

Abstract
The development of an efficient and stereoselective trans-Diels-Alder paradigm is described. A central element of this transformation is the introduction of a temporary dienophilic functionality (A), which serves both to activate the substrate for Diels-Alder cycloaddtion and, through its subsequent removal, to facilitate conversion of the cis-fused cycloadducts to the trans-fused series.
The importance of the Diels-Alder reaction can hardly be exaggerated. Its many features have been widely discussed.1 The ability to reach cis-fused bicyclic systems by a Diels-Alder pathway has been a bulwark of complex target-oriented synthesis.2 We well recognize that strategy in chemical synthesis is primarily beholden to capabilities arising from advances in methodology. This said, strategy also involves an intrinsic cognitive element. It connects a particular target to the huge database of organic chemistry, seeking to apply its most relevant and salient features to the challenge at hand. Central in this regard has been the logic of retrosynthetic analysis, which prioritizes various bond disconnections in an inverse sequential manner.3 The attainments resulting from strategic bond retrosynthetic analysis are legion.
Complementary to the powerful thought process of bond disconnections, we have been entertaining a different approach which we term “pattern analysis.”4 Although these approaches have some commonality, pattern analysis emphasizes a holistic view of the target structure, seeking connectivity between its key substructural characteristics (sometimes obvious but sometimes quite subtle) and established or prospectively implementable pathways. For instance, in pattern analysis, the possibility of a Diels-Alder (DA) application is provoked by the recognition of a cis junction. By contrast, trans junctions tend to teach away5 from a Diels-Alder universe.
It goes without saying that the scope of pattern analysis would be dramatically expanded if targets containing trans junctions could also be encompassed in the general Diels-Alder logic. The simplest scenario would contemplate an antarafacial cycloaddition.6 Not having any constructive suggestions in this regard, we turned to the next best thing. As outlined in Scheme 1, we envisioned equipping an otherwise unreactive dienophile (such as cyclohexene) with a temporary, readily removable activating group (A). Following cycloaddition, a product of the type 2 would be obtained. The resultant cis-fused substructure would be diverted to the trans series (1) through excision of the activating moiety and its controlled replacement. The traceless function (A) would first serve the purpose of activating the dienophile for cycloaddition, thereby affording a cis junction. Subsequent chemistry would provide entry to the trans-fused series. We selected a nitro group as (A), based on its dienophile activating properties,7 its known success in controlling DA regioselectivity,8 and its ability to generate free radical intermediates.9 We describe herein the use of nitrocycloalkenes as dienophiles, pointing toward an emerging trans-Diels-Alder paradigm.
Scheme 1.

Trans-Diels-Alder Paradigm.
We began by studying the dienophilicity of compound 3. At the outset, there had been only one evaluation of it as a dienophile in a Diels-Alder reaction.10 In the event, exploitation of its reactivity in cycloadditions with hydrocarbon dienes (see entry 1-4, Table 1) proved to be a particularly challenging problem. At temperatures below ∼120-130 °C, there was little or no indication of cycloaddition. At temperatures above 140 °C there seemed to be very serious deterioration of the dienophile, resulting in extremely low yields of cycloadduct. We were, however, able to identify a narrow temperature range in which marginally useful cycloaddition could be achieved. Still, in hydrocarbon solvents such as toluene or on THF, the yields of isolated cycloadducts were quite low, apparently reflecting competition between the innate destruction of 3, versus its cycloaddition.
Table 1.
Diels-Alder Reaction of Nitrocycloalkenes.
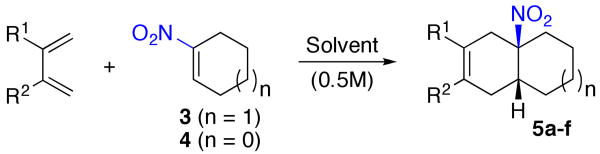 | |||||||
|---|---|---|---|---|---|---|---|
| entry | 5 | n | diene | solvent | T (°C) | T (h) | yield (%)a |
| 1 | a | 1 | R1=R2=CH3 | tolueneb | 110 | 12 | trace |
| 2 | a | 1 | R1=R2=CH3 | tolueneb | 140 | 36 | 18 |
| 3 | a | 1 | R1=R2=CH3 | tolueneb | 150 | 36 | trace |
| 4 | a | 1 | R1=R2=CH3 | THF | 130 MWc | 12 | 17 |
| 5 | a | 1 | R1=R2=CH3 | CH3CH2OH | 130 MWc | 14 | 36 |
| 6 | a | 1 | R1=R2=CH3 | CF3CH2OH | 130 | 24 | 70 |
| 7 | a | 1 | R1=R2=CH3 | CF3CH2OH | 130 MWc | 12 | 78 |
| 8 | b | 1 | R1=H, R2=CH3 | CF3CH2OH | 125 MWc | 14 | 57d,e |
| 9 | c | 1 | R1=R2=(CH2)4 | CF3CH2OH | 130 MWc | 10 | 75 |
| 10 | d | 1 | R1=R2=Ph | tolueneb,f | 150 | 48 | 45 |
| 11 | e | 0 | R1=R2=CH3 | CF3CH2OH | 100 | 12 | 78 |
| 12 | f | 0 | R1=H, R2=CH3 | CF3CH2OH | 110 | 12 | 77g |
Isolated yield.
In the presence of 2,6-di-tert-butylcresol (5 mol%).
MW=microwave.
p-directed/m-directed=15:1.
The reaction in toluene gave a 1:1mixture of regioisomers as a 5% yield
The diene was insoluble in CF3CH2OH.
p-directed/m-directed=11:1.
During the course of these studies, we did note some improvement of cycloaddition yield in alcoholic solvents11 (see entry 5, Table 1). Fortunately a more substantial increase in yield was realized when we turned to 2,2,2-trifluoroethanol as solvent, particularly when thermolysis was conducted under microwave conditions (entry 7). We emphasize, however, that the hydroxylic solvent effect, including 2,2,2-trifluoroethanol, apparently does not arise from catalysis (through hydrogen bonding or via other means). Thus, we see no indication that cycloaddition occurs at lower temperatures with 2,2,2-trifluoroethanol. Rather, the differences in yield seem to reflect a more favorable distribution of cycloaddition relative to decomposition. While the basis of this effect remains to be explored, it was very helpful to our program. Reasonable yields of cycloaddition from compound 3 could now be achieved. Happily, the protocol which was worked out for 2,3-dimethyl-1,3-butadiene was extendable to several other dienes (see Table 1). Moreover, we prepared 1-nitrocyclopentene (4)10 and evaluated its dienophilicity with the same dienes. Yields for these reactions are also included in Table 1. Here, we note that the temperatures required for cycloaddition were substantially lower (ca 100-110 °C) for 4 than for 3, though, for the moment, the isolated yields tend to be similar. Table 1 summarizes yields for various conditions under which cycloaddition of dienophiles 3 and 4 with various acyclic dienes could be achieved.
Having substantially advanced the practicality of cycloadditions with nitrocycloalkene dienophiles, we next faced the central issue of denitration. We well recognized that if reductive denitration would lead to a cis junction, the nitrocycloalkene dienophile would have functioned as an equivalent of otherwise unreactive cyclohexene or cyclopentene.12 However, if the excision-replacement sequence would afford a trans junction, an overall trans-Diels-Alder type paradigm would have been demonstrated.
In the event, we began by treating compound 5a with tri-n-butyltin hydride in the presence of AIBN. We noted an 8:1 ratio of trans-6a to cis-7a, as established by comparison with authentic reference compounds prepared in multistep sequences.13 In a similar way, the isoprene adduct 5b also gave rise to an 8:1 ratio of trans-6b to cis-7b. Other examples are shown in Table 2. Thus, while one could hope for even stronger stereoselectivity, these data encourage application of Diels-Alder logic to the synthesis of trans octalins.
Table 2.
Radical Denitration.
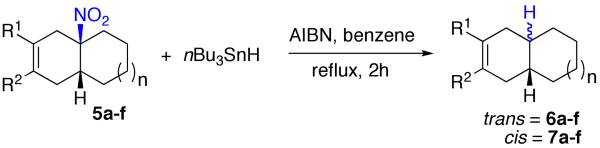 | |||||
|---|---|---|---|---|---|
| entry | 6,7 | n | diene | yield (%)a | 6:7b |
| 1 | a | 1 | R1=R2=CH3 | 67 | 8:1 |
| 2 | b | 1 | R1=H, R2=CH3 | 53 | 8:1 |
| 3 | c | 1 | R1=R2=(CH2)4 | 81 | 12:1c |
| 4d | d | 1 | R1=R2=Ph | 83 | 7:1 |
| 5 | e | 0 | R1=R2=CH3 | 61 | 1.3:1 |
| 6 | f | 0 | R1=H, R2=CH3 | 35d | 1.9:1 |
Isolated yield.
Determined by 1H-NMR.
Determined by GC.
The compound is volatile.
Perhaps, this selectivity pattern represents a preference for conformer type A relative to conformer B thereby accounting for the selective formation of trans product (Figure 1). However, in the hydrindene series, the ratio of the denitration products, was much closer to unity, presumably reflecting altered trans:cis preferences.
Figure 1.

Structures of the tertiary bridge head radical.
In the next step in our exploration, we evaluated the consequences of using a more functionalized diene. In that way, additional exploitable functionality would be delivered to the eventual denitration product. We began by studying the cycloaddition reaction of compound 3 with diene 8.14 Fortunately, our conditions described above for cycloaddition with acyclic hydrocarbon dienes work quite well with diene 8. In 2,2,2-trifluoroethanol at 80 °C, a 61% yield of adduct 9 was obtained.15 With 1-nitrocyclopentene (4), cycloaddition under comparable conditions afforded adduct 12 (Scheme 2).
Scheme 2a.

aKey: (a) CF3CH2OH, 80 °C, MW, 12 h, 61%. (b) HF, CH3CN, rt, 30 min. (c) nBu3SnH, AIBN, benzene, reflux, 2 h. (d) CF3CH2OH, 80 °C, MW, 6 h, 72%.
We then turned to denitration reactions. For each compound, this process was studied in two different sequences. In one arm, the silyl enol ethers were hydrolyzed and the subsequent ketones were denitrated. In this case, a 5:1 ratio of trans compound 10 to cis compound 11 was obtained. A very similar result pertained when we reversed the order of steps, wherein denitration was carried out at the stage of adduct 9 and followed by hydrolysis of the silyl enol ether, to provide a 6:1 ratio of trans-10 : cis-11.16
We applied the same methodology to adduct 12, which arose from 4. Once again, both denitration sequences were pursued. When denitration was conducted at the stage of the silyl enol ether, followed by hydrolysis, a 1.4:1 ratio of trans-1417 to cis-1318 was obtained. By contrast, when denitration was conducted at the ketone stage (i.e. after hydrolysis), a 15:1 ratio of cis-13 to trans-14 was produced. Thus, once again, in the octalin series a stereoselective route to trans-fused ketone 10 has been established. By contrast, in the hydrindane series, a stereoselective route to the cis fusion has been realized. Unfortunately, no corresponding protocol now available to us provides a trans hydrindanone junction with useful levels of stereoselection (vide infra).
Finally, we turned our attention to the cycloaddition reaction of diene 1519 with 1-nitrocyclohexene (3). As it turned out, this reaction could not be conducted in 2,2,2-trifluoroethanol because of rapid conversion of the diene to the corresponding ketone, methoxybutenone. Accordingly, the reaction was conducted in xylene under reflux, as shown in Scheme 3. This treatment gave rise to a mixture of endo and exo Diels-Alder product, indicated as 16. Again, we examined the stereochemical outcome of the denitration reaction when conducted at one of the two different stages. Thus, when reduction was conducted at the stage of the ketone 16 (obtained by prior hydrolysis of the silyl enol ether function), a 2:1 ratio of trans:cis octalones was obtained. In contrast, when the denitration was conducted at the silyl enol ether stage and the denitration product subjected to acid hydrolysis of the silyl enol ether, an 8:1 ratio of trans-17 to cis-18 was produced.20 Once again, we see how the chemistry described above points the way to introduction of trans ring junctions in Diels-Alder driven constructions.
Scheme 3a.

aKey: (a) xylene, reflux, 36 h. (b) 0.05 M HCl, THF, 2 h, 67% for 2 steps. (c) nBu3SnH, AIBN, benzene, reflux, 2 h, 89%. (d) TFAA, benzene, Dean-Stark, 24 h, 60%. (e) toluene, 130 °C, 36 h, 90%. (f) nBu3SnH, AIBN, benzene, reflux, 2 h. (g) HF, CH3CN, rt, 10 min, 53% for 2 steps.
In summary then, while many provocative possibilities remain to be explored, the chemistry described above already holds promise for application to two types of situations. In the hydrindane series, denitration can be used to generate a cis fusion, thereby establishing a Diels-Alder equivalency for the otherwise inert cyclopentene. By contrast in the octalin series, it is possible to take advantage of this chemistry to produce, selectively, trans junctions. In that sense, the nitrocyclohexene will have served in a Diels-Alder context as an equivalent of the otherwise unavailable dienophile, E-cyclohexene. Studies addressing a menu of follow-up possibilities suggested by these findings are in progress.
Supplementary Material
Acknowledgments
Support was provided by the NIH (HL25848 and CA103823 to SJD). W.H.K. is grateful for a Korea Research Foundation Grant funded by the Korean government (KRF-2007-357-c00060). We thank Rebecca Wilson and Dana Ryan for assistance with the preparation of the manuscript and Prof. W. F. Berkowitz and Dr. Pavel Nagorny for helpful discussions. We also thank Dr. George Sukenick, Ms. Hui Fang, Sylvi Rusli (NMR Core Facility, Sloan-Kettering Institute) and Dr. Yasuhiro Itagaki (Mass Spectral Core Facility, Columbia University) for mass spectral and NMR spectroscopic analysis.
Footnotes
Supporting Information Available: Experimental procedures, copies of spectral data, and characterization (PDF). This material is available free of charge via the Internet at http://pubs.acs.org
References
- 1.(a) Corey EJ. Angew Chem Int Ed. 2009;48:2100–2117. doi: 10.1002/anie.200805374. [DOI] [PubMed] [Google Scholar]; (b) Corey EJ. Angew Chem Int Ed. 2002;41:1650–1667. doi: 10.1002/1521-3773(20020517)41:10<1650::aid-anie1650>3.0.co;2-b. [DOI] [PubMed] [Google Scholar]; (c) Bear BR, Sparks SM, Shea KJ. Angew Chem Int Ed. 2001;40:820–849. [PubMed] [Google Scholar]; (d) Williams RM, Stocking EM. Angew Chem Int Ed. 2003;42:3078–3115. doi: 10.1002/anie.200200534. [DOI] [PubMed] [Google Scholar]; (e) Jørgensen KA. Angew Chem Int Ed. 2000;39:3558–3588. doi: 10.1002/1521-3773(20001016)39:20<3558::aid-anie3558>3.0.co;2-i. [DOI] [PubMed] [Google Scholar]
- 2.(a) Nicolaou KC, Snyder SA, Montagnon T, Vassilikogiannakis G. Angew Chem Int Ed. 2002;41:1668–1698. doi: 10.1002/1521-3773(20020517)41:10<1668::aid-anie1668>3.0.co;2-z. [DOI] [PubMed] [Google Scholar]; (b) Danishefsky S. Acc Chem Res. 1981;14:400–406. [Google Scholar]
- 3.(a) Corey EJ. Pure Appl Chem. 1967:19–37. [Google Scholar]; (b) Corey EJ, Wipke WT. Science. 1969;166:178–192. doi: 10.1126/science.166.3902.178. [DOI] [PubMed] [Google Scholar]; (c) Corey EJ. Angew Chem Int Ed. 1991;30:455–612. [Google Scholar]; (d) Corey EJ, Chelg X. The Logic of Chemical Synthesis. Wiley-VCH; New York: 1995. [Google Scholar]
- 4.(a) Wilson RM, Danishefsky SJ. Acc Chem Res. 2006;39:539–549. doi: 10.1021/ar068018n. [DOI] [PubMed] [Google Scholar]; (b) Wilson RM, Danishefsky SJ. J Org Chem. 2007;72:4293–4305. doi: 10.1021/jo070871s. [DOI] [PubMed] [Google Scholar]
- 5.The possibility of epimerizing a cis junction, obtained from a Diels Alder reaction, to the trans series was well recognized and practiced by Woodward and co-workers in their total synthesis of steroids. Other instances of epimerization are also cited herein. Woodward RB, Sondheimer F, Taub D, Heusler K, McLamore WM. J Am Chem Soc. 1952;74:4223–4251.Stevens RV, Angle SR, Kloc K, Mak KF, Trueblood KN, Liu YX. J Org Chem. 1986;51:4347–4353. and references therein.Mukhopadhyay A, Ali SM, Husain M, Suryawanshi SN, Bhakuni DS. Tetrahedron Lett. 1989;30:1853–1856.Of course, this approach requires a vicinal keto group to enable epimerization and the capacity to apply strict thermodynamic or kinetic controls to govern the junction stereochemistry.
- 6.Woodward RB, Hoffmann R. Angew Chem Int Ed. 1969;8:781–932. [Google Scholar]
- 7.Ono N. The Nitro Group in Organic Synthesis. Wiley-VCH; New York: 2001. and pertinent references cited therein. [Google Scholar]
- 8.(a) Danishefsky S, Prisbylla MP, Hiner S. J Am Chem Soc. 1978;100:2918–2920. [Google Scholar]; (b) Corey EJ, Estreicher H. Tetrahedron Lett. 1981;22:603–606. [Google Scholar]
- 9.(a) Ono N, Miyake H, Tamura R, Kaji A. Tetrahedron Lett. 1981;22:1705–1708. [Google Scholar]; (b) Ono N, Miyake H, Kamimura A, Hamamoto I, Tamura R, Kaji A. Tetrahedron. 1985;41:4013–4023. [Google Scholar]; (c) Ono N, Miyake H, Kaji A. J Chem Soc Chem Commun. 1982:33–34. [Google Scholar]
- 10.Corey EJ, Estreicher H. J Am Chem Soc. 1978;100:6294–6295. [Google Scholar]
- 11.Breslow R, Guo TJ. Am Chem Soc. 1988;110:5613–5617. [Google Scholar]
- 12.Following completion of this work, we noted that in footnote (18) in reference (10), Professor Corey futuristically envisioned potential value of reductive denitration at the junction. However no results are provided. Nonetheless the kernel of the idea can be found in Corey's comment.
- 13.Lee JH, Kim WH, Danishefsky SJ. Tetrahedron Lett. 2009 doi: 10.1016/j.tetlet.2009.07.068. In Press. [DOI] [Google Scholar]
- 14.Jung ME, McCombs CA. Tetrahedron Lett. 1976;17:2935–2938. [Google Scholar]
- 15.Cycloaddition of compound 3 with several 2-silyloxy-1,3-butadienes as well as diene 8 in toluene under the variety of conditions did not proceed at all.
- 16.(a) Oritani T, Yamashita K. J Org Chem. 1984;49:3689–3694. [Google Scholar]; (b) Rao HSP, Reddy KS. Tetrahedron Lett. 1994;35:171–174. [Google Scholar]; (c) Augustine RL. J Org Chem. 1958;23:1853–1856. [Google Scholar]; (d) Macdonald TL, Mahalingam S. J Am Chem Soc. 1980;102:2113–2115. [Google Scholar]
- 17.Krawczyk AR, Jones JB. J Org Chem. 1989;54:1795–1801. [Google Scholar]
- 18.Okuda Y, Morizawa Y, Oshima K, Nozaki H. Tetrahedron Lett. 1984;25:2483–2486. and pertinent references cited therein. [Google Scholar]
- 19.Danishefsky S, Kitahara T. J Am Chem Soc. 1974;96:7807–7808. [Google Scholar]
- 20.(a) Corey EJ, Boger DL. Tetrahedron Lett. 1978;19:4597–4600. [Google Scholar]; (b) Angell EC, Fringuelli F, Pizzo F, Taticchi A, Wenkert E. J Org Chem. 1988;53:1424–1426. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.