initiation in young adults. Biol Psychiatry. 2008;63(11):1039–46. doi: 10.1016/j.biopsych.2007.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schochet TL, Kelley AE, Landry CF. Differential expression of arc mRNA and other plasticity-related genes induced by nicotine in adolescent rat forebrain. Neuroscience. 2005;135(1):285–97. doi: 10.1016/j.neuroscience.2005.05.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeman P, Bzowej NH, Guan HC, Bergeron C, Becker LE, Reynolds GP, Bird ED, Riederer P, Jellinger K, Watanabe S. Human brain dopamine receptors in children and aging adults. Synapse. 1987;1(5):399–404. doi: 10.1002/syn.890010503. [DOI] [PubMed] [Google Scholar]
- Semba K, Fibiger HC. Time of origin of cholinergic neurons in the rat basal forebrain. J Comp Neurol. 1988;269(1):87–95. doi: 10.1002/cne.902690107. [DOI] [PubMed] [Google Scholar]
- Sexton M, Fox NL, Hebel JR. Prenatal exposure to tobacco: II. Effects on cognitive functioning at age three. Int J Epidemiol. 1990;19(1):72–7. doi: 10.1093/ije/19.1.72. [DOI] [PubMed] [Google Scholar]
- Shacka JJ, Fennell OB, Robinson SE. Prenatal nicotine sex-dependently alters agonist-induced locomotion and stereotypy. Neurotoxicol Teratol. 1997;19(6):467–76. doi: 10.1016/s0892-0362(97)00063-9. [DOI] [PubMed] [Google Scholar]
- Shearman E, Fallon S, Sershen H, Lajtha A. Nicotine-induced monoamine neurotransmitter changes in the brain of young rats. Brain Res Bull. 2008;76(6):626–39. doi: 10.1016/j.brainresbull.2008.03.017. [DOI] [PubMed] [Google Scholar]
- Shram MJ, Funk D, Li Z, Lê AD. Acute nicotine enhances c-fos mRNA expression differentially in reward-related substrates of adolescent and adult rat brain. Neurosci Lett. 2007;418(3):286–91. doi: 10.1016/j.neulet.2007.03.034. [DOI] [PubMed] [Google Scholar]
- Shram MJ, Funk D, Li Z, Lê AD. Periadolescent and adult rats respond differently in tests measuring the rewarding and aversive effects of nicotine. Psychopharmacology (Berl) 2006;186(2):201–8. doi: 10.1007/s00213-006-0373-8. [DOI] [PubMed] [Google Scholar]
- Shram MJ, Siu EC, Li Z, Tyndale RF, Lê AD. Interactions between age and the aversive effects of nicotine withdrawal under mecamylamine-precipitated and spontaneous conditions in male Wistar rats. Psychopharmacology (Berl) 2008;198(2):181–90. doi: 10.1007/s00213-008-1115-x. [DOI] [PubMed] [Google Scholar]
- Slawecki CJ, Gilder A, Roth J, Ehlers CL. Increased anxiety-like behavior in adult rats exposed to nicotine as adolescents. Pharmacol Biochem Behav. 2003;75(2):355–61. doi: 10.1016/s0091-3057(03)00093-5. [DOI] [PubMed] [Google Scholar]
- Slawecki CJ, Thorsell AK, El Khoury A, Mathé AA, Ehlers CL. Increased CRF- like and NPY-like immunoreactivity in adult rats exposed to nicotine during adolescence: relation to anxiety-like and depressive-like behavior. Neuropeptides. 2005;39(4):369–77. doi: 10.1016/j.npep.2005.06.002. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, Cousins MM, Seidler FJ. Administration of nicotine to adolescent rats evokes regionally selective upregulation of CNS alpha 7 nicotinic acetylcholine receptors. Brain Res. 2004;1030(1):159–63. doi: 10.1016/j.brainres.2004.10.009. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, Lappi SE, McCook EC, Lorber BA, Seidler FJ. Loss of neonatal hypoxia tolerance after prenatal nicotine exposure: implications for sudden infant death syndrome. Brain Res Bull. 1995;38(1):69–75. doi: 10.1016/0361-9230(95)00073-n. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, MacKillop EA, Rudder CL, Ryde IT, Tate CA, Seidler FJ. Permanent, sex-selective effects of prenatal or adolescent nicotine exposure, separately or sequentially, in rat brain regions: indices of cholinergic and serotonergic synaptic function, cell signaling, and neural cell number and size at 6 months of age. Neuropsychopharmacology. 2007;32(5):1082–97. doi: 10.1038/sj.npp.1301231. [DOI] [PubMed] [Google Scholar]
- Slotkin TA, Orband-Miller L, Queen KL. Development of [3H]nicotine binding sites in brain regions of rats exposed to nicotine prenatally via maternal injections or infusions. J Pharmacol Exp Ther. 1987a;242(1):232–7. [PubMed] [Google Scholar]
- Slotkin TA, Orband-Miller L, Queen KL, Whitmore WL, Seidler FJ. Effects of prenatal nicotine exposure on biochemical development of rat brain regions: maternal drug infusions via osmotic minipumps. J Pharmacol Exp Ther. 1987b;240(2):602–11. [PubMed] [Google Scholar]
- Slotkin TA, Seidler FJ. A unique role for striatal serotonergic systems in the withdrawal from adolescent nicotine administration. Neurotoxicol Teratol. 2007;29(1):10–6. doi: 10.1016/j.ntt.2006.06.001. [DOI] [PubMed] [Google Scholar]
- Slotkin TA. Fetal nicotine or cocaine exposure: which one is worse? J Pharmacol Exp Ther. 1998;285(3):931–45. [PubMed] [Google Scholar]
- Small DH, Reed G, Whitefield B, Nurcombe V. Cholinergic regulation of neurite outgrowth from isolated chick sympathetic neurons in culture. J Neurosci. 1995;15(1 Pt 1):144–51. doi: 10.1523/JNEUROSCI.15-01-00144.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith LN, McDonald CG, Bergstrom HC, Brielmaier JM, Eppolito AK, Wheeler TL, Falco AM, Smith RF. Long-term changes in fear conditioning and anxiety- like behavior following nicotine exposure in adult versus adolescent rats. Pharmacol Biochem Behav. 2006;85(1):91–7. doi: 10.1016/j.pbb.2006.07.014. [DOI] [PubMed] [Google Scholar]
- Son JH, Winzer-Serhan UH. Postnatal expression of alpha2 nicotinic acetylcholine receptor subunit mRNA in developing cortex and hippocampus. J Chem Neuroanat. 2006;32(2–4):179–90. doi: 10.1016/j.jchemneu.2006.09.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spear LP. The adolescent brain and age-related behavioral manifestations. Neurosci Biobehav Rev. 2000;24(4):417–63. doi: 10.1016/s0149-7634(00)00014-2. [DOI] [PubMed] [Google Scholar]
- Stanwood GD, McElligot S, Lu L, McGonigle P. Ontogeny of dopamine D3 receptors in the nucleus accumbens of the rat. Neurosci Lett. 1997;223(1):13–6. doi: 10.1016/s0304-3940(97)13396-1. [DOI] [PubMed] [Google Scholar]
- Tapia L, Kuryatov A, Lindstrom J. Ca2+ permeability of the (alpha4)3(beta2)2 stoichiometry greatly exceeds that of (alpha4)2(beta2)3 human acetylcholine receptors. Mol Pharmacol. 2007;71(3):769–76. doi: 10.1124/mol.106.030445. [DOI] [PubMed] [Google Scholar]
- Tarazi FI, Tomasini EC, Baldessarini RJ. Postnatal development of dopamine D1- like receptors in rat cortical and striatolimbic brain regions: An autoradiographic study. Dev Neurosci. 1999;21(1):43–9. doi: 10.1159/000017365. [DOI] [PubMed] [Google Scholar]
- Teicher MH, Andersen SL, Hostetter JC., Jr Evidence for dopamine receptor pruning between adolescence and adulthood in striatum but not nucleus accumbens. Brain Res Dev Brain Res. 1995;89(2):167–72. doi: 10.1016/0165-3806(95)00109-q. [DOI] [PubMed] [Google Scholar]
- Tercyak KP, Lerman C, Audrain J. Association of attention-deficit/hyperactivity disorder symptoms with levels of cigarette smoking in a community sample of adolescents. J Am Acad Child Adolesc Psychiatry. 2002;41(7):799–805. doi: 10.1097/00004583-200207000-00011. [DOI] [PubMed] [Google Scholar]
- Thapar A, Fowler T, Rice F, Scourfield J, van den Bree M, Thomas H, Harold G, Hay D. Maternal smoking during pregnancy and attention deficit hyperactivity disorder symptoms in offspring. Am J Psychiatry. 2003;160(11):1985–9. doi: 10.1176/appi.ajp.160.11.1985. [DOI] [PubMed] [Google Scholar]
- Tizabi Y, Perry DC. Prenatal nicotine exposure is associated with an increase in [125I]epibatidine binding in discrete cortical regions in rats. Pharmacol Biochem Behav. 2000;67(2):319–23. doi: 10.1016/s0091-3057(00)00379-8. [DOI] [PubMed] [Google Scholar]
- Thompson RJ, Jackson A, Nurse CA. Developmental loss of hypoxic chemosensitivity in rat adrenomedullary chromaffin cells. J Physiol. 1997;498(Pt 2):503–10. doi: 10.1113/jphysiol.1997.sp021876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomsen MS, Mikkelsen JD, Timmermann DB, Peters D, Hay-Schmidt A, Martens H, Hansen HH. The selective alpha7 nicotinic acetylcholine receptor agonist A-582941 activates immediate early genes in limbic regions of the forebrain: Differential effects in the juvenile and adult rat. Neuroscience. 2008;154(2):741–53. doi: 10.1016/j.neuroscience.2008.03.083. [DOI] [PubMed] [Google Scholar]
- Toga AW, Thompson PM, Sowell ER. Mapping brain maturation. Trends Neurosci. 2006;29:148–59. doi: 10.1016/j.tins.2006.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres OV, Tejeda HA, Natividad LA, O’Dell LE. Enhanced vulnerability to the rewarding effects of nicotine during the adolescent period of development. Pharmacol Biochem Behav. 2008 doi: 10.1016/j.pbb.2008.05.009. Epub on May 23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trauth JA, Seidler FJ, McCook EC, Slotkin TA. Adolescent nicotine exposure causes persistent upregulation of nicotinic cholinergic receptors in rat brain regions. Brain Res. 1999;851(1–2):9–19. doi: 10.1016/s0006-8993(99)01994-0. [DOI] [PubMed] [Google Scholar]
- Trauth JA, Seidler FJ, Slotkin TA. Persistent and delayed behavioral changes after nicotine treatment in adolescent rats. Brain Res. 2000;880(1–2):167–72. doi: 10.1016/s0006-8993(00)02823-7. [DOI] [PubMed] [Google Scholar]
- Tredway TL, Guo JZ, Chiappinelli VA. N-type voltage-dependent calcium channels mediate the nicotinic enhancement of GABA release in chick brain. J Neurophysiol. 1999;81(2):447–54. doi: 10.1152/jn.1999.81.2.447. [DOI] [PubMed] [Google Scholar]
- Tribollet E, Bertrand D, Marguerat A, Raggenbass M. Comparative distribution of nicotinic receptor subtypes during development, adulthood and aging: an autoradiographic study in the rat brain. Neuroscience. 2004;124(2):405–20. doi: 10.1016/j.neuroscience.2003.09.028. [DOI] [PubMed] [Google Scholar]
- Tseng KY, O’Donnell P. Post-pubertal emergence of prefrontal cortical up states induced by D1-NMDA co-activation. Cereb Cortex. 2005;15(1):49–57. doi: 10.1093/cercor/bhh107. [DOI] [PubMed] [Google Scholar]
- Tseng KY, O’Donnell P. Dopamine modulation of prefrontal cortical interneurons changes during adolescence. Cereb Cortex. 2007;17(5):1235–40. doi: 10.1093/cercor/bhl034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. Department of Health and Human Services. Reducing the Health Consequences of Smoking: 25 Years of Progress. U.S. Department of Health and Human Services, Centers for Disease Control and Prevention, National Center for Chronic Disease Prevention and Health Promotion, Office on Smoking and Health; 1989. [Google Scholar]
- Vastola BJ, Douglas LA, Varlinskaya EI, Spear LP. Nicotine-induced conditioned place preference in adolescent and adult rats. Physiol Behav. 2002;77(1):107–14. doi: 10.1016/s0031-9384(02)00818-1. [DOI] [PubMed] [Google Scholar]
- Vázquez DM, Akil H. Pituitary-adrenal response to ether vapor in the weanling animal: characterization of the inhibitory effect of glucocorticoids on adrenocorticotropin secretion. Pediatr Res. 1993;34(5):646–53. doi: 10.1203/00006450-199311000-00017. [DOI] [PubMed] [Google Scholar]
- Viemari JC. Noradrenergic modulation of the respiratory neural network. Respir Physiol Neurobiol. 2008 doi: 10.1016/j.resp.2008.06.016. Epub on Jun 27. [DOI] [PubMed] [Google Scholar]
- von Ziegler NI, Schlumpf M, Lichtensteiger W. Prenatal nicotine exposure selectively affects perinatal forebrain aromatase activity and fetal adrenal function in male rats. Brain Res Dev Brain Res. 1991;62(1):23–31. doi: 10.1016/0165-3806(91)90186-m. [DOI] [PubMed] [Google Scholar]
- Voorn P, Kalsbeek A, Jorritsma-Byham B, Groenewegen HJ. The pre- and postnatal development of the dopaminergic cell groups in the ventral mesencephalon and the dopaminergic innervation of the striatum of the rat. Neuroscience. 1988;25(3):857–87. doi: 10.1016/0306-4522(88)90041-3. [DOI] [PubMed] [Google Scholar]
- Wang F, Gerzanich V, Wells GB, Anand R, Peng X, Keyser K, Lindstrom J. Assembly of human neuronal nicotinic receptor alpha5 subunits with alpha3, beta2, and beta4 subunits. J Biol Chem. 1996;271(30):17656–65. doi: 10.1074/jbc.271.30.17656. [DOI] [PubMed] [Google Scholar]
- Wang J, Wang X, Irnaten M, Venkatesan P, Evans C, Baxi S, Mendelowitz D. Endogenous acetylcholine and nicotine activation enhances GABAergic and glycinergic inputs to cardiac vagal neurons. J Neurophysiol. 2003;89(5):2473–81. doi: 10.1152/jn.00934.2002. [DOI] [PubMed] [Google Scholar]
- Weissman MM, Warner V, Wickramaratne PJ, Kandel DB. Maternal smoking during pregnancy and psychopathology in offspring followed to adulthood. J Am Acad Child Adolesc Psychiatry. 1999;38(7):892–9. doi: 10.1097/00004583-199907000-00020. [DOI] [PubMed] [Google Scholar]
- Welker WI. Analysis of Sniffing of the Albino Rat. Behaviour. 1964;22(3–4):223–244. [Google Scholar]
- Wilmouth CE, Spear LP. Adolescent and adult rats’ aversion to flavors previously paired with nicotine. Ann N Y Acad Sci. 2004;1021:462–4. doi: 10.1196/annals.1308.065. [DOI] [PubMed] [Google Scholar]
- Winzer-Serhan UH, Leslie FM. Codistribution of nicotinic acetylcholine receptor subunit alpha3 and beta4 mRNAs during rat brain development. J Comp Neurol. 1997;386(4):540–54. doi: 10.1002/(sici)1096-9861(19971006)386:4<540::aid-cne2>3.0.co;2-2. [DOI] [PubMed] [Google Scholar]
- Winzer-Serhan UH, Leslie FM. Expression of alpha5 nicotinic acetylcholine receptor subunit mRNA during hippocampal and cortical development. J Comp Neurol. 2005;481(1):19–30. doi: 10.1002/cne.20357. [DOI] [PubMed] [Google Scholar]
- Wu LT, Anthony JC. Tobacco smoking and depressed mood in late childhood and early adolescence. Am J Public Health. 1999;89(12):1837–40. doi: 10.2105/ajph.89.12.1837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Seidler FJ, Cousins MM, Slikker W, Jr, Slotkin TA. Adolescent nicotine administration alters serotonin receptors and cell signaling mediated through adenylyl cyclase. Brain Res. 2002;951(2):280–92. doi: 10.1016/s0006-8993(02)03174-8. [DOI] [PubMed] [Google Scholar]
- Zehr JL, Todd BJ, Schulz KM, McCarthy MM, Sisk CL. Dendritic pruning of the medial amygdala during pubertal development of the male Syrian hamster. J Neurobiol. 2006;66(6):578–90. doi: 10.1002/neu.20251. [DOI] [PubMed] [Google Scholar]
- Zhang X, Liu C, Miao H, Gong ZH, Nordberg A. Postnatal changes of nicotinic acetylcholine receptor alpha 2, alpha 3, alpha 4, alpha 7 and beta 2 subunits genes expression in rat brain. Int J Dev Neurosci. 1998;16(6):507–18. doi: 10.1016/s0736-5748(98)00044-6. [DOI] [PubMed] [Google Scholar]
- Zheng JQ, Felder M, Connor JA, Poo MM. Turning of nerve growth cones induced by neurotransmitters. Nature. 1994;368(6467):140–4. doi: 10.1038/368140a0. [DOI] [PubMed] [Google Scholar]
- Zoli M, Le Novère N, Hill JA, Jr, Changeux JP. Developmental regulation of nicotinic ACh receptor subunit mRNAs in the rat central and peripheral nervous systems. J Neurosci. 1995;15(3 Pt 1):1912–39. doi: 10.1523/JNEUROSCI.15-03-01912.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zoli M, Moretti M, Zanardi A, McIntosh JM, Clementi F, Gotti C. Identification of the nicotinic receptor subtypes expressed on dopaminergic terminals in the rat striatum. J Neurosci. 2002 Oct 15;22(20):8785–9. doi: 10.1523/JNEUROSCI.22-20-08785.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]