

Abstract
Our aims were to examine whether oxidative DNA damage was elevated in brain cells of male C57BL/6 mice after oxidative stress, and to determine whether neuronal nitric oxide synthase (nNOS) was involved in such damage. Oxidative stress was induced by occluding both common carotid arteries for 90 min, followed by reperfusion. Escherichia coli exonuclease III (Exo III) removes apyrimidinic or apurinic (AP) sites and 3′-phosphate termini in single-strand breaks, and converts these lesions to 3′OH termini. These ExoIII-sensitive sites (EXOSS) can then be postlabeled using digoxigenin-11-dUTP and Klenow DNA polymerase-I, and detected using fluorescein isothiocyanate-IgG against digoxigenin. Compared with the non-ischemia controls, the density of EXOSS-positive cells was elevated at least 20-fold (P < 0.01) at 15 min of reperfusion, and remained elevated for another 30 min. EXOSS mainly occurred in the cell nuclei of the astrocytes and neurons. Signs of cell death were detected at 24 h of reperfusion and occurred mostly in the neurons. Both DNA damage and cell death in the cerebral cortical neurons were abolished by treatment with 3-bromo-7-nitroindazole (30 mg/kg, intraperitoneal), which specifically inhibited nNOS. Our results suggest that nNOS, its activator (calcium), and peroxynitrite exacerbate oxidative DNA damage after brain ischemia.
Keywords: aging, apoptosis, oxidative DNA damage, DNA repair, stroke
THE INITIATION OF cell death in the central nervous system (CNS) has been associated with an elevation in calcium influx, glutamate release, and oxidative stress (1). Among the three, oxidative DNA damage is the least studied mechanism, in part because of the difficulties in its detection. Recently, an increase in the immunoreactivity of 8-hydroxyl guanine (oh8G, or its deoxy form, oh8dG), a marker for oxidative stress-related damage to nucleic acids, has been found in brain cells from patients with Alzheimer’s disease (2) after injection of NMDA (3) or MPTP (4), and after brain injury caused by cerebral ischemia-reperfusion (5, 6), age-associated oxidative damage (7, 8), and certain forms of stress (9). Some of these experimental conditions have been known to cause cell death in the CNS, and mimic several human diseases including Parkinson’s disease, cardiac arrest, and stroke. Although the immunoreactivity of oh8G/oh8dG is a reporter phenotype for oxygen radicals and can be used to survey vulnerable cell populations at stress, the antibody reacts to oh8G in RNA and oh8dG in DNA. A method to locate in situ DNA lesions is essential to experimental and clinical biologists.
There is a need to define critical events in pathological cascades resulting from trigger points of DNA damage, and to differentiate reversible from irreversible DNA damage in the CNS. DNA fragmentation due to apoptosis (programmed cell death) or necrosis results from an irreversible process, whereas the majority of oxidative DNA injuries due to hydroxyl radicals can be removed via various DNA repair mechanisms (5, 6). The reversibility of oxidative DNA lesions as a result of hydroxyl radicals is useful for drug development. Hydroxyl radicals can be from nitric oxide (NO) and superoxide, and are known to generate AP sites and DNA strand breaks. DNA strand breaks induced by hydroxyl radicals include those bearing 3′-hydroxy (3′-OH), 3′-phosphate (3′-PO4), and 3′-phosphoglycolate (3′-PG) termini (10). It has been, however, extremely difficult to detect oxidative DNA damage of these types in situ. Among DNA strand breaks due to ischemic injury in the CNS, only strand breaks bearing 3′-OH termini that are associated with cell death have been reported (11-15). DNA strand breaks bearing 3′-PO4 and 3′-PG termini often escape detection with terminal transferase or DNA polymerases, which have been used in the assay to detect TUNEL staining for any sign of cell death.
The purposes of this study were to examine 1) the presence of AP sites and single strand breaks in DNA bearing 3′-PO4 termini after forebrain ischemiareperfusion (FbIR), and 2) whether nNOS contributes to the formation of these oxidative DNA lesions in the brain after FbIR. In the method we described here, we removed 3′-PO4 termini in DNA single-strand breaks and AP sites in DNA through the use of Escherichia coli exonuclease III (ExoIII) (16). The 3′-OH termini that are generated after ExoIII treatment could then be extended using Klenow DNA polymerase-I and digoxigenin-11-dUTP (dig-dUTP). The incorporated dig-nucleotide was then detected through the use of fluorescein isothiocyanate (FITC) conjugates of antibodies against digoxigenin. In addition, we examined the possible role of nNOS in the contribution of NO and the formation of oxidative DNA damage using the specific inhibitor of nNOS, 3-bromo-7-nitroindazole (3BR7NI).
MATERIALS AND METHODS
Brain injury model
Oxidative stress was induced in 72 male mice (C57BL6/20–25 g, Taconic Farms, Germantown, N.Y.) with an FbIR model consisting of bilateral occlusion of the common carotid arteries for 90 min followed by various lengths of reperfusion (up to 1 day) before they were killed (5). Mice were anesthetized with ketamine [100 mg/kg, intraperitoneally (i.p.)] plus xylazine (13 mg/kg, i.p.) before surgery. The control group (n=10) received the same anesthesia and underwent the same surgical procedure, but without FbIR. In the group of mice treated with (n=6) or without (n=4) nNOS inhibitor, 3BR7NI (Alexis, San Diego, Calif.), 3BR7NI (30 mg/kg) in soybean oil, or soybean oil alone (4 ml/kg) was given intraperitoneally 5 min after vessel occlusion (6). Body temperature was maintained at 37 ± 0.5°C during surgery and the postoperative period until the animals recovered fully from anesthesia. Housing and anesthesia were in accordance with the Guide for the Care and Use of Laboratory Animals, USDA regulations, and the guidelines of the American Veterinary Medical Association Panel on Euthanasia. At the designated reperfusion time points, mice were killed for brain tissue preparation using 40 ml of saline, followed by the same volume of 4% paraformaldehyde in 0.1 M phosphate buffer (pH 7.4). Brain tissue was removed and was either flash-frozen or embedded in paraffin blocks (5). Nicotin-amide adenine dinucleotide phosphate diaphorase (NADPHd) staining was the same as reported by Cork et al. (17). Cell death was determined using terminal UTP nick-end labeling (TUNEL) staining (Apop Tag Detection Kit, Oncor, Gaithersburg, Md.) as has been previously described (5, 11-15). Astrocytes were identified using polyclonal antibodies against GFAP (Sigma Chemical Co., St. Louis, Mo.).
In situ detection of exonuclease III enzyme-sensitive sites (EXOSS)
Four 20-μm coronal sections from each brain were taken at 100-μm intervals posterior to the bregma (1–3 mm) and were examined for each determination. The sections were dried in a vacuum jar at room temperature for 2–16 h, and then digested by proteinase K (0.02 mg/ml) at 37°C for 30 min, followed by three washes in phosphate-buffered saline (PBS, pH 7.4). The sections were incubated with E. coli ExoIII (30 U/slide, Life Technology, Gaithersburg, Md.) in a humidified chamber at 37°C for 3 h, followed by washes in PBS. Control tissue included brain sections from non-FbIR animals and from FbIR animals not treated with ExoIII. The fragments generated by ExoIII were labeled with dig-dUTP using Klenow DNA polymerase-I (0.15 U/slide, Boehringer Mannheim, Indianapolis, Ind.) at 37°C for 1 h. After washing in PBS, the slides were incubated with the antibody against dig-dUTP-FITC (Oncor) under dim light at room temperature for 1 h. The nuclear DNA was stained with propidium iodide (PI, 0.05 μg/ml) in the presence of heat-treated RNase A (0.5 μg/ml) for 5 min at room temperature. There is no nonspecific exonuclease present because we fail to observe incorporation of dig-dUTP into nuclear DNA in the cerebellum of the animals treated with FbIR.
The specific activity of E. coli ExoIII was tested using its ability to excise an AP site in a double-stranded (DS) oligomer of the c-fos gene (custom-made by Sigma Genosys, The Woodlands, Tex.). Primer D (5′-CATCATGGTCXTGGTTTGGGCA-3′ (18), where X was an AP site) was labeled on the 5′ terminus using [γ-32P]ATP and T4 polynucleotide kinase, followed by purification using a Sephadex G-25 column. The resultant 32P-primer D was hybridized to its complementary template primer A (5′tgcccaaaccaYgaccatgatg-3′, where Y was an adenosine), and heated to 70°C, followed by cooling to 25°C. A single-stranded break bearing a 3′PO4 terminus on the oh8dG site of primer Z was generated in 32P-DS oligomer Z, in which the X and Y pair in the c-fos oligomer was an oh8dG and cytosine pair, using E. coli formamidopyrimidine glycosylase (Fpg protein) (5, 6). Before use in the EXOSS assay, we tested the inability of Klenow DNA polymerase I to resynthesize the 3′-PO4 terminus on excised oligomer Z using the template primer C. The reaction products were resolved in sequencing polyacrylamide gel electrophoresis (PAGE; 10%) using electrophoresis and autoradiography.
Data acquisition and statistical analysis
For fluorescent dig-dUTP detection in the EXOSS and TUNEL assay, the brain sections were examined under a microscope using a mercury light source with a Leica I3 filter (450–510 nm). Photographs were taken under red and green digitized spectra using a Cooled Color Digital Camera (the SPOT camera, Diagnostic Instruments, Sterling Heights, Mich.). The orange (captured under dual red and green spectra) or red (captured under red spectrum alone) coloration due to PI staining of the nuclear DNA indicated the location of nuclei. Cells with at least a fourfold increase in FITC signal (the green signal) over the background (30 ± 18, mean ± se), as quantified using Adobe PhotoShop, were defined as positive cells for EXOSS or TUNEL staining (6). For an animal to be scored as positive or negative, the same response had to be observed in at least two separate determinations in samples taken from the same brain. Each determination consisted of examining four coronal sections (each separated at a distance of 100 μm) per brain. We defined any animal with an average density of ≥ 150 positive cells/mm2 (excluding brain surface epithelial cells) in all four tissue sections (the cerebral cortex, hippocampal formation, striatum, and arcuate nuclei of the hypothalamus) of the brain to be a positive animal.
Preparation of cell extracts for nNOS activity
Mice were injected intraperitoneally with 3BR7NI (30 mg/ kg) in soybean oil. At various times after injection (four animals each for 0, 30, 60, 90, and 120 min of reperfusion), the animals were decapitated under anesthesia. The cerebellum and the cerebral cortex were separated from other structures, flash frozen in liquid nitrogen, and then transferred to a freezer (-80°C) for storage. Brain tissue was extracted by homogenization in an ice-cold buffer (100 mg of tissue/ml buffer) containing 25 mM Tris (pH 7.4), 1 mM EDTA, and 1 mM ethylene glycol-bis (β-aminoethylether)-N,N,N′,N′-tetraacetic acid (EGTA) in a tissue grinder (18). The cytoplasmic homogenate was then separated from the nuclear fraction by centrifugation (Eppendorf Centrifuge 5415C) at 16,000 g for 5 min at 4°C. The supernatant was incubated in a 100-μl resin suspension (Dowex 50 cation exchanger) to remove endogenous arginine. The protein content in each sample was determined in triplicate using the Bio-Rad Protein Assay (Hercules, Calif.). The cytoplasmic homogenate was stored in aliquots at -80°C.
NOS assay
NOS activity was determined using the NOS Detect Assay Kit (Stratagene, La Jolla, Calif.). The arginine-free extract was incubated in the presence of 2× reaction buffer (50 mM Tris-HCl, pH 7.4, 6 μM BH4, 2 μM FAD, and 2 μM FMN), 10 mM NADPH, [3H]arginine (1 μCi/μl), and 6 mM CaCl2 in a final volume of 40 μl at room temperature for 30 min. The reaction was stopped by adding 400 μl of a stop buffer (5 mM EDTA and 50 mM HEPES, pH 5.5). The L-[3H]citrulline in the reaction mix was separated using resin suspension. After centrifuging, the L-[3H]citrulline in the supernatant was removed and its radioactivity was determined using liquid scintillation counting. The brain NOS activity in each animal was determined three times. The average NOS activity was determined from a minimum of three animals in each time point and its mean ± SE was presented. The amount of L-[3H]citrulline was expressed as pmol/mg cellular protein/30 min.
RESULTS
An increase in EXOSS after FbIR
We measured the presence of two types of oxidative DNA injury in situ, AP sites and DNA strand breaks bearing a 3′-PO4 terminus, using E. coli ExoIII. This method allowed identification of the location of oxidative DNA damage. Moreover, this method detects only substrates in DNA. A barely detectable green fluorescent signal was observed in the cortical cell cytoplasm in all 10 non-FbIR control animals (Fig. 1A). The fluorescent signal in the cortical nuclei of animals that received 90 min of forebrain ischemia (FbI) and no reperfusion (90/0, Fig. 1B, n=3) was notably increased but was diffuse and mainly was present in the cytoplasm. The signal became clearly defined and was significantly increased in intensity (P < 0.001) in the cerebral cortex of FbIR animals with 15 min of reperfusion (90/15, Fig. 1C, n=10). The green signal was located with the orange PI stain. In some cells, the green signal stained toward the nuclear periphery but within the boundary of the nucleus, and in other cells, the signal intensely filled the entire nucleus. In FbIR animals not treated with ExoIII, no significant dig-dUTP incorporation at 15 min or less of reperfusion was observed (Fig. 1D). Indeed, ExoIII excised the AP sites on the 32P-DS oligomer D in test tubes (Fig. 2). Klenow DNA polymerase-I synthesized the excised primer D (Fig. 2A), or the Fpg-excised primer Z to its original length when the Fpg-excised DS oligomer Z was further treated with ExoIII before primer extension (Fig. 2B). 32P-DS oligomer Z after Fpg protein without ExoIII was not used by Klenow DNA polymerase-I, indicating that the 3′-PO4 termini in DS oligomer Z generated by E. coli Fpg protein could not be used for extension by Klenow DNA polymerase I. Taken together, the data in Fig. 1D and Fig. 2 demonstrate that 3′-OH termini were below detection because Klenow DNA polymerase-I did not incorporate the dig-dUTP or fluorescent substrates. Presumably, these 3′-OH termini are rapidly repaired. Therefore, the EXOSS shown as green fluorescent signals represented incorporation of dig-dUTP onto the sites of DNA damage as made accessible by ExoIII.
Figure 1.

In situ EXOSS detection using E. coli ExoIII and Klenow DNA polymerase-I. Cerebral cortices from animals with no FbIR (A), or FbIR of 90/0 (B), 90/15 (C, D). All panels were from samples treated with ExoIII, except panel D, before incorporation of dig-dUTP using Klenow DNA polymerase-I. The green fluorescence represents signs of EXOSS, and the orange signal indicates nuclei as counter-stained using PI. Bar = 50 μm.
Figure 2.

Extension synthesis by Klenow DNA polymerase-I on single-stranded DNA breaks. The AP site in 32P-DS oligomer D was treated with (lanes 2–5) or without (lane 1) E. coli ExoIII (A). The reaction mixtures were then incubated either with buffer (lanes 1–3) or dNTP (40 μM) plus endonuclease-free Klenow DNA polymerase-I (lanes 4 and 5 for two different lots of enzymes) at 37°C for 5 min. The oh8dG removal in 32P-DS oligomer Z was treated with (lanes 2–6) or without (lane 1) E. coli Fpg protein at 37°C for 10 min (B). The PO4 terminus in 32P-DS oligomer Z was incubated with (lanes 3, 5, 6) or without (lanes 1, 2, 4) E. coli Exo III at 37°C for 10 min before primer extension using Klenow DNA polymerase-I. The reaction was stopped by heating and further analyzed in sequencing PAGE (10%). +, Enzyme added, -, no enzyme (buffer only). Note that DNA polymerase-I could not perform primer extension synthesis (lane 4) unless the Fpg-protein products (3′-PO4 termini) were pretreated with ExoIII (lanes 5 and 6, for two different lots of DNA pol-I).
Using the EXOSS detection, the FITC intensity increased and became uniformly distributed in the nuclei in the ischemic cortices reperfused for 30 and 45 min (90/30 and 90/45, Fig. 3A, B, n=7 and 6, respectively). At ∼60 min of reperfusion, the intensity started declining (90/60, Fig. 3C, n=8). Moreover, the signal that remained in the cortex at 90/60 FbIR was mostly perinuclear. Therefore, the cellular location of EXOSS at this time point was visibly different from that observed at 15, 30, and 45 min of reperfusion. The data from these 44 animals were summarized, statistically analyzed, and presented in Fig. 4. The density of EXOSS-positive cells per square millimeter (mean ± se) in the cerebral cortex increased from 29 ± 6 in the control non-FbIR brains to 121 ± 71 in the brains after FbIR (90/0), and further significantly increased (P < 0.01) to 512 ± 38 at 90/15 FbIR, 662 ± 43 at 90/30 FbIR, and 600 ± 40 at 90/45 FbIR. At 90/15 FbIR, nine of ten (90%) animals were EXOSS-positive, as defined in Materials and Methods, but none of the ten non-FbIR controls was EXOSS-positive (P < 0.01, Fisher’s exact test). The number of EXOSS-positive cells was reduced to 104 ± 71 by 90/60 FbIR, and was not significantly different from the controls (Fig. 4).
Figure 3.

In situ EXOSS detection using E. coli ExoIII and Klenow DNA polymerase-I. Cerebral cortices from animals as in Fig. 1, but with FbIR 90/30 (A), 90/45 (B), and 90/60 (C). All panels were from samples treated with ExoIII before incorporation of dig-dUTP using Klenow DNA polymerase-I. The animals (90/15 FbIR) in panel D were treated with 3BR7NI after vessel occlusion. The green fluorescent represents EXOSS, and the orange signal indicates nuclei as counter-stained using PI. Bar = 50 μm.
Figure 4.

EXOSS in mouse brain after FbIR. Quantitative analysis of cerebral cortical cells (EXOSS-positive per mm2) at 0, 15, 30, 45, and 60 min after 90 min of forebrain ischemia. Intraperitoneal soybean oil control (open bars, n=44 total, see text) or 3BR7NI in oil (closed bars, n=4 for each of 15, 30, and 45 min). ***P < 0.01.
Cerebral cortical EXOSS mediated by nNOS
To examine whether nNOS mediated DNA damage, we injected 3BR7NI into 12 animals, 5 min after initiating bilateral carotid artery occlusion. 3BR7NI significantly reduced the intensity of the EXOSS signal in the entire brain of the mouse after FbIR, but the effect was most obvious in the cortex (Fig. 3D). The cerebral cortex of 90/15 FbIR animals treated with 3BR7NI showed no EXOSS-positive cells. At 90/30 and 90/45 FbIR, we observed a re-appearance of EXOSS-positive cells in 3BR7NI animals, but the number of positive cells was not significantly different than in the non-FbIR controls (P > 0.05, t test; Fig. 4). The EXOSS in the arcuate nuclei of the hypothalamus were reduced, but not abolished, in animals treated with 3BR7NI (data not shown).
To examine whether 3BR7NI inhibited NOS activity in the mouse brain, we measured brain NOS activity in the cytosolic extract from the brains of 19 mice. After the administration of 3BR7NI, we observed that brain NOS activity (pmol/mg protein/30 min) in the cerebral cortex was significantly reduced (P < 0.001, one-way analysis of variance) for at least 90 min after injection (Fig. 5). By 120 min, the brain NOS activity had returned to a value that was not significantly different from the control. The inhibition of nNOS activity by one single injection at 5 min after vessel occlusion correlated positively with the suppression of EXOSS-positive cells at the 90/30 FbIR time point (Fig. 4), i.e., ∼120 min after 3BR7NI injection.
Figure 5.
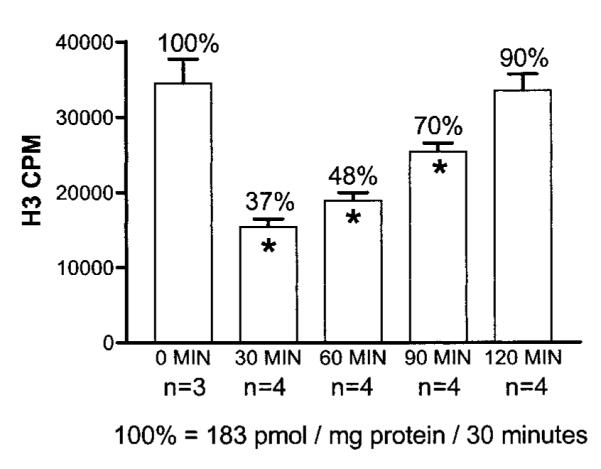
The effect of 3BR7NI (30 mg/kg) on brain NOS activity in cytoplasm from the mouse brain. The mean ± SE of NOS activity (three determinations in each animal) from three or four animals (n) are shown. The time after drug injection is indicated. *p < 0.001.
EXOSS in neurons and astrocytes
In the FbIR animals, the cells that contained nuclear EXOSS were mostly non-astrocytes (neurons), and a few astrocytes, in the cortex, striatum, hippocampal formation, and arcuate nuclei of the hypothalamus (Fig. 6). We also noticed that the distribution of EXOSS-positive cells was not uniform throughout the brain: the number of EXOSS-positive cells and the intensity of EXOSS staining in the arcuate nuclei of the hypothalamus in all 90/15 FbIR animals was higher than in the surrounding hypothalamus (Fig. 7). The density of EXOSS-positive cells (1585 ± 47 cells/mm2) in the arcuate nuclei was significantly higher (P < 0.01, t test) than in the surrounding areas (233 ± 23 cells/mm2).
Figure 6.

EXOSS in neurons and astrocytes after FbIR. Coronal sections with double staining for EXOSS (top, green signals) or GFAP (bottom, red signals) immunoreactivity from animals that underwent 90/15 FbIR. Boxes, GFAP-negative (neurons); arrows, GFAP-positive (astrocytes); ARN, arcuate nuclei of the hypothalamus. Bars = 25 μm.
Figure 7.

EXOSS in the arcuate nuclei of the hypothalamus after FbIR. The surrounding area of the third cerebral ventricle shows clusters of EXOSS-positive signal (the white signal) in the arcuate nuclei of the hypothalamus (FbIR, 90/15), whereas the adjacent hypothalamus showed less fluorescent signal. Bar = 100 μm.
EXOSS in NADPHd-negative cells
Because we did not observe a significant elevation in the peptide of nNOS within 30 min of reperfusion in this C57BL6 mouse FbIR model using Western blot assay (data not shown), we examined whether the arcuate nuclei of hypothalamus contained NADPHd-positive cells. The brain cells that express NOS are known to be NADPHd-positive (19). Two types of NADPHd-positive cells were found in the mouse brain (Fig. 8). One stained intensely throughout the cell soma, dendrites, and axon (Fig. 8A) and could be found in the frontal cortex, the piriform cortex, the striatum, and the thalamus. The other NADPHd-positive cell demonstrated pale granular staining and could be found in the molecular layer of dentate gyrus, the anterior amygdaloid nuclei, and the para-central thalamic nuclei (Fig. 8B). The distribution of NADPHd-positive cells in a C57BL6 mouse brain was similar to that reported by Cork et al. (17). We detected little NADPHd staining in the arcuate nuclei of the hypothalamus (Fig. 8C).
Figure 8.

NADPHd-staining in the arcuate nuclei of the hypothalamus after FbIR. Composite low-magnification image (bar = 200 μm) of NADPHd staining (the dark signals) in the right hemisphere of a mouse brain. A, B) Higher magnification of two types of NADPHd-positive staining (bar = 20 μm). C) Negative staining in the projection of arcuate nuclei of the hypothalamus (bar = 120 μm).
TUNEL-positive staining as a sign of cell death
Because the effect of 3BR7NI on EXOSS-positive signal was different in the cerebral cortex and arcuate nuclei, we used TUNEL staining to examine for signs of cell death in these two areas. We observed TUNEL-positive signs in the cortex (Fig. 9A, B), hippocampus, striatum, hypothalamus, and amygdaloid nuclei from all 12 animals that underwent 90 min of ischemia and 1 day of reperfusion (90/1-day). In the cerebral cortex and hippocampus of the FbIR animals, TUNEL-positive staining was located mainly in the neurons (Fig. 10, arrows). The astrocytes, as shown by GFAP-positive staining (Fig. 10, arrowheads) showed no TUNEL-positive staining. Cortical DNA from 90/1-day FbIR animals contained multiple DNA ladders of 180 base pairs (not shown). No TUNEL-positive cells, as defined in Materials and Methods, were observed in any of the six non-FbIR controls (Fig. 9C). In 11 of the 12 FbIR (90/1-day) animals that received 3BR7NI, we did not observe any TUNEL-positive staining (Fig. 9D, P < 0.005, Fisher’s exact test). The number of TUNEL-positive cells in the arcuate nuclei of the hypothalamus was not abolished by 3BR7NI.
Figure 9.

TUNEL-positive staining after FbIR with and without 3BR7NI. Typical TUNEL-positive brain tissue (the white fluorescent signal) in the cerebral cortex 1 day after 90 min of ischemia (FbIR, two magnifications were shown in panels A and B), compared with the same tissue from a no-FbIR control (C) and an FbIR animal with 3BR7NI (D). Because we could not obtain images at the low magnification for the animals with no FbIR and with FbIR + 3BR7NI, we obtained images with a higher magnification for panels B—D. Bar = 100 μm in panel A and 25 μm in panels B—D.
Figure 10.

Coronal sections with double staining for TUNEL and GFAP immunoreactivity in the cerebral cortex and the hippocampal formation from animals with 1 reperfusion day after 90 min FbIR (similar to Fig. 8, A & B). Arrows, neurons; arrowheads, astrocytes. Most TUNEL-positive staining was found in non-astrocytes. Bar = 20 μm.
DISCUSSION
The molecular mechanisms underlying neuronal death after ischemic injury have been under investigation. Our current study morphologically identifies AP sites and 3′-PO4 termini in single-strand breaks as EXOSS in DNA of the cell after FbIR. Previous studies using chromatographic methods (4, 5, 8, 9), terminal transferase or DNA polymerase-I (12), or using antibodies against oh8dG were unable to identify these types of DNA damage (2, 4, 6). Moreover, we have not observed excessive EXOSS or signs of cell death (TUNEL staining) in the sham-controls, and the data suggest that drugs used for anesthesia have a minimal impact on EXOSS.
The DNA damage we localized could be the result of excessive oxidative stress in the brain. We have reported a significant elevation in cortical oh8G/oh8dG, a marker of oxidative stress, within 30 min of reperfusion after cerebral ischemia in the mouse (5) and in the rat (6). Our current study using the FbIR mouse model suggests that nuclear EXOSS could also represent an interaction of hydroxyl radicals and cellular DNA (20). DNA strand breaks have been known to activate poly(ADP-ribose)polymerase (PARP) during DNA repair (21, 22). Polymerization of ADP-ribose could prevent recombination of DNA strand breaks (23a). Activation of PARP and subsequent energy failure as a result of ATP depletion could be responsible for further ischemic brain injury (24). Indeed, PARP inhibitors have been shown to prevent secondary energy failure, and to produce neuroprotection.
In the majority of patients who suffer a major heart attack or thrombolic stroke, the interruption in the flow of oxygenated blood to the brain introduces energy failure in the brain at the cellular level. Subsequent restoration of blood flow creates an excess of electrons at the molecular level (25). The electron imbalance creates an increase in oxygen radicals (oxidative stress), which can damage proteins, membrane, and nucleic acids in a larger population of brain cells. A similar process may be involved in patients who undergo small, often sub-clinical, thrombolic strokes that gradually destroy brain tissue and that may present clinically as senile dementia in later life. A better understanding of the pathophysiological process involved may facilitate the development of effective oxygen scavengers to be given acutely or prophylatically to reduce the damage associated with excess oxidative stress. Alternatively or in combination with oxygen scavenger therapy, enzyme inhibitors or antisense DNA-mediated gene targeting may allow us to transiently and locally interrupt the cascade of molecular events that lead to secondary brain damage after stroke.
Two types of DNA strand breaks can be identified, either by interaction with hydroxyl radicals (oxidative DNA damage) or by endonuclease digestion (apoptotic DNA damage) after experimental brain ischemia. Because we did not observe significant incorporation of dig-dUTP without ExoIII at 90/15 FbIR, we concluded that strand breaks during the first 15 min after forebrain ischemia were mainly oxidative DNA damage bearing the 3′PO4 terminus, and did not contain a significant amount of apoptotic DNA damage. This conclusion is also supported by the fact that EXOSS decreased within 60 min of reperfusion. This reversibility of oxidative DNA damage as a result of the DNA repair process has been reported previously (5, 6, 26). Therefore, the majority of oxidative DNA damage detected by EXOSS is repaired via DNA repair mechanisms, in contrast to the irreversible DNA fragmentation detected using TUNEL staining. The importance of DNA repair is illustrated by the fact that an impaired ability to repair oxidative DNA damage may have a role in human diseases of neurological dysfunction (27-29), and in experimental brain attack (stroke) leading to neuronal apoptosis (30).
Nitric oxide, as a radical of oxygen, is one of many chemicals that have been implicated in cell death (31, 32). Our results suggest that oxidative DNA damage is partially mediated by the product of nNOS: NO or its derivatives (3, 14, 15, 33-35). This conclusion also suggests that calcium influx, which activates nNOS, can be an initiating factor for the formation of EXOSS in the cortex. The decrease of EXOSS-positive cells in animals treated with 90/15 FbIR and 3BR7NI, and the reappearance of EXOSS-positive cells in animals with the same inhibitor over the next 30 min correlated positively with the return of NOS activity. On the other hand, our data do not exclude the role of other factors, such as glutamate release and inducible NOS (iNOS) in initiating cell death (36, 37). That 3BR7NI did not completely abolish EXOSS and TUNEL staining in the arcuate nuclei of hypothalamus, and the lack of NOS-positive cells in the arcuate nuclei of the hypothalamus support this notion. Activity of calcium-independent iNOS in the presence of 3BR7NI may be responsible for the gradual reappearance of EXOSS staining observed at longer reperfusion time points. Nevertheless, the formation of EXOSS in the ischemic cerebral cortex during the first 30 min of reperfusion appears to correlate positively with the activation of neuronal death in the cerebral cortex because inhibiting nNOS activities with 3BR7NI can abolish both cortical EXOSS and TUNEL staining.
AP sites and DNA single-strand breaks bearing 3′-PO4 termini that have been detected after FbIR occur concurrently with DNA base modifications in the nuclear genes after forebrain ischemia (5). Because NO appears to cause deamination of deoxynucleosides (38), one would expect an elevated C-to-T transition after FbIR if NO was the initiator of DNA damage. The mutation spectrum after FbIR in the mouse does not, however, contain significant elevated C-to-T transitions (5, 39). Together, the data suggest that DNA damage by nNOS could be initiated by the derivatives of NO, most likely by peroxynitrite. The peroxynitrite molecule may be cleaved to form the hydroxyl radical and NO2. Therefore, localization of EXOSS may, in effect, detect the results of increased NO and superoxide molecules in the DNA injury after FbIR (40).
The detection of oh8dG and its open ring derivatives after FbIR, which we reported earlier (5), suggest that hydroxyl radicals initiate the damage (32, 41). Together with the 3BR7NI data, our findings support the idea that hydroxyl radicals are most likely generated from peroxynitrite. This finding suggests strongly that peroxynitrite could penetrate the nuclei, such that the hydroxyl radicals produced by peroxynitrite are in close proximity to nuclear DNA. Moreover, that superoxide reacts with NO to form peroxynitrite, and the observation of cytosolic EXOSS in 90/0 FbIR animals suggest that mitochondria could also contain DNA damage (6-9, 42, 43). However, the majority of EXOSS in our study detected at 15 min of reperfusion were found in the nuclei, suggesting that the repair of mitochondrial DNA (mtDNA) damage is rapid in our model. This is supported by studies that have shown that the repair of mtDNA damage is dependent on the dose of hydroxyl radicals (43, 44). In our study the DNA injury was being actively repaired within 60 min of reperfusion. This observation agrees with the time frame we have observed of the repair of oxidative DNA damage in several nuclear genes of the brain after ischemic injury (5, 6).
Less clear are the different fates of neurons and astrocytes, both of which contain nuclear EXOSS. Although DNA repair is efficient, signs of cell death one day after 90 min of ischemia were observed mostly in neurons, and only rarely in astrocytes. One possible explanation is that a more effective DNA repair mechanism may be present in astrocytes (45). Because our understanding of the enzymology of DNA repair in the brain is rudimentary at best, the reason for this difference in cellular survival is not clear. Furthermore, we do not know whether the neurons that show EXOSS are the same as those that show apoptotic cell death later. The greater vulnerability of neurons to oxidative stress, however, is consistent with the observation that reduced DNA repair efficiency is associated with premature neuronal death in persons with amyotrophic lateral sclerosis (27). It remains to be determined whether diminished DNA repair capacity and/or the fidelity of repair accuracy in the neurons would lead to cell death in the FbIR model (46). Alternatively, these neurons may not be NASPHd-positive and, therefore, are particularly sensitive to death (47). Our results support the notion that early damage in cortical nuclei may be exacerbated by the presence of nNOS.
In conclusion, the results presented in these studies indicate that DNA injury marked by AP sites and/or single-strand breaks bearing 3′-PO4 termini is an early sign of oxidative stress, and that many of the lesions are reversible within 60 min of reperfusion. The elimination of EXOSS by 3BR7NI leads to a reduction of apoptotic cell death in the cortical neurons. However, nNOS activity is only one of many factors that cause DNA injury in the brain after FbIR, because the specific inhibitor of nNOS did not completely abolish EXOSS in the entire brain. The absence of NOS-positive cells in the arcuate nuclei of the hypothalamus may reduce the effectiveness of neuroprotection by 3BR7NI.
Acknowledgments
We thank Drs. S. T. Cao, for excellent technical assistance, R. Grossman, for suggestions, Winifred Hamilton (Baylor College of Medicine), for an editorial review of this manuscript, and Micky Fry (University of Washington, Seattle, WA), for technical discussion. This work was supported in part by an Established Investigator Award (9640202N) from the American Heart Association, and by a grant from NINDS (NS34810).
REFERENCES
- 1.Choi DW. Calcium: Still center-stage in hypoxicischemic neuronal death. Trends Neurosci. 1995;18:58–60. [PubMed] [Google Scholar]
- 2.Nunomura A, Perry G, Pappolla MA, Wade R, Hirai K, Chiba S, Smith MA. RNA oxidation is a prominent feature of vulnerable neurons in Alzheimer’s disease. J. Neurosci. 1999;19:1959–1964. doi: 10.1523/JNEUROSCI.19-06-01959.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ayata C, Ayata G, Hara H, Matthews RT, Beal MF, Ferrante RJ, Endres M, Kim A, Christie RH, Waeber C, Huang PL, Hyman BT, Moskowitz MA. Mechanisms of reduced striatal NMDA excitotoxicity in type I nitric oxide synthase knock-out mice. J. Neurosci. 1997;17:6908–6917. doi: 10.1523/JNEUROSCI.17-18-06908.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schulz JB, Mathews RT, Jenkins BG, Ferrante RJ, Siwek D, Henshaw DR, Cipolloni PB, Mecocci P, Kowall NW, Rosen BR, Beal MF. Blockade of neuronal nitric oxide synthase protects against excitotoxicity in vivo. J. Neurosci. 1995;15:8419–8429. doi: 10.1523/JNEUROSCI.15-12-08419.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu PK, Hsu CY, Dizdaroglu M, Floyd RA, Kow YW, Karakaya A, Rabow LE, Cui JK. Damage, repair and mutagenesis in nuclear genes after mouse forebrain ischemia-reperfusion. J. Neurosci. 1996;16:6795–6806. doi: 10.1523/JNEUROSCI.16-21-06795.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cui J, Holmes EH, Liu PK. Oxidative damages to the c-fos gene and reduction of its transcription after focal cerebral ischemia in the rat brain. J. Neurochem. 1999;73:1–11. doi: 10.1046/j.1471-4159.1999.0731164.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De la Asuncion JG, Millan A, Pla R, Bruseghini L, Esteras A, Pallarde FV, Sastre J, Vina DJ. Mitochondrial glutathione oxidation correlates with age-associated oxidative damage to mitochondrial DNA. FASEB J. 1996;10:333–338. doi: 10.1096/fasebj.10.2.8641567. [DOI] [PubMed] [Google Scholar]
- 8.Mecocci P, MacGarvey U, Kaufman AE, Koontz D, Shoffner JM, Wallace DC, Beal MF. Oxidative damage to mitochondrial DNA shows marked age-dependent increases in human brain. Ann. Neurol. 1993;34:609–616. doi: 10.1002/ana.410340416. [DOI] [PubMed] [Google Scholar]
- 9.Liu J, Wang X, Shigenaga MK, Yeo HC, Mori A, Ames BN. Immobilization stress causes oxidative damage to lipid, protein, and DNA in the brain of rats. FASEB J. 1996;10:1532–1538. [PubMed] [Google Scholar]
- 10.Henner WD, Grunberg SM, Haseltine WA. Sites and structure of γ radiation-induced DNA strand breaks. J. Biol. Chem. 1982;257:11750–11754. [PubMed] [Google Scholar]
- 11.Gavrieli Y, Sherman Y, Ben-Sasson SA. Identification of programmed cell death in situ via specific labeling of nuclear DNA fragmentation. J. Cell Biol. 1992;119:493–501. doi: 10.1083/jcb.119.3.493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen J, Jin K, Chen M, Pei W, Kawaguchi K, Greenberg DA, Simon RP. Early detection of DNA strand breaks in the brain after transient focal ischemia: Implications for the role of DNA damage in apoptosis and neuronal cell death. J. Neurochem. 1997;69:232–245. doi: 10.1046/j.1471-4159.1997.69010232.x. [DOI] [PubMed] [Google Scholar]
- 13.Du C, Hu R, Csernansky CA, Hsu CY, Choi DW. Very delayed infarction after mild focal cerebral ischemia: A role for apoptosis? J. Cereb. Blood Flow Metab. 1996;16:195–201. doi: 10.1097/00004647-199603000-00003. [DOI] [PubMed] [Google Scholar]
- 14.Yoshida T, Limmroth V, Irikura K, Moskowitz MA. The NOS inhibitor, 7-nitroindazole, decreases focal infarct volume but not the response to topical acetylcholine in pial vessels. J. Cereb. Blood Flow Metab. 1994;14:924–929. doi: 10.1038/jcbfm.1994.123. [DOI] [PubMed] [Google Scholar]
- 15.Kamii H, Mikawa S, Murakami K, Kinouchi H, Yoshimoto T, Reola L, Carlson E, Epstein CJ, Chan PH. Effects of nitric oxide synthase inhibition on brain infarction in SOD-1-transgenic mice following transient focal cerebral ischemia. J. Cereb. Blood Flow Metab. 1994;16:153–1157. doi: 10.1097/00004647-199611000-00009. [DOI] [PubMed] [Google Scholar]
- 16.Kornberg A. DNA Replication. Freeman; San Francisco: 1980. Exonuclease III; pp. 324–327. [Google Scholar]
- 17.Cork RJ, Perrone ML, Bridges D, Wandell J, Scheiner CA, Mize RR. A web-accessible digital atlas of the distribution of nitric oxide synthase in the mouse brain. Prog. Brain Res. 1998;118:37–50. doi: 10.1016/s0079-6123(08)63199-4. [DOI] [PubMed] [Google Scholar]
- 18.Liu PK, Salminen A, He YY, Jiang MH, Xue JJ, Liu JS, Hsu CY. Suppression of ischemia-induced Fos expression and AP-1 activity by an antisense oligodeoxynucleotide to c-fos mRNA. Ann. Neurol. 1994;36:566–576. doi: 10.1002/ana.410360405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dawson TM, Bredt DS, Fotuhi M, Hwang PM, Snyder SH. Nitric oxide synthase and neuronal NADPH diaphorase are identical in brain and peripheral tissues. Proc. Natl. Acad. Sci. USA. 1991;8:7797–7801. doi: 10.1073/pnas.88.17.7797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Inoue S, Kawanishe S. Oxidative DNA damage induced by simultaneous generation of nitric oxide and superoxide. FEBS Lett. 1995;371:86–88. doi: 10.1016/0014-5793(95)00873-8. [DOI] [PubMed] [Google Scholar]
- 21.De Murcia G, Menisser-de Murcia J, Schreiber V. Poly(ADP-ribose)polymerase: a molecular nick sensor. Trends Biochem. Sci. 1992;19:172–176. doi: 10.1016/0968-0004(94)90280-1. [DOI] [PubMed] [Google Scholar]
- 22.Lindahl T, Satoh MS, Poirier GG, Klungland A. Post-translational modification of poly(ADP-ribose)polymerase induced by DNA strand breaks. Trends Biochem. Sci. 1995;20:405–411. doi: 10.1016/s0968-0004(00)89089-1. [DOI] [PubMed] [Google Scholar]
- 23.Shall S. ADP-ribosylation reactions. Biochemie. 1995;77:313–318. doi: 10.1016/0300-9084(96)88140-5. [DOI] [PubMed] [Google Scholar]
- 23a.Lindahl T, Wood RD. Frontiers in cell biology: quality control by DNA repair. Science. 1999;286:1897–1905. doi: 10.1126/science.286.5446.1897. [DOI] [PubMed] [Google Scholar]
- 24.Endres M, Wang Z, Namura S, Waeber C, Moskowitz MA. Ischemia brain injury is mediated by the activation of poly(ADP-ribose)polymerase. J. Cereb. Blood Flow Metab. 1997;17:1143–1151. doi: 10.1097/00004647-199711000-00002. [DOI] [PubMed] [Google Scholar]
- 25.Liu PK, Hamilton WJ, Hsu CY. Apoptosis: DNA damage and repair in stroke. In: Miller LP, editor. Stroke Therapy: Basic, Preclinical, and Clinical Direction. Wiley-Liss; New York: 1999. pp. 299–320. [Google Scholar]
- 26.Lin L, Cao S, Cui J, Hamilton WJ, Liu PK. Upregulation of base excision repair activity for oh8dG in the mouse brain after forebrain ischemia-reperfusion. J. Neurochem. 2000 doi: 10.1046/j.1471-4159.2000.741098.x. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kisby GE, Milne J, Sweatt C. Evidence of reduced DNA repair in amyotrophic lateral sclerosis brain tissue. Neuroreport. 1997;8:1337–1340. doi: 10.1097/00001756-199704140-00004. [DOI] [PubMed] [Google Scholar]
- 28.Readon JT, Bessho T, Kung HC, Bolton PH, Sancar A. In vitro repair of oxidative DNA damage by human nucleotide excision repair system: possible explanation for neurodegeneration in Xeroderma pigmentosum patients. Proc. Natl. Acad. Sci. USA. 1997;94:9463–9468. doi: 10.1073/pnas.94.17.9463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Parshad R, Sanford KK, Price FM. Fluorescence light-induced chromatid breaks distinguish Alzheimer disease cells from normal cells in tissue culture. Proc. Natl. Acad. Sci. USA. 1996;93:5146–5150. doi: 10.1073/pnas.93.10.5146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kawase M, Fujimura M, Morita-Fujimura Y, Chan PH. Reduction of apurinic/apyrimidinic endonuclease expression after transient global cerebral ischemia in rats. Stroke. 1999;30:441–449. doi: 10.1161/01.str.30.2.441. [DOI] [PubMed] [Google Scholar]
- 31.Lipton SA, Choi YB, Pan ZH, Lei SZ, Chen HS, Singel DJ, Stamler JS. A redox-based mechanism for the neuroprotective and neurodestructive effects of NO and related nitroso compounds. Nature (London) 1993;364:626–632. doi: 10.1038/364626a0. [DOI] [PubMed] [Google Scholar]
- 32.Beckman JS, Beckman TW, Chen J, Marshall PA, Freeman BA. Apparent hydroxyl radical production by peroxynitrite: Implications for endothelial injury from nitric oxide and superoxide. Proc. Natl. Acad. Sci. USA. 1990;87:1620–1624. doi: 10.1073/pnas.87.4.1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Nanri K, Montecot C, Springhetti V, Seylaz J, Pinard E. The selective inhibitor of neuronal nitric oxide synthase, 7-nitroindazole, reduces the delayed neuronal damage due to forebrain ischemia in rats. Stroke. 1998;29:1248–1253. doi: 10.1161/01.str.29.6.1248. [DOI] [PubMed] [Google Scholar]
- 34.O’Neill MJ, Hicks C, Ward M. Neuroprotective effects of 7-nitroindazole in the gerbil model of global cerebral ischemia. Eur. J. Pharmacol. 1996;310:115–122. doi: 10.1016/0014-2999(96)00387-1. [DOI] [PubMed] [Google Scholar]
- 35.Huang Z, Huang PL, Panahian N, Dalkara T, Fishman MC, Moskowitz MA. Effects of cerebral ischemia in mice deficient in neuronal nitric oxide synthase. Science. 1994;265:1883–1885. doi: 10.1126/science.7522345. [DOI] [PubMed] [Google Scholar]
- 36.Shimizu-Sasamata M, Bosque-Hamilton P, Huang PL, Moskowitz MA, Lo EH. Attenuated neurotransmitter release and spreading depression-like depolarizations after focal ischemia in mutant mice with disrupted type I nitric oxide synthase gene. J. Neurosci. 1998;18:9564–9571. doi: 10.1523/JNEUROSCI.18-22-09564.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iadecola C, Zhang F, Casey R, Nagayama M, Ross ME. Delayed reduction of ischemia brain injury and neurological deficits in mice lacking the inducible nitric oxide synthase gene. J. Neurosci. 1997;23:9157–9164. doi: 10.1523/JNEUROSCI.17-23-09157.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Wink DA, Kasprzak KS, Maragos CM, Elespuru RK, Misra M, Dunams TM, Cebula TA, Koch WH, Andrews AW, Allen JS, Keefer LK. DNA deaminating ability and genotoxicity of nitric oxide and its progenitors. Science. 1991;254:1001–1003. doi: 10.1126/science.1948068. [DOI] [PubMed] [Google Scholar]
- 39.Zhuang JC, Lin C, Lin D, Wogan GN. Mutagenesis associated with nitric oxide production in macrophages. Proc. Natl. Acad. Sci. USA. 1998;95:8286–8291. doi: 10.1073/pnas.95.14.8286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liu TH, Beckman JS, Freeman BA, Hogan EL, Hsu CY. Polyethylene glycol-conjugated superoxide dismutase and catalase reduce ischemic brain injury. Am. J. Physiol. 1989;256:H589. doi: 10.1152/ajpheart.1989.256.2.H589. [DOI] [PubMed] [Google Scholar]
- 41.Hall ED, Braughler JM. Central nervous system trauma and stroke. II. Physiological and pharmacological evidence for involvement of oxygen radicals and lipid peroxidation. Free Rad. Med. Biol. 1989;6:303–313. doi: 10.1016/0891-5849(89)90057-9. [DOI] [PubMed] [Google Scholar]
- 42.Murakami K, Kondo T, Kawase M, Li Y, Sato S, Chen SF, Chan PH. Mitochondrial susceptibility to oxidative stress exacerbates cerebral infarction that follows permanent focal cerebral ischemia in mutant mice with manganese superoxide dismutase deficiency. J. Neurosci. 1993;18:205–213. doi: 10.1523/JNEUROSCI.18-01-00205.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Yakes FM, Van Houten B. Mitochondrial DNA damage is more extensive and persists longer than nuclear DNA damage in human cells following oxidative stress. Proc. Natl. Acad. Sci. USA. 1997;94:514–519. doi: 10.1073/pnas.94.2.514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Taffe BG, Larminat F, Laval J, Croteau DL, Anson RM, Bohr VA. Gene-specific nuclear and mitochondrial repair of formamidopyrimidine DNA glycosylase-sensitive sites in Chinese hamster ovary cells. Mut. Res. 1996;364:183–192. doi: 10.1016/s0921-8777(96)00031-6. [DOI] [PubMed] [Google Scholar]
- 45.Gobbel GT, Bellinzona M, Vogt AR, Gupta N, Fike JR, Chan P. Response of postmitotic neurons to x-irradiation: Implications for the role of DNA damage in neuronal apoptosis. J. Neurosci. 1998;18:147–155. doi: 10.1523/JNEUROSCI.18-01-00147.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Suzuki M, Avicola AK, Hood L, Loeb LA. Low fidelity mutants in the O-helix of Thermus aquaticus DNA polymerase I. J. Biol. Chem. 1997;272:11228–11235. doi: 10.1074/jbc.272.17.11228. [DOI] [PubMed] [Google Scholar]
- 47.Koh J, Choi D. Vulnerability of cultured cortical neurons to damage by excitotoxins: differential susceptibility of neurons containing NADPH-diaphorase. J. Neurosci. 1988;8:2153–2163. doi: 10.1523/JNEUROSCI.08-06-02153.1988. [DOI] [PMC free article] [PubMed] [Google Scholar]