

Abstract
Multiple sclerosis is an inflammatory disease of the central nervous system that begins as a relapsing-remitting disease (RRMS) and is followed by a progressive phase (SPMS). The progressive phase causes the greatest disability and has no effective therapy, but the processes that drive SPMS are mostly unknown. Here we found higher serum concentrations of 15α-hydroxicholestene (15-HC) in patients with SPMS and in mice with secondary progressive experimental autoimmune encephalomyelitis (EAE) but not in patients with RRMS. In mice, 15-HC activated microglia, macrophages and astrocytes through a pathway involving Toll-like receptor 2 (TLR2) and poly(ADP-ribose) polymerase 1 (PARP-1). PARP-1 activity was higher in monocytes of patients with SPMS, and PARP-1 inhibition suppressed the progression of EAE. Thus, the TLR2–PARP-1 pathway is a potential new therapeutic target in SPMS.
Multiple sclerosis is the leading cause of neurological disability in young adults1. In 85% of patients, multiple sclerosis initially follows a relapsing-remitting course (RRMS) in which acute autoimmune attacks against the central nervous system (CNS) are followed by a complete recovery2. Most patients with RRMS go on to develop secondary progressive multiple sclerosis (SPMS), characterized by the progressive and irreversible accumulation of neurological disability2. The progressive disease course that characterizes SPMS generally occurs in the absence of new inflammatory lesions, which suggests that other mechanisms are involved in this stage of multiple sclerosis3. Indeed, treatments that halt the adaptive inflammatory response have beneficial effects on RRMS but are usually ineffective in SPMS4. Thus, it is important to characterize the pathological processes that drive SPMS and to identify new therapies for progressive multiple sclerosis and biomarkers to monitor disease progression.
Both acute and chronic CNS inflammation and demyelination result in axonal damage5 that can be detected early in the course of RRMS6. Progressive irreversible disability, however, does not occur during these early stages of RRMS, probably as a result of compensatory mechanisms operating in the brain5. Thus, it has been suggested that the clinical transition to SPMS occurs when the accumulated neuronal loss reaches a threshold that cannot be compensated for by the plasticity of the CNS.
A change in the nature of the CNS inflammation has also been linked to the transition to SPMS. Dendritic cells from patients with SPMS secrete more interleukin 12 (IL-12) and IL-18 than do dendritic cells from healthy control subjects or patients with RRMS7–10. Moreover, dendritic cells from patients with SPMS have higher expression of the costimulatory molecule CD80 and lower expression of the inhibitory molecule PDL1 and are more efficient in activating T helper type 1 cells11. On the basis of those observations and the limited efficacy of therapies that target the adaptive immune response on SPMS, it has been proposed that whereas both adaptive and innate immune responses drive RRMS, mainly sustained innate immunity is involved in SPMS12.
Studies using antigen microarrays13,14 have detected higher concentrations of antibodies to 15-oxysterols, oxidized derivatives of cholesterol, in the serum of patients with multiple sclerosis15. It has been shown that 7-ketocholesterol (7-KC), an oxidized derivative of cholesterol that is not a 15-oxysterol, activates microglia by a mechanism dependent on the nuclear enzyme PARP-1 (poly(ADP-ribose) polymerase 1) that results in neuronal damage in vitro16. As neuronal loss is thought to contribute to the pathogenesis of progressive multiple sclerosis5, we investigated the function of 15-oxysterols in multiple sclerosis and experimental autoimmune encephalomyelitis (EAE). We found higher serum concentrations of the 15-oxysterol 15α-hydroxicholestene (15-HC) in SPMS and in mouse secondary progressive EAE. In vitro and in vivo, 15-HC activated macrophages, microglia and astrocytes by a signaling pathway dependent on Toll-like receptor 2 (TLR2; A002294) and PARP-1. Moreover, inhibition of PARP-1 suppressed axonal loss and disability in secondary progressive EAE. Thus, 15-HC is a potential biomarker for monitoring disease progression in multiple sclerosis, and the TLR2–PARP-1 axis is a potential new therapeutic target for SPMS.
Results
More 15-HC in SPMS and secondary progressive EAE
To investigate the function of 15-oxysterols in multiple sclerosis, we measured the concentrations of 15-ketocholestene (15-KE), 15-keto-cholestane (15-KA) and 15-HC in sera from patients with RRMS or SPMS and healthy control subjects (clinical characteristics, Supplementary Table 1). We detected higher concentrations of 15-KE and 15-KA in RRMS samples (Fig. 1a) and a further upregulation of 15-KE and 15-KA in SPMS samples (Fig. 1a). Notably, 15-HC concentrations were higher only in SPMS serum samples (Fig. 1a).
Figure 1.

Higher 15-HC serum concentrations in human SPMS and secondary progressive EAE. (a) Gas chromatography–mass spectrometry analysis of the concentration of 15-oxysterols in healthy control subjects (HC; n = 10), patients with RRMS (n = 12) and patients with SPMS (n = 10). (b) Concentration of 15-oxysterols at the peak of the acute attack of NOD EAE (day 18) or during the progressive phase of NOD EAE (day 55). P values, one-way analysis of variance (ANOVA). Data are representative of two experiments (mean and s.e.m.).
Ten-week-old nonobese diabetic (NOD) mice immunized with a peptide of amino acids 35–55 of myelin oligodendrocyte glycoprotein (MOG(35–55)) develop a form of EAE that resembles SPMS17. In this model, a self-limited acute neurological syndrome peaks at day 18 after immunization, followed by a phase of irreversible progressive neurological impairment17. We used this model of secondary progressive EAE to analyze the concentrations of 15-oxysterols at the peak of the acute EAE attack (day 18) and during the progressive phase of EAE (day 55). We found that both 15-KA and 15-KE were upregulated both during the acute attack and the progressive phase of EAE (Fig. 1b). However, 15-HC concentrations were upregulated only during the progressive phase of EAE (Fig. 1b), which suggests that 15-HC is linked to progressive neurological impairment both in human SPMS and mouse secondary progressive EAE.
PARP-1 activation promotes EAE progression
The ketocholesterol 7-KC damages DNA and activates PARP-1 (ref. 16). Thus, we investigated the effect of 15-HC on the activity of PARP-1 in a cell-free system. Incubation of recombinant PARP-1 with either undamaged DNA or 15-HC alone did not have a significant effect on PARP-1 activity. However, preincubation of DNA with 15-HC resulted in activation of PARP-1 to a degree similar to that achieved with 7-KC or DNA with single-strand breakage (Fig. 2a). Thus, 15-HC can activate PARP-1 by a DNA damage–dependent mechanism.
Figure 2.
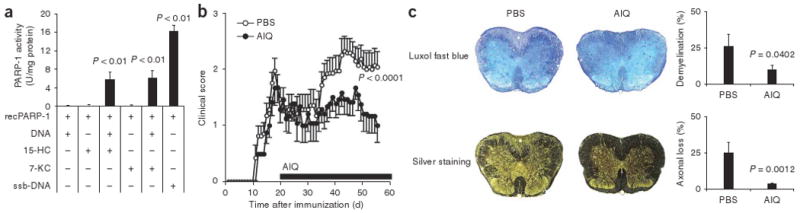
Inhibition of PARP-1 suppresses axonal loss and progressive disability in secondary progressive EAE. (a) Activity of recombinant PARP-1 (recPARP-1) incubated with DNA pretreated with 15-HC (+; row 3) or 7-KC (+; row 4). DNA without pretreatment (N) serves as a negative control; DNA with single-strand breakage (ssb-DNA) serves as a positive control. P values, one-way ANOVA. Data are representative of three independent experiments (mean and s.e.m.). (b,c) Clinical scores (b) and histopathological analysis of EAE (c) in 10-week-old NOD mice in which secondary progressive EAE was induced by immunization with MOG(35–55) in CFA (100 μg per mouse), with pertussis toxin (150 ng per mouse) given at the time of immunization and 48 h later, and AIQ (3 mg per kg body weight) or PBS (control) administered daily beginning on day 20. (c) Spinal cord sections at day 55, stained with Luxol fast blue or Bielschowsky's silver stain (left) for analysis of demyelination or axonal loss (right), respectively. P values, Student's t-test. Data are representative of two independent experiments (mean and s.e.m.).
To investigate the function of PARP-1 in the progressive multiple sclerosis model, we used the PARP-1-specific inhibitor 5-aminoiso-quinolinone (AIQ)18 in NOD secondary progressive EAE. We administered AIQ intraperitoneally at a dose of 3mg per kg body weight daily from day 20 (after the mice recovered from the first peak of disease) to the end of the experiment. Administration of AIQ resulted in inhibition of the clinical signs of secondary progressive EAE (Fig. 2b and Supplementary Table 2).
In the NOD model of secondary progressive EAE, the progressive accumulation of clinical disability is linked to extensive demyelination and axonal loss17. Histological analysis of control and AIQ-treated mice showed that inhibition of PARP-1 with AIQ resulted in significantly less demyelination and axonal loss (Fig. 2c). Thus, activation of PARP-1 promotes the progression of secondary progressive EAE in NOD mice.
Activation of PARP-1 during progressive disease
To understand the mechanistic basis for the function of PARP-1 in secondary progressive EAE, we first studied the adaptive encephalitogenic response in AIQ-treated mice. Splenocytes from control and AIQ-treated mice showed similar proliferative responses after activation with MOG(35–55) or mitogenic antibody to CD3 (anti-CD3; Supplementary Fig. 1a). Furthermore, we found no differences in the secretion of proinflammatory cytokines (interferon-γ and IL-17) or anti-inflammatory cytokines (IL-10 and transforming growth factor-β1) triggered by MOG(35–55) or mitogenic anti-CD3 (Supplementary Fig. 1b). Thus, suppression of the progressive phase of NOD EAE by AIQ does not result from its influence on the adaptive encephalitogenic response.
Activation of microglia, CNS-infiltrating macrophages and astrocytes has been linked to the progressive phase of NOD EAE17; thus, we analyzed the expression of Iba1 and GFAP (markers of microglia and CNS-infiltrating macrophages, and of astrocytes, respectively) in the CNS of NOD mice during the progressive phase of NOD EAE. Similar to what has been described before17, the progressive phase of NOD EAE was associated with intense staining for Iba1 and GFAP in the CNS (Fig. 3a). Administration of AIQ resulted in significantly fewer Iba1+ microglia and CNS-infiltrating macrophages and GFAP+ astrocytes (Fig. 3a).
Figure 3.
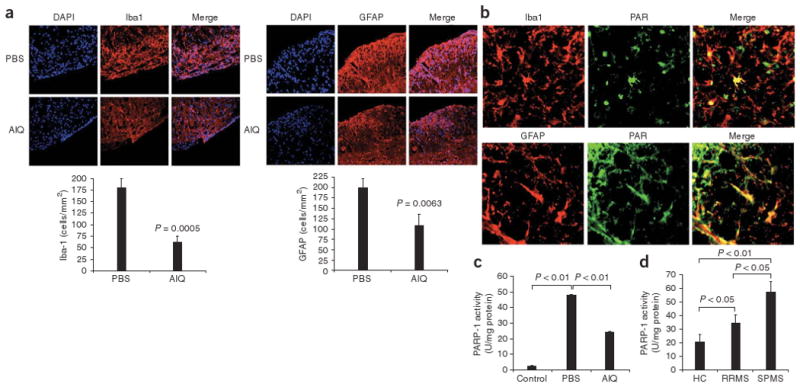
Activation of PARP-1 during human SPMS and secondary progressive EAE. (a) Microsopy of spinal cords from PBS- or AIQ-treated mice for analysis of the presence of microglial cells, CNS-infiltrating macrophages (Iba1) and astrocytes (GFAP); the DNA-intercalating dye DAPI stains nuclei. Original magnification, ×200. Below, quantification of stained cells. P values, Student's t-test. (b) Activation of PARP-1 in the spinal cords of NOD mice during the progressive phase of NOD EAE, assessed as the accumulation of PAR (green) in Iba1+ or GFAP+ (red) cells. Original magnification, ×200. (c) Enzymatic activity of PARP-1 in CD11b+ CNS cells isolated from AIQ- or PBS-treated mice at day 55, during the progressive phase of NOD EAE. P values, Student's t-test. (d) Enzymatic activity of PARP-1 in peripheral blood monocytes isolated from healthy control subjects and patients with RRMS or SPMS. P values, one-way ANOVA. Data are representative of two experiments (c) or two independent experiments (a,b,d; mean and s.e.m.).
Activated microglia, CNS-infiltrating macrophages and astrocytes produce tumor necrosis factor (TNF) and nitric oxide, which have direct neurotoxic effects19–21 that have been linked to axonal loss in secondary progressive EAE17. The chemokine CCL2 contributes to this pathogenic process by recruiting peripheral macrophages to the CNS22. We thus analyzed the expression of TNF, inducible nitric oxide synthase (iNOS) and CCL2 in the CNS of NOD mice during the progressive phase of NOD EAE. We detected substantial quantities of TNF, iNOS and CCL2 in the CNS, which were diminished by treatment with AIQ (Supplementary Fig. 2). Hence, inhibition of PARP-1 resulted in substantial suppression of the activation of microglia, CNS-infiltrating macrophages and astrocytes during the progressive phase of NOD EAE.
PARP-1 activation results in the poly(ADP) ribosylation of its target proteins16; thus, PARP-1 activity can be studied in situ with antibodies to poly(ADP-ribose) (PAR). To study the function of PARP-1 in the activation of microglia, CNS-infiltrating macrophages and astrocytes during the progressive phase of NOD EAE, we used anti-PAR in combination with anti-Iba1 and anti-GFAP. We found that PARP-1 was activated in microglia, CNS-infiltrating macrophages and astrocytes during the progressive phase of NOD EAE (Fig. 3b); PARP-1 activation was lower in AIQ-treated mice (data not shown). We obtained similar results when we measured PARP-1 enzymatic activity: CD11b+ CNS cells isolated from PBS-treated mice during the progressive phase of secondary progressive EAE showed significant upregulation in PARP-1 activity (Fig. 3c) which was significantly diminished by treatment with AIQ (Fig. 3c). Together these results indicate that PARP-1 activity is linked to the activation of microglia, CNS-infiltrating macrophages and astrocytes during the progressive phase of NOD EAE.
To investigate the relevance of our findings for human multiple sclerosis, we measured PARP-1 activity in patients with multiple sclerosis. We found that PARP-1 activity was higher in the peripheral blood monocytes of patients with RRMS or SPMS, with significantly higher activity in patients with SPMS (Fig. 3d). Thus, PARP-1 activation is linked to both human SPMS and secondary progressive EAE.
PARP-1 activation by 15-HC promotes neuroinflammation
We next studied the direct effects of 15-HC on microglia, macrophages and astrocytes. Incubation of macrophages, microglia or astrocytes with 15-HC resulted in a dose-dependent activation of PARP-1 that could be inhibited with AIQ (Fig. 4a,b). We obtained similar results when we used 7-KC as a positive control (Supplementary Fig. 3 and Supplementary Table 3). Incubation of microglia, macrophages or astrocytes with 15-HC resulted in the secretion of nitric oxide, CCL2 and TNF that was inhibited by coincubation with AIQ (Supplementary Table 4). Thus, 15-HC activates microglia, macrophages and astrocytes by a PARP-1-dependent mechanism.
Figure 4.
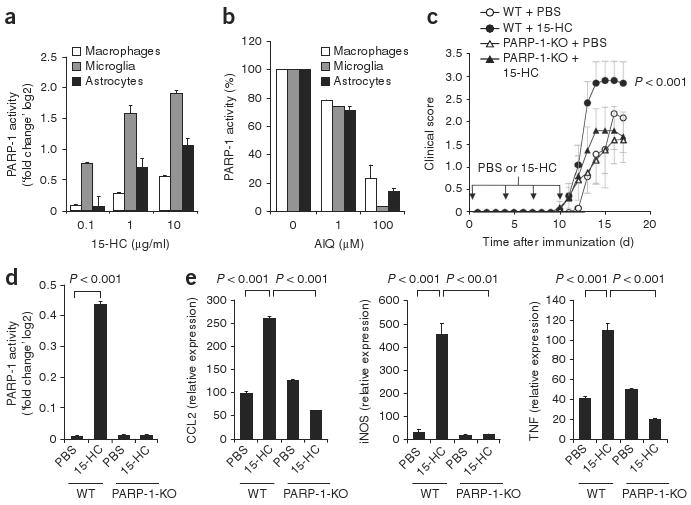
Activation of microglia, macrophages and astrocytes by 5-HC via PARP-1. (a) PARP-1 activity in mouse microglia, macrophages or astrocytes incubated with 15-HC, presented relative to activity in vehicle-treated cells. (b) Inhibition of PARP-1 activity in mouse microglia, macrophages or astrocytes activated with 15-HC (1 μg/ml) in the presence of AIQ (concentration, horizontal axis), presented relative to that in cells not treated with AIQ, set as 100%. (c) Disease scores of PARP-1-deficient (PARP-1-KO) mice and wild-type (WT) mice treated with PBS or 15-HC (downward arrows) during the course of EAE. (d) PARP-1 activity in CD11b+ CNS cells isolated from PARP-1-deficient and wild-type mice treated with PBS or 15-HC during the course of EAE. (e) Expression of TNF, iNOS and CCL2 mRNA in CD11b+ CNS cells isolated from PARP-1-deficient and wild-type mice treated with PBS or 15-HC during the course of EAE. Expression is presented relative to that in PBS-treated cells (d) or naive mice (e). P values, one-way ANOVA. Data are representative of three (a,b) or two (c–e) independent experiments (mean and s.e.m.).
To determine if 15-HC contributed to neuroinflammation in a PARP-1-dependent way in vivo, we studied PARP-1-deficient mice. We induced EAE in PARP-1-deficient and wild-type mice by injecting an emulsion of MOG(35–55) and complete Freund's adjuvant (CFA) and administered 15-HC during the induction of EAE and then again intraperitoneally on days 4, 7 and 10 as described23. We then monitored the mice for clinical signs of the disease. The administration of 15-HC resulted in significant worsening of EAE in wild-type mice but not in PARP-1-deficient mice (Fig. 4c and Supplementary Table 5). CD11b+ cells isolated from the CNS of 15-HC-treated wild-type mice showed significant upregulation in PARP-1 activity (Fig. 4d). The activation of PARP-1 in CD11b+ CNS cells correlated with higher expression of CCL2, iNOS and TNF mRNA, as measured by real-time PCR (Fig. 4e); this upregulation was absent in 15-HC-treated PARP-1-deficient mice (Fig. 4e). We detected no effects of 15-HC on the encephalitogenic recall response to MOG(35–55) or anti-CD3 in wild-type or PARP-1-deficient mice (data not shown). Thus, 15-HC enhanced neuroinflam-mation in vivo by a PARP-1-dependent mechanism.
Activation of PARP-1 by 15-HC via a TLR2-dependent pathway
We used reverse-phase protein array24 to characterize the signaling pathways involved in the activation of PARP-1 by 15-HC. Preliminary kinetic studies showed that incubation of macrophages or microglia with 15-HC for 5 min resulted in PARP-1 activation, whereas longer incubation times were required for activation induced by 7-KC (Fig. 5a). Thus, we constructed reverse-phase protein arrays by spotting microarrays with cell lysates prepared at various times (0–30 min) after the activation of microglia or macrophages with 15-HC. We then probed the arrays with antibodies specific for proteins or phosphorylated proteins linked to signaling pathways of interest (Supplementary Table 6). Treatment of microglia or macrophages with 15-HC resulted in the phosphorylation of proteins known to mediate TLR signaling, including TAK1, IRAK, Akt, p38, Pyk2, PKC, PI(3)K and Erk1/2 (ref. 25; Fig. 5b); we obtained similar results when we analyzed these proteins by immunoblot (Supplementary Fig. 4). We then used specific inhibitors to investigate the functional relevance of the protein kinases identified above in the activation of PARP-1 by 15-HC. Treatment of microglia or macrophages with 15-HC in the presence of specific inhibitors of Erk1/2, Akt, PI(3)K, PKC or p38 resulted in significant inhibition of PARP-1 activation (Fig. 5c).
Figure 5.
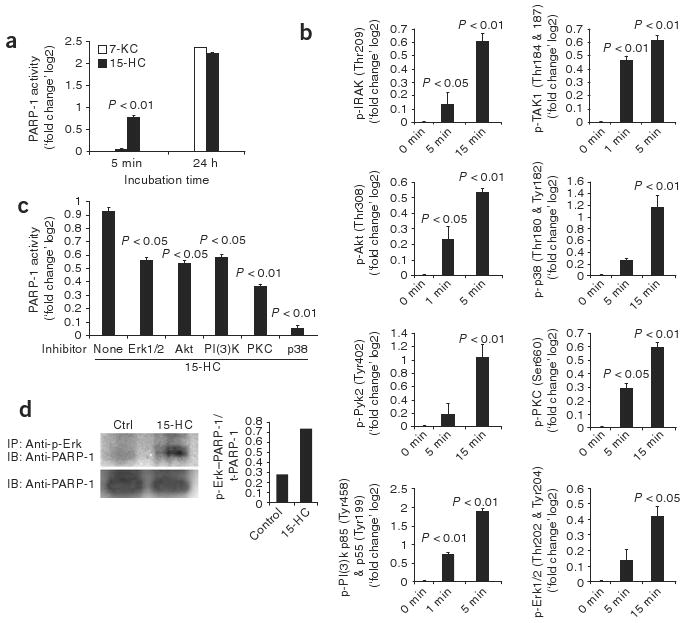
Signaling events mediating the activation of PARP-1 by 15-HC. (a) Time course of microglial PARP-1 activation by 15-oxysterols or 7-KC. P value, two-way ANOVA. (b) Signaling pathways activated in microglial cells stimulated with 15-HC and lysed at 1, 5 or 15 min; signal-transduction arrays prepared with the lysates were probed with phosphorylation-specific antibodies. Phosphorylated (p-) residues are in parentheses on vertical axes. P values, one-way ANOVA. (c) Activation of microglial PARP-1 by 15-HC in the presence of kinase inhibitors. P values, one-way ANOVA. (d) Coimmunoprecipitation of phosphorylated Erk1/2 and PARP-1 in lysates of microglial cells treated with PBS (control) or 15-HC, immunoprecipitated (IP) with antibodies to phosphorylated Erk1/2 and then analyzed by immunoblot (IB) with anti-PARP-1. Below, immunoblot analysis of whole-cell lysates with anti-PARP-1 (loading control). Right, ratio of PARP-1 immunoprecipitated with antibody to phosphorylated Erk1 (p-Erk–PARP-1) to total PARP-1 (t-PARP-1) in lysates. Data are representative of two (b) or three (a,c,d) independent experiments (mean and s.e.m., a–c).
The kinases Erk1 and Erk2 physically interact with PARP-1 and induce PARP-1 activation through a mechanism independent of DNA damage26. Thus, we did coimmunoprecipitation studies to better characterize the mechanisms by which 15-HC activates PARP-1. We activated microglia and macrophages with 15-HC, used antibodies specific for phosphorylated Erk1/2 for immunoprecipitation and probed the immunoprecipitated material with anti-PARP-1. Treatment of microglia and macrophages with 15-HC resulted in substantial interaction of Erk1/2 with PARP-1 (Fig. 5d).
To confirm the involvement of TLR signaling in the activation of PARP-1 by 15-HC, we used TLR-blocking antibodies. Anti-TLR2 inhibited the activation of PARP-1 by 15-HC, but anti-TLR4 did not; we found no effect when we used isotype-matched control antibodies (Fig. 6a). This inhibition of the activation of PARP-1 by 15-HC correlated with complete suppression of the phosphorylation of TAK1, IRAK, Akt, p38, Pyk2, PKC, PI(3)K and Erk1/2 (Supplementary Fig. 5).
Figure 6.

Activation of PARP-1 by 15-HC is mediated by TLR2. (a) PARP-1 activity in microglia, macrophages and astrocytes activated with 15-HC in the presence of blocking anti-TLR2 or anti-TLR4 or isotype-matched control antibody (IC). (b) PARP-1 activity in HEK293 cells left untransfected (293-null) or transfected with plasmid encoding TLR2 (293-TLR2), then activated with 15-HC or with LPS from Porphyromonas gingivalis (pgLPS) or Escherichia coli (ecLPS). (c) Disease scores of TLR2-deficient (TLR2-KO) and wild-type mice treated with PBS or 15-HC during the induction and course of EAE. (d) PARP-1 activity in CD11b+ CNS cells isolated from TLR2-deficient and wild-type mice treated with PBS or 15-HC during the course of EAE. P values, test. (e) Expression of TNF, iNOS and CCL2 mRNA by CD11b+ CNS cells isolated from TLR2-deficient and wild-type mice treated with PBS or 15-HC during the course of EAE. P values, one-way ANOVA (a,b,d,e) or two-way ANOVA (c). Data are representative of three independent experiments (mean and s.e.m.).
To further confirm the involvement of TLR2 in the activation of PARP-1 by 15-HC, we used the human embryonic kidney cell line HEK293, which expresses PARP-1 but does not express TLR2. Treatment of HEK293 cells with 15-HC had no significant effect on PARP-1 activity (Fig. 6b). However, transfection of HEK293 cells with a plasmid encoding TLR2 resulted in significant activation of PARP-1 by 15-HC (Fig. 6b).
To investigate the function of TLR2 in the promotion of PARP-1 activation and neuroinflammation by 15-HC in vivo, we studied TLR2-deficient mice. We administered 15-HC to TLR2-deficient and wild-type mice during the induction of EAE (induced with an emulsion of MOG(35–55) and CFA) and then again intraperitoneally on days 4, 7 and 10 as described23; we then monitored the mice for clinical signs of disease. The administration of 15-HC resulted in significant worsening of EAE in wild-type mice but not in TLR2-deficient mice (Fig. 6c and Supplementary Table 2b). CD11b+ CNS cells isolated from 15-HC-treated TLR2-deficient mice showed no upregulation of PARP-1 activity (Fig. 6d). Furthermore, treatment of TLR2-deficient mice with 15-HC did not result in upregulation of the expression of CCL2, iNOS or TNF by CD11b+ CNS cells (Fig. 6e). We detected no differences in the encephalitogenic recall response to MOG(35–55) or anti-CD3 (data not shown). Thus, in vivo, 15-HC might contribute to neuropathology by activating microglia, CNS-infiltrating macrophages and astrocytes in a TLR2-dependent way.
Discussion
In this work we have investigated the function of 15-oxysterols in SPMS in humans and secondary progressive EAE in NOD mice. Published studies have detected higher serum and cerebrospinal fluid concentrations of the oxysterol 7-KC in human and experimental multiple sclerosis16,27, but 7-KC values are in the normal range in the chronic phase of EAE16. We found higher serum concentrations of the 15-oxysterols 15-KA and 15-KE in RRMS and further upregulation of their serum concentrations in SPMS. Notably, the serum concentrations of 15-HC were upregulated only in the progressive phase of human SPMS and NOD EAE, which suggests that stage-specific processes control the production of 15-HC. The transition to SPMS has been linked to extensive microglia activation28. As the degradation of myelin by microglia is an important source of oxysterols16,29, we speculate that changes in the CNS innate immune response alter the local catabolism of cholesterol and favor the generation of 15-HC in SPMS. Regardless of the specific mechanisms that lead to the greater abundance of 15-HC in the serum in SPMS, its association with human SPMS and secondary progressive EAE and its contribution to disease pathogenesis by a TLR2–PARP-1 pathway suggest that higher serum concentrations of 15-HC identify patients in the progressive phase of the disease. Because 15-HC can be detected in serum, the measurement of 15-HC can be easily applied to clinical samples.
Secondary progressive EAE recapitulates several features of SPMS. As in multiple sclerosis, a self-limited acute neurological syndrome is followed by a phase of irreversible progressive neurological impairment in secondary progressive EAE17. This clinical course resembles the progressive accumulation of neurological disability that follows the relapsing-remitting phase in patients with SPMS2. The spinal cords of NOD mice are characterized by pronounced axonal loss and demyelination during the progressive phase of EAE but not during the acute phase of EAE17. These pathological findings resemble the accumulation of neuronal loss found in SPMS5. Moreover, both SPMS27 and the progressive phase of NOD EAE17 are characterized by extensive microglial activation. Nonetheless, although NOD EAE resembles SPMS, differences do exist. The progressive phase of NOD EAE follows a relatively short period of acute CNS inflammation17, whereas the clinical transition to SPMS usually follows a relapsing-remitting phase that generally spans 10–20 years (ref. 2). Furthermore, the progressive phase of NOD EAE is linked to prominent axonal loss in the spinal cord but not in the brain17, whereas there is brain axonal loss in SPMS. Thus, although NOD EAE does not mimic SPMS in all ways, it models salient features of the clinical course and spinal cord pathology.
The administration of 15-HC led to worsening of EAE in a PARP-1-and TLR2-dependent way, concomitant with higher expression of TNF, iNOS and CCL2 by CD11b+ CNS cells. In contrast, treatment with the PARP-1-specific inhibitor AIQ suppressed the progressive phase of NOD EAE and was linked to lower abundance of TNF, iNOS and CCL2 in the CNS. AIQ and 15-HC, however, did not affect the adaptive encephalitogenic immune response. Together these results suggest that activation of PARP-1 by 15-HC contributes to the progression of multiple sclerosis by acting on the innate immune response in the CNS. Indeed, given the higher 15-HC serum concentrations and monocyte PARP-1 activity found in human SPMS, our data suggest that 15-HC might contribute to the extensive activation of microglia that characterizes human with SPMS and secondary progressive EAE17,28.
The oxysterol 7-KC can activate microglial PARP-1 by a DNA damage–dependent pathway16. Although we found that 15-HC can use the same pathway to activate PARP-1 in a cell-free system, our results have shown that TLR2 was required for the activation of PARP-1 by 15-HC in living cells in vitro and in vivo. Our experiments did not address whether 15-HC binds directly to TLR2 or if it signals through TLR2 by an indirect mechanism. Nevertheless, our experiments suggest that the DNA damage–dependent pathway of PARP-1 activation is unlikely to be important in activation of the CNS innate immune response by 15-HC during the course of neuroinflammation. Additional studies are needed to define the function in vivo by TLRs and other surface receptors in the activation of the CNS innate immune response by oxysterols other than 15-HC.
Astrocytes, microglia and CNS-infiltrating monocytes influence the course of multiple sclerosis through several mechanisms that include the direct destruction of neurons by cytokines19,20 or nitric oxide21, the recruitment of peripheral cells to the CNS22 and the secretion of cytokines that modulate peripheral adaptive immunity30. We found that activation of microglia, macrophages and astrocytes by 15-HC resulted in the production of TNF, nitric oxide and CCL2. TNF and nitric oxide have neurotoxic activities that might contribute directly to axonal degeneration and disease progression19–21. The ensuing myelin degradation would result in the production of new 15-HC and additional axonal destruction, facilitating the perpetuation of innate neuroinflammation. This cycle might be further amplified by the production of CCL2, leading to the recruitment of additional proinflammatory cells to the CNS22.
Our observations suggest that dysregulated CNS innate immunity promotes neurodegeneration and the accumulation of disability in SPMS. Thus, new therapeutic approaches for SPMS should target both specific mechanisms of neurodegeneration, such as glutamate toxicity17,31,32, and the innate immune response that supports them. Here we have shown that PARP-1 is a potential therapeutic target for halting the CNS innate immune response that promotes axonal loss and the accumulation of disability in human SPMS. Because the progressive phase of multiple sclerosis causes the greatest disability and is the least responsive to therapy3,4, investigation of the processes that drive SPMS is a priority in multiple sclerosis research.
Methods
Methods and any associated references are available in the online version of the paper at http://www.nature.com/natureimmunology/.
Supplementary Material
Acknowledgments
We thank A.S. Basso (Universidade Federal de São Paulo) for discussions, and M. Oukka (Harvard Medical School) for TLR2-deficient mice. Supported by the US National Institutes of Health (AI435801 and NS38037), the US National Multiple Sclerosis Society (PP1289 to H.L.W.) and the Human Frontiers of Science Program Organization (F.J.Q.).
Footnotes
Author Contributions: M.F.F and F.J.Q. did experiments and analyzed data; R.G. provided purified human monocytes; G.I and M.L. contributed samples; and M.F.F., F.J.Q. and H.L.W. wrote the manuscript.
Accession code UCSD-Nature Signaling Gateway (http://www.signaling-gateway.org): A002294.
Note: Supplementary information is available on the Nature Immunology website.
Reprints and permissions information is available online at http://npg.nature.com/reprintsandpermissions/.
References
- 1.McFarland HF, Martin R. Multiple sclerosis: a complicated picture of autoimmunity. Nat Immunol. 2007;8:913–919. doi: 10.1038/ni1507. [DOI] [PubMed] [Google Scholar]
- 2.Compston A, Coles A. Multiple sclerosis. Lancet. 2008;372:1502–1517. doi: 10.1016/S0140-6736(08)61620-7. [DOI] [PubMed] [Google Scholar]
- 3.Rovaris M, et al. Secondary progressive multiple sclerosis: current knowledge and future challenges. Lancet Neurol. 2006;5:343–354. doi: 10.1016/S1474-4422(06)70410-0. [DOI] [PubMed] [Google Scholar]
- 4.Lopez-Diego RS, Weiner HL. Novel therapeutic strategies for multiple sclerosis—a multifaceted adversary. Nat Rev Drug Discov. 2008;7:909–925. doi: 10.1038/nrd2358. [DOI] [PubMed] [Google Scholar]
- 5.Trapp BD, Nave KA. Multiple sclerosis an immune or neurodegenerative disorder? Annu Rev Neurosci. 2008;31:247–269. doi: 10.1146/annurev.neuro.30.051606.094313. [DOI] [PubMed] [Google Scholar]
- 6.Trapp BD, et al. Axonal transection in the lesions of multiple sclerosis. N Engl J Med. 1998;338:278–285. doi: 10.1056/NEJM199801293380502. [DOI] [PubMed] [Google Scholar]
- 7.Balashov KE, Comabella M, Ohashi T, Khoury SJ, Weiner HL. Defective regulation of IFNγ and IL-12 by endogenous IL-10 in progressive MS. Neurology. 2000;55:192–198. doi: 10.1212/wnl.55.2.192. [DOI] [PubMed] [Google Scholar]
- 8.Balashov KE, Smith DR, Khoury SJ, Hafler DA, Weiner HL. Increased interleukin 12 production in progressive multiple sclerosis induction by activated CD4+ T cells via CD40 ligand. Proc Natl Acad Sci USA. 1997;94:599–603. doi: 10.1073/pnas.94.2.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Comabella M, et al. Elevated interleukin-12 in progressive multiple sclerosis correlates with disease activity and is normalized by pulse cyclophosphamide therapy. J Clin Invest. 1998;102:671–678. doi: 10.1172/JCI3125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Karni A, Koldzic DN, Bharanidharan P, Khoury SJ, Weiner HL. IL-18 is linked to raised IFN-γ in multiple sclerosis and is induced by activated CD4+ T cells via CD40–CD40 ligand interactions. J Neuroimmunol. 2002;125:134–140. doi: 10.1016/s0165-5728(02)00018-8. [DOI] [PubMed] [Google Scholar]
- 11.Karni A, et al. Innate immunity in multiple sclerosis: myeloid dendritic cells in secondary progressive multiple sclerosis are activated and drive a proinflammatory immune response. J Immunol. 2006;177:4196–4202. doi: 10.4049/jimmunol.177.6.4196. [DOI] [PubMed] [Google Scholar]
- 12.Weiner HL. A shift from adaptive to innate immunity a potential mechanism of disease progression in multiple sclerosis. J Neurol. 2008;255:3–11. doi: 10.1007/s00415-008-1002-8. [DOI] [PubMed] [Google Scholar]
- 13.Quintana FJ, et al. Functional immunomics: microarray analysis of IgG autoantibody repertoires predicts the future response of mice to induced diabetes. Proc Natl Acad Sci USA. 2004;101:14615–14621. doi: 10.1073/pnas.0404848101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Robinson WH, et al. Protein microarrays guide tolerizing DNA vaccine treatment of autoimmune encephalomyelitis. Nat Biotechnol. 2003;21:1033–1039. doi: 10.1038/nbt859. [DOI] [PubMed] [Google Scholar]
- 15.Quintana FJ, et al. Antigen microarrays identify unique serum autoantibody signatures in clinical and pathologic subtypes of multiple sclerosis. Proc Natl Acad Sci USA. 2008;105:18889–18894. doi: 10.1073/pnas.0806310105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Diestel A, et al. Activation of microglial poly(ADP-ribose)-polymerase-1 by cholesterol breakdown products during neuroinflammation: a link between demyelination and neuronal damage. J Exp Med. 2003;198:1729–1740. doi: 10.1084/jem.20030975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Basso AS, et al. Reversal of axonal loss and disability in a mouse model of progressive multiple sclerosis. J Clin Invest. 2008;118:1532–1543. doi: 10.1172/JCI33464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Suto MJ, Turner WR, Arundel-Suto CM, Werbel LM, Sebolt-Leopold JS. Dihydroisoquinolinones the design and synthesis of a new series of potent inhibitors of poly(ADP-ribose) polymerase. Anticancer Drug Des. 1991;6:107–117. [PubMed] [Google Scholar]
- 19.Jack C, Ruffini F, Bar-Or A, Antel JP. Microglia and multiple sclerosis. J Neurosci Res. 2005;81:363–373. doi: 10.1002/jnr.20482. [DOI] [PubMed] [Google Scholar]
- 20.Nair A, Frederick TJ, Miller SD. Astrocytes in multiple sclerosis a product of their environment. Cell Mol Life Sci. 2008;65:2702–2720. doi: 10.1007/s00018-008-8059-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Smith KJ, Lassmann H. The role of nitric oxide in multiple sclerosis. Lancet Neurol. 2002;1:232–241. doi: 10.1016/s1474-4422(02)00102-3. [DOI] [PubMed] [Google Scholar]
- 22.Charo IF, Ransohoff RM. The many roles of chemokines and chemokine receptors in inflammation. N Engl J Med. 2006;354:610–621. doi: 10.1056/NEJMra052723. [DOI] [PubMed] [Google Scholar]
- 23.Kanter JL, et al. Lipid microarrays identify key mediators of autoimmune brain inflammation. Nat Med. 2006;12:138–143. doi: 10.1038/nm1344. [DOI] [PubMed] [Google Scholar]
- 24.Chan SM, Ermann J, Su L, Fathman CG, Utz PJ. Protein microarrays for multiplex analysis of signal transduction pathways. Nat Med. 2004;10:1390–1396. doi: 10.1038/nm1139. [DOI] [PubMed] [Google Scholar]
- 25.Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. doi: 10.1016/j.cell.2006.02.015. [DOI] [PubMed] [Google Scholar]
- 26.Cohen-Armon M, et al. DNA-independent PARP-1 activation by phosphorylated ERK2 increases Elk1 activity: a link to histone acetylation. Mol Cell. 2007;25:297–308. doi: 10.1016/j.molcel.2006.12.012. [DOI] [PubMed] [Google Scholar]
- 27.Leoni V, Lutjohann D, Masterman T. Levels of 7-oxocholesterol in cerebrospinal fluid are more than one thousand times lower than reported in multiple sclerosis. J Lipid Res. 2005;46:191–195. doi: 10.1194/jlr.C400005-JLR200. [DOI] [PubMed] [Google Scholar]
- 28.Kutzelnigg A, et al. Cortical demyelination and diffuse white matter injury in multiple sclerosis. Brain. 2005;128:2705–2712. doi: 10.1093/brain/awh641. [DOI] [PubMed] [Google Scholar]
- 29.Bongarzone ER, Pasquini JM, Soto EF. Oxidative damage to proteins and lipids of CNS myelin produced by in vitro generated reactive oxygen species. J Neurosci Res. 1995;41:213–221. doi: 10.1002/jnr.490410209. [DOI] [PubMed] [Google Scholar]
- 30.Luo J, et al. Glia-dependent TGF-β signaling, acting independently of the TH17 pathway, is critical for initiation of murine autoimmune encephalomyelitis. J Clin Invest. 2007;117:3306–3315. doi: 10.1172/JCI31763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Pitt D, Werner P, Raine CS. Glutamate excitotoxicity in a model of multiple sclerosis. Nat Med. 2000;6:67–70. doi: 10.1038/71555. [DOI] [PubMed] [Google Scholar]
- 32.Smith T, Groom A, Zhu B, Turski L. Autoimmune encephalomyelitis ameliorated by AMPA antagonists. Nat Med. 2000;6:62–66. doi: 10.1038/71548. [DOI] [PubMed] [Google Scholar]
- 33.Quintana FJ, et al. Control of Treg and TH17 cell differentiation by the aryl hydrocarbon receptor. Nature. 2008;453:65–71. doi: 10.1038/nature06880. [DOI] [PubMed] [Google Scholar]
- 34.Meng G, et al. Antagonistic antibody prevents toll-like receptor 2-driven lethal shock-like syndromes. J Clin Invest. 2004;113:1473–1481. doi: 10.1172/JCI20762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Qi HY, Shelhamer JH. Toll-like receptor 4 signaling regulates cytosolic phospholipase A2 activation and lipid generation in lipopolysaccharide-stimulated macrophages. J Biol Chem. 2005;280:38969–38975. doi: 10.1074/jbc.M509352200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.