

Abstract
Plasmodium faliciparum causes 1–2 million deaths annually, yet current drug therapies are compromised by resistance. We previously described potent and selective triazolopyrimidine-based inhibitors of P. falciparum dihydroorotate dehydrogenase (PfDHODH) that inhibited parasite growth in vitro, however they showed no activity in vivo. Here we show that lack of efficacy against P. berghei in mice resulted from a combination of poor plasma exposure, and reduced potency against P. berghei DHODH. For compounds containing naphthyl (DSM1) or anthracenyl (DSM2), plasma exposure was reduced upon repeated dosing. Phenyl-substituted triazolopyrimidines were synthesized leading to identification of analogs with low predicted metabolism in human liver microsomes and which showed prolonged exposure in mice. Compound 21 (DSM74), containing p-trifluoromethylphenyl, suppressed growth of P. berghei in mice after oral administration. This study provides the first proof of concept that DHODH inhibitors can suppress Plasmodium growth in vivo, validating DHODH as a new target for anti-malarial chemotherapy.
Introduction
Malaria remains one of the greatest global health challenges despite intensive efforts to control the disease. 1, 2 It is endemic in 87 countries, putting at risk 2.4 billion people, with the greatest burden found in the tropical regions of the poorest countries in Africa, Asia and Latin America. Plasmodium falciparum is responsible for most of the severe clinical malaria, resulting in 1 – 2 million deaths per year. Effective vaccines have yet to be developed so anti-malaria control programs focus on chemotherapy. While an array of anti-malarial agents have been developed, widespread drug resistance has severely limited the effectiveness of many of these agents. 3 Combination chemotherapy using artemisinin derivatives and longer acting anti-malarials have provided new avenues to combat the disease, 4 however despite these advances the discovery and development of new drugs to treat and prevent malaria remains essential if the global burden of malaria is to be successfully reduced.
The completion of the malaria genome project 5 has motivated the search for novel targets in the parasite. Despite the identification of many essential genes, and extensive efforts to understand the biology of the parasite, very few new validated targets have been identified in malaria. 6 Indeed no new clinically proven targets have emerged since the discovery of the mechanism of action of atovaquone. 7, 8
Pyrimidine biosynthesis provides a significant opportunity for the development of new chemotherapeutic agents against the malaria parasite. While mammalian cells contain enzymes for both de novo biosynthesis and salvage of preformed pyrimidine bases and nucleosides, the parasite relies entirely on de novo synthesis. 9,10 Plasmodium dihydroorotate dehydrogenase (DHODH) is an essential mitochondrial enzyme that catalyzes the flavin mononucleotide-dependent formation of orotic acid, a key step in de novo pyrimidine bioysynthesis. 11,12 The primary function of the mitochondrial electron transport chain in the parasite appears to be to provide oxidized ubiquinone (CoQ) as the physiological oxidant in the DHODH reaction, further demonstrating the importance of DHODH function to parasite growth. 13 Inhibitors of human DHODH are used for the treatment of rheumatoid arthritis, illustrating that small molecule inhibitors of DHODH can have drug-like properties. 14, 15 Finally, both X-ray structural studies of DHODH bound to inhibitors, 16,17 and biochemical studies 18–22 on DHODH from different species, have demonstrated that inhibitors such as 1 (Figure 1A) bind to a species-variable pocket that is adjacent to the flavin cofactor (Figure 1B). These findings provide the structural basis for the identification of species-specific inhibitors. The species-variable site has also been proposed to bind CoQ, however, studies from our lab suggest that the CoQ site does not overlap the inhibitor-binding site. Instead inhibitors block the electron transfer between CoQ, bound at an adjacent site, and the flavin cofactor. 21
Figure 1. Species-variable inhibitor binding site of PfDHODH.

A. Structure of the DHODH inhibitor 2-cyano-3-hydroxy-N-[4-(trifluoromethyl)phenyl]-2-butenamide (A77 1726) 1. B. Amino acid residues within 4.2 Å of the co-crystallized inhibitor 1 are shown. The inhibitor (pink) is displayed showing the Van der Waals surface, the FMN cofactor (yellow) and orotate (pink) are displayed as sticks. Residues in grey are conserved between PfDHODH and hDHODH, while residues in green are variable. The two amino acid residues that differ between PfDHODH and PbDHODH are indicated by *. The figure was generated using PyMol from the file 1TV5.pdb.
We previously identified a potent triazolopyrimidine-based inhibitor 2 ( 5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-naphthalen-2-yl-amine DSM1) of PfDHODH that showed greater than 4,000-fold selectivity for the malarial enzyme when compared to human DHODH, and which showed similarly potent activity against P. falciparum in whole cell assays (EC50 = 80 nM). 22 Despite the potent activity against P. falciparum, we show in this report that these compounds have no measurable activity against P. berghei infection in mice. Using an integrated lead optimization strategy combining medicinal chemistry, enzymology and ADME evaluation we first identified the liabilities of the initial compound series. Based on that data we then designed a modified analog that showed good exposure in vivo after oral dosing, and which was able to suppress parasitemia in the P. berghei mouse model. These data represent the first report of a DHODH inhibitor with anti-malarial activity in an animal model, further validating DHODH as a promising new target for the development of much needed new anti-malarial agents.
Results
Compounds 2 and 3 do not suppress parasite levels in the P. berghei mouse model
The two most potent compounds identified in our initial SAR analysis of the triazolopyrimidine series contained a naphthyl 2 or anthracenyl 3 (5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-anthracen-2-yl-amine DSM2) functional group22 (Table 1). In order to determine if these compounds showed activity against P. berghei in vivo, we tested these compounds by dosing orally at 100 mg/kg/dose every 4 hours × 10 doses, then every 6 hours × 6 doses. There were no major ill effects in the animals observed from the compound administration, however neither compound had any impact on parasite blood levels (data not shown). Based on these results a systematic series of studies were undertaken to identify the liabilities of the initial compounds to circumvent the problems.
Table 1.
Activity of triazolopyrimidine-based derivatives against DHODH and P. falciparum
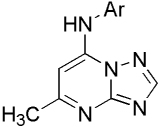 | ||||||
|---|---|---|---|---|---|---|
| Compd | Name | Ar | IC50(µM)b PfDHODH |
IC50(µM)b PbDHODH |
IC50(µM)b hDHODH |
EC50(µM)c Pf 3D7 cells |
| 2 a | DSM1 | 2-Naphthyl | 0.047 ± 0.02 | 0.23 ± 0.04 | >200 | 0.076 ± 0.04b |
| 3 a | DSM2 | 2-Anthracenyl | 0.056 ± 0.02 | 3.7 ± 0.14 | >200 | 0.19 ± 0.12b |
| 4a | DSM12 | Ph | >200 | >10 | >200 | >100 |
| 8 | DSM106 | 2-F-Ph | >100 | >100 | ND | >50 |
| 9 | DSM95 | 2-Cl-Ph | >100 | >100 | ND | 30 ± 0.01 |
| 10 | DSM93 | 2-CH3-Ph | >100 | >100 | ND | >50 |
| 11 | DSM91 | 2-CF3-Ph | >100 | >100 | ND | ND |
| 12 | DSM92 | 2-CN-Ph | >100 | >100 | ND | 25 ± 0.002 |
| 13 | DSM72 | 3-F-Ph | 9.2 ± 0.2 | >100 | ND | 34 ± 0.03 |
| 14 | DSM75 | 3-Cl-Ph | 1.4 ± 0.13 | 31 ± 3.2 | >100 | 8.5 ± 0.002 |
| 15 | DSM94 | 3-CH3-Ph | 4.9 ± 0.52 | >100 | ND | 37 ± 0.02 |
| 16 | DSM73 | 3-CF3-Ph | 14 ± 0.0 | 55 ± 10 | ND | 19 ± 0.01 |
| 17 | DSM131 | 4-F-Ph | 19 ± 2.0 | >100 | ND | 50 ± 1.1 |
| 18 | DSM89 | 4-Cl-Ph | 1.6 ± 0.24 | 5.7 ± 0.9 | >100 | 12 ± 0.002 |
| 19 | DSM98 | 4-Br-Ph | 0.78 ± 0.25 | 1.3 ± 0.3 | >100 | 4.2 ± 0.07 |
| 20 | DSM97 | 4-CH3-Ph | 4.2 ± 0.35 | 16 ± 2.5 | ND | 6.4 ± 0.004 |
| 21 | DSM74 | 4-CF3-Ph | 0.28 ± 0.02 | 0.38 ± 0.02 | >100 | 0.34 ± 0.04b |
| 22 | DSM100 | 4-OCH3-Ph | 4.6 ± 0.14 | >50 | ND | 14 ± 0.005 |
| 23 | DSM87 | 4-OCF3-Ph | 1.2 ± 0.21 | 4 ± 0.7 | >100 | 3.8 ± 0.01 |
| 24 | DSM88 | 4-OCHF2-Ph | 2.8 ± 0.28 | 25 ± 2.4 | ND | 8.2 ± 0.01 |
| 25 | DSM99 | 4-NO2-Ph | 4.3 ± 0.14 | 3.7 ± 0.7 | ND | ND |
| 26 | DSM69 | 4-C6H5-Ph | 5.3 ± 0.8 | ND | ND | >50 |
| 27 | DSM84 | 4-C6H5CH2-Ph | 2.2 ± 0.32 | >100 | ND | 5.0 ± 0.0 |
| 28 | DSM110 | 2,3-Di-F-Ph | 39 ± 7.5 | >100 | ND | ND |
| 29 | DSM107 | 2,5-Di-F-Ph | >50 | >100 | ND | >50 |
| 30 | DSM108 | 2,6-Di-F-Ph | >100 | >100 | ND | >50 |
| 31 | DSM109 | 2,3,4-Tri-F-Ph | 29 ± 4.4 | >100 | ND | >50 |
| 32 | DSM111 | 2,4,5-Tri-F-Ph | 17 ± 3.6 | >100 | ND | >50 |
| 33 | DSM120 | 2,4,6-Tri-F-Ph | >100 | >100 | ND | ND |
| 34 | DSM132 | 2,3,5,6-Tetra-F-Ph | >100 | >100 | ND | >50 |
| 35 | DSM133 | 2,3,4,5,6-Penta-F-Ph | >100 | >100 | ND | >50 |
| 36 | DSM85 | 2-F-4-CH3-Ph | 27 ± 2.8 | >100 | ND | 35 ± 0.002 |
| 37 | DSM112 | 3,4-Di-F-Ph | 8.0 ± 1.1 | 10 ± 5.6 | ND | >50 |
| 38 | DSM121 | 3,5-Di-F-Ph | 9.4 ± 1.2 | 21 ± 3.5 | ND | ND |
| 39 | DSM126 | 3,5-Di-CF3-Ph | >100 | >100 | ND | >5 |
| 40 | DSM82 | 3-CH3-4-F-Ph | 4.6 ± 0 | 21 ± 1.4 | ND | 8.7 ± 0.001 |
| 41 | DSM124 | 3-CF3-4-Cl-Ph | 0.8 ± 0.28 | 2.4 ± 0.2 | >100 | >5 |
| 42 | DSM128 | 3-CF3-4-Br-Ph | 0.45 ± 0.07 | 1.5 ± 0.4 | >100 | 2.6 ± 0.002 |
| 43 | DSM122 | 3-CF3-4-CH3-Ph | 1.55 ± 0.49 | 7.3 ± 1.2 | >100 | 8.1 ± 0.1 |
| 44 | DSM123 | 3,4-Di-CH3-Ph | 0.35 ± 0.21 | 4.0 ± 0.4 | >100 | 4.8 ± 2.3b |
| 45 | DSM81 | 3-CF3-4-CN-Ph | 4.9 ± 0.7 | 11 ± 1.2 | ND | 8.8 ± 0.001 |
| 46 | DSM83 | 3-F-4-CH3-Ph | 0.86 ± 0.09 | 3.2 ± 0.4 | >100 | 3.6 ± 0.004 |
| 47 | DSM125 | 3-F-4-CF3-Ph | 0.077 ± 0.03 | 0.26 ± 0.05 | >100 | 1.3 ± 0.1b |
Compounds reported previously22. ND, not determined.
standard error of the mean.
standard error of the fit.
Species differences in inhibitor potency between PfDHODH and PbDHODH
Previous mutational studies localized the binding-site of the triazolopyrimidine inhibitor series to the species-variable pocket that is adjacent to the flavin cofactor 22 (Figure 1B), providing a structural basis for the observed species selectivity in binding. Comparison of the amino acid sequence of this binding site between the human malaria PfDHODH and the mouse malaria PbDHODH identifies two amino acid sequence differences between the enzymes from these two malaria parasites (M536 and G181 in PfDHODH are V536 and S181 in PbDHODH). In order to determine if these sequence variations contribute to differences in binding affinity of the triazolopyrimidine compound series between PfDHODH and PbDHODH, we cloned and expressed PbDHODH from our standard vector and determined the IC50’s for 2 and 3 (Table 1). PbDHODH is significantly less sensitive to inhibition by both compounds; the IC50 for compounds 2 and 3 are 5.3 - and 66 - fold higher for PbDHODH than for PfDHODH. These differences in inhibitor potency explain, in part, the lack of efficacy of 2 and 3 in the P. berghei model.
Compounds 2 and 3 showed reduced plasma exposure upon repeat dosing
In order to determine if poor plasma exposure contributed to the lack of efficacy of 2 and 3 in the P. berghei model we measured plasma concentrations in uninfected mice after oral dosing of each compound (Figure 2). After a single oral dose (50 mg/kg) plasma levels of 2 and 3 reached the low micromolar range (2 – 3 µM), with compound 2 concentrations reaching a maximum at 30 min after dosing and rapidly declining by 240 min after dosing. Compound 3 showed more prolonged exposure after a single oral dose and concentrations were maintained between 1 – 3 µM over a period of 240 min. In contrast, when mice were treated with compound 2 or 3 orally for a period of 2 to 4 days, the plasma concentrations measured after the last dose were significantly reduced relative to the concentrations measured after a single dose of 2 or 3 (Figure 2). For 2 the effects were particularly striking, where plasma levels at 240 min on day 4 where nearly undetectable. No adverse reactions were observed following the doses used in this study. These data suggest that 2 and 3 may either be metabolic inducers, or they may induce efflux transport proteins thus reducing oral drug absorption in mice. Finally, while plasma concentrations of 3 were higher than 2, the observed plasma levels (~1 µM) are clearly insufficient to clear parasites from the blood of the infected mice, given the significantly higher IC50 for 3 against PbDHODH (IC50 = 3.7 µM for PbDHODH vs 0.056 µM for PfDHODH). Thus we conclude that the lack of efficacy of 2 in the P. berghei model results primarily from reduced exposure upon repeat administration, while for compound 3 reduced inhibitor potency for PbDHODH compared to PfDHODH is likely to be the primary contributing factor.
Figure 2. Plasma exposure of 2 and 3 after repeat dosing in mice.

Control mice (designated C) were administered blank vehicle for 4 days, followed by a single oral dose of 2 (panel A) or 3 (panel B) at 50 mg/kg (n=3 mice per time point) on day 5. For the repeat dosing groups, an oral dose of 50 mg/kg was administered 1x daily using two schedules: a) compound was administered on days 3, 4 and 5 with vehicle administered on days 1 and 2, or b) compound was administered on days 1–5. Plasma concentrations were measured at 30 and 240 min after the day 5 dosing for all groups.
Synthesis and evaluation of substituted phenyl derivatives as naphthyl replacements
Cytochrome P450 (CYP) induction has been previously demonstrated for structurally similar compounds containing the naphthyl functional group. 23 Thus we hypothesized that the reduced plasma concentrations of 2 upon repeat administration could be due to the naphthyl functional group, although we can not rule out other explanations such as induction of efflux transporters. We initiated a chemistry effort to replace the naphthyl group with a series of commercially available aromatic amines. Components of the Topliss series24, 25 were systematically evaluated to test the electronic and steric requirements for activity at this position. Compounds were synthesized as shown in Scheme 1. In total, 40 new compounds were synthesized with substituted phenyl groups replacing the naphthyl moiety (Table 1). These compounds were tested for activity against three species of recombinant enzyme (PfDHODH, PbDHODH and hDHODH) and in whole cell P. falciparum parasite assays. As previously reported, the unsubstituted phenyl compound (4) is completely inactive. 22 Single position ortho substituted phenyl compounds (8–12) are likewise inactive, while meta substitutions (13–16) significantly improve activity over the unsubstituted phenyl derivative. Compounds that contain ortho substitution in combination with substitutions at the meta or para position typically have poor activity, although it is improved over ortho substitution alone (28–36). The most active single substituted compounds in the series contained para substituents (17–27), with larger hydrophobic residues being preferred: CF3, > Br>OCF3>CH3, NO2, F, Cl. Compounds 21 (DSM74) (IC50 PfDHODH = 0.3 µM) was the most active in this set. Finally, combinations of para and meta substitutions (40–47) yielded compounds with the best activity against PfDHODH as exemplified by 47 (DSM125) (IC50 PfDHODH = 0.077 µM), which has similar potency on the enzyme to the original lead compound 2. All compounds tested were inactive against hDHODH, showing that the selectivity relative to the host enzyme is maintained upon replacement of the naphthyl moiety with a substituted phenyl ring. As was previously observed, most of the new compounds in the series also showed preferential binding towards PfDHODH in comparison to PbDHODH. The one exception to this was compound 21, which showed equal potency against PfDHODH and PbDHODH.
Scheme 1.

Synthetic strategy for the compounds 8–47a.
a Reagents and conditions:(i) CH3COCH2CO2C2H5, AcOH, 3.5 h, reflux, 55%.; (ii) POCI3, 45 min., reflux, 58%.; (iii) Ar-NH2, EtOH, 2–30 h, room temp., 80–87%.
A good correlation was observed over 3 log units of activity between activity on PfDHODH and activity in whole cell P. falciparum assays (Figure 3; r2 = 0.73, slope = 0.78 ± 0.087). This analysis included data from Table 1 as well as our previously reported triazolopyrimidine derivatives. 22 Additionally, without exception compounds that were inactive on the enzyme were also inactive against P. falciparum in the whole cell assay (IC50 and EC50’s >100 µM).
Figure 3. Comparison of the series activity on PfDHODH vs P. falciparum whole cell assays.

The log of the PfDHODH IC50 data are plotted versus the log of the P. falciparum EC50 data (in micromolar). The plotted data include compounds 2, 3, 12, 13, 16, 18, 21, 24, 27, 36, 40, 42 and 47 described in Table 1 and those previously reported from the series (compounds 8–12 and 14–17 from 22). Compounds for which solubility limits prevented the quantitative determination of either the IC50/EC50 have been left off the plot. Data were fitted by linear regression analysis where r2 = 0.73, slope = 0.78 ± 0.087.
Metabolic analysis of the substituted phenyl derivatives
In vitro metabolism data in human liver microsomes were collected on a subset of the active compounds in Table 1 to assess their metabolic stability (Table 2). Based on the measured in vitro degradation 2 and 3 were found to have high rates of metabolism and high predicted hepatic extraction ratios (i.e. the fraction of compound cleared by the liver on the first pass). Both 2 and 3 showed evidence for putative hydroxylation products (P+16) while in addition putative hydroxylation followed by glucuronidation (P+16+176) was also observed for 2. In contrast, compounds with halogenated side chains in either the para or meta position showed a reduced rate of metabolism and only a single metabolite was identified (putative hydroxylation product), suggesting that they will have significantly better metabolic stability than the lead compound 2. Compounds that show the greatest metabolic stability (21, 45, 47) contain para or meta substituents that include a halogen, and in the case of 45 and 47 no metabolites were detected. While the potential for species differences in metabolism can not be ruled out given the available data, the increased metabolic stability in human microsomes was a key characteristic for compound progression.
Table 2.
In vitro human microsomal metabolism
| Compd | Degradation half-life (min) |
In vitro ClInt (mL/min/mg protein) |
Predicted EH | Putative metabolites |
|---|---|---|---|---|
| 2 | 18 | 0.096 | 0.84 | P+16; P+16+18; P+16+176 |
| 3 | 43 | 0.037 | 0.70 | P+16 |
| 13 | 97 | 0.018 | 0.50 | P+16 |
| 14 | 45 | 0.038 | 0.68 | P+16 |
| 16 | 60 | 0.029 | 0.62 | P+16 |
| 21 | 230 | 0.0075 | 0.29 | P+16 |
| 40 | 56 | 0.031 | 0.64 | P+16 |
| 41 | 130 | 0.013 | 0.42 | P+16 |
| 42 | 130 | 0.014 | 0.43 | P+16 |
| 43 | 19 | 0.093 | 0.84 | P+16 |
| 44 | 23 | 0.077 | 0.81 | P+16; P+32 |
| 45 | 280 | 0.0062 | 0.26 | None detected |
| 46 | 36 | 0.096 | 0.73 | P+16 |
| 47 | 360 | 0.0048 | 0.21 | None detected |
EH was estimated based on the measured rate of degradation of the test compound in vitro.
In vivo studies in mice with 21 were undertaken to determine if it would show better plasma exposure than 2. Mice were dosed orally at a single dose of either 20 mg/kg or 50 mg/kg (Figure 4A). Compound 21 reached peak plasma concentrations of 7.9 and 27.6 µM (2.3 and 8.1 µg/mL, respectively), respectively after these doses. Plasma concentrations were maintained above 1 µM (0.29 µg/mL) for 16 h after the 50 mg/kg dose which represents a significant improvement over the exposure observed for for 2 or 3. No adverse reactions were observed following oral administration of 21 at the doses used in this study. The apparent half-life of 21 was 1.8 h at 20 mg/kg and 3 h at 50 mg/kg, however these values are approximations as the terminal phase may not have been fully defined by the available data. Some evidence of nonlinear (dose-dependent) pharmacokinetics for 21 was observed in a number of PK parameters: Cmax increased 3.5-fold for a 2.5-fold increase in dose (from 7.9 µM or 2.3 µg/mL at 20 mg/kg to 27.6 µM or 8.1 µg/mL at 50 mg/kg), and the AUC0-last, increased 4.4-fold for a 2.5-fold increase in dose (from 2690 µM.min or 13.1 µg.h/mL at 20 mg/kg to 11757 µM.min or 57.5 µg.h/mL at 50 mg/kg).
Figure 4. In vivo mouse efficacy studies for compound 21.

A. Plasma concentrations after a single oral dose (20 mg/kg or 50 mg/kg) of 21 in mice. Peak plasma concentrations were 7.9 and 27.6 µM (2.3 and 8.1 µg/mL) and the apparent half-life was 1.8 h and 3 h at 20 mg/kg and 50 mg/kg, respectively. B. Efficacy in the standard P. berghei mouse model. Mice were infected with parasites on day 0, and compound 21 dosing began on day 1 after the presence of parasites was established. Dosing was by the oral route at either 50 mg/kg q.d. for 4 days (squares, solid lines) or 50 mg/kg b.i.d. for 4 days (triangles, solid lines) (days 1–4). No drug control data for each group is displayed with a broken line.
In vivo efficacy data for 21 in the P. berghei mouse model
The combined observations that 21 showed good plasma exposure in mice and that it is equally potent against PfDHODH and PbDHODH, suggested that this compound might be suitable for a proof of concept experiment to demonstrate in vivo efficacy of a DHODH inhibitor. Further, the IC50 on the parasite enzymes (0.3 µM) was virtually identical to the EC50 measured in whole cell P. falciparum assays (Table 1). Mice were infected with P. berghei by IP injection 1 day before dosing with compound. Parasitemia was confirmed in all mice before dosing began. Mice were dosed orally with either vehicle, or with 50 mg/kg 21 q.d. (once daily) or b.i.d. (twice daily) for 4 days, resulting in 4 or 8 total doses respectively (Figure 4B). Both dosage schemes resulted in significant suppression of parasite growth. The twice a day dosing scheme provided better parasite suppression than the single dose per day regiment (95% parasite suppression vs 71% suppression, respectively, on day 5 post infection). The greater efficacy observed for b.i.d. dosing is consistent with the observation that by 15 h after a single dose of 21 (Fig. 4A) plasma concentrations of 21 are less than 10-fold above the IC50 measured in the in vitro whole cell P. falciparum parasite assay (Table 1). However the lack of a reliable whole cell P. berghei assay makes unambiguous analysis of plasma levels vs parasite EC50 difficult. Despite the finding that the IC50 for 21 on P. berghei and P. falciparum DHODH are similar, it remains possible that P. berghei cells are less sensitive to 21 than P. falciparum. No adverse reactions were observed as a result of 21 administration at the doses used in the study.
Discussion
The development of new anti-malarial chemotherapies is needed to combat the formidable challenge of drug resistance that continues to develop against existing drugs. The relatively small number of validated targets in malaria limits the identification of new chemical entities. The de novo pyrimidine biosynthetic pathway, and in particular PfDHODH, has been the focus of significant research effort to identify new anti-malarial agents, however full validation of the target has been lacking.18, 19, 22, 26, 27 In this report we demonstrate for the first time that PfDHODH inhibitors can inhibit the growth of malaria parasites in animals, providing strong validation of PfDHODH as a new target for the development of novel anti-malarial compounds.
The initial triazolopyrimidine-based lead compound 2 identified from our high-throughput screen showed excellent potency against both PfDHODH and P. falciparum in culture, however it showed no activity in the P. berghei mouse model. This compound was shown to exhibit reduced exposure in mice upon repeat administration most likely contributing to the lack of in vivo efficacy. In order to overcome this liability we synthesized a series of new derivatives with naphthyl replacements, and within this series we identified a number of analogs with similar potency to 2. These compounds showed excellent correlation between the activity against PfDHODH and P. falciparum cells in vitro. These data provide convincing evidence that the mechanism of cell killing by the triazolopyrimidine compound series is mediated by inhibition of PfDHODH.
The substitution of the naphthyl moiety with a substituted phenyl containing trifluoromethyl at the para position 21 led to a compound that was more metabolically stable in human liver microsomes in vitro, and provided significantly higher plasma concentrations in vivo in mice. Compound 21 exhibited good plasma exposure after oral dosing in mice, with significant plasma concentrations remaining for a sufficient time to allow once daily or twice daily dosing to suppress parasite growth. The efficacy of 21 was further enhanced by the similar potency of 21 against both PfDHODH and PbDHODH, allowing the P. berghei mouse model to be used for evaluation of this compound.
Comparison of the activity of the triazolopyrimidine-based series (Table 1 and Table 2) between enzymes from the two Plasmodium species, P. falciparum and P. berghei, demonstrated that there is considerable species selectivity of binding with most inhibitors showing 7 – 70-fold greater potency against the P. falciparum enzyme. This difference can be traced to the observation that two amino acids in the species-specific inhibitor-binding site are variable between these two Plasmodium species enzymes (Figure 1B). We conclude that the standard P. berghei mouse model will not always be suitable for lead optimization programs to develop DHODH inhibitors for humans. These data underscores the importance of developing additional in vivo models for these studies.
In conclusion, we have identified triazolopyrimidine-based PfDHODH inhibitors with good metabolic stability in human liver microsomes, prolonged plasma exposure in mice, and the ability to inhibit parasite growth in whole animals. Compound 21 has an IC50 in the mid nM range, which is less potent than many effective anti-malarial agents that are used clinically. The challenge remains to identify derivatives that show greater potency, while maintaining metabolic stability, oral bioavailability, and good plasma exposure, thus leading to the identification of compounds that will provide a sterilizing cure of parasite infections in vivo.
Experimental Section
Cloning of P. berghei DHODH
The polymerase chain reaction was used to amplify a truncated soluble form of P.berghei DHODH containing amino acids 130 – 517 from P.berghei strain ANKA genomic DNA (kindly provided by Andy Waters). Primers 5’-TAGTAGGATTACATA TGTATTTTGAATCATATAACCCCG-3’ and 5’-CTCGAGCACTTTCAAAGATTT TGC-3’), which introduce NdeI and XhoI restriction sites, respectively, were used to generate an 1194-bp DNA fragment. The PCR fragment was ligated into Zero Blunt TOPO vector (Invitrogen). QuickChange site-directed mutagenesis kit (Stratagene) was used to remove two internal NdeI sites from P.berghei DHODH using primers 5’-TGGATTATTACCCTATGATACAAG-3’ and 5’-CTT GTA TCA TAG GGT AATAAT CCA-3’ for the first NdeI site, and primers 5’-TGTTTAG TATCGTATGATAAATGG-3’and 5’CCATTTATCATACGATACTAAACA-3’ for the second NdeI site. The NdeI free P.berghei DHODH was digested with NdeI and XhoI and ligated into pET22b expression vector (Novagen) to produce the C-terminal His-tagged P. berghei recombinant protein. The N-terminal start sequence of the coding region is 130-MYFESYN-136.
Protein purification and steady-state kinetic analysis
PfDHODH, PbDHODH, and hDHODH were expressed in their truncated soluble form in E. coli, and purified as previously described. 18, 19 Steady-state kinetic assays were performed as previously described. 18, 19, 28 The reduction of 2,6-dichloroindophenol (DCIP; 0.12 mM) was followed at 600 nm (ε=18.8 mM−1cm−1) using enzyme (ET = 5 – 10 nM) and substrates (0.2 mM L-dihydroorotate and 0.02 mM CoQD) in assay buffer (100 mM HEPES, pH 8.0, 150 mM NaCl, 10% Glycerol, 0.1%Triton) at 20 °C. For potent compounds activity was confirmed by the direct detection of orotic acid at 296 nm (ε296 = 4.30 mM−1cm−1). Data were fitted to Eq. 1 to determine IC50 values. 29
| Eq. 1 |
P. falciparum cell culture
Parasite clones 3D7, FCR3, K1, Dd2, HB3 and D6 were propagated in Gibco-Invitrogen RPMI-1640 supplemented with 20% human type A+ plasma and 2% (w/v) red blood cells. 30 Low-passage L1210 mouse leukemia cells (American Type Culture Collection) were also grown in plasma-supplemented RPMI-1640. Blood products were obtained from Biochemed Services, Virginia. To study inhibition of cell proliferation [3H]-hypoxanthine uptake was measured in drug-treated P. falciparum infected-erythrocytes and L1210 cells described previously.31 Data were fitted to Eq. 2 to determine EC50
| Eq. 2 |
Curve fitting and error analysis
Enzyme and parasite IC50 (EC50) data were determined over a range of inhibitor concentrations using triplicate data points at each concentration. IC50 values were determined using the graphing program Prism (GraphPad).
In vitro human microsomal metabolism
Metabolic stability was evaluated by incubating test compounds (1 µM) individually at 37 °C with pooled human liver microsomes (BD Gentest, Woburn, MA) at a microsomal protein concentration of 0.4 mg/mL. The metabolic reaction was initiated by the addition of an NADPH-regenerating buffer system containing 1 mg/mL NADP, 1 mg/mL glucose-6-phosphate, 1 U/mL glucose-6-phosphate dehydrogenase and 0.67 mg/mL MgCl2, and reactions were quenched at various time points over the incubation period by the addition of ice-cold acetonitrile. The relative loss of parent compound and formation of metabolic products was monitored by LC/MS using a Waters/Micromass QTOF mass spectrometer.
Test compound concentrations were determined by comparison to a calibration curve prepared in microsomal matrix and concentration versus time data were fitted to an exponential decay function to determine the first-order rate constant for substrate depletion. This rate constant was then used to calculate the degradation half-life, in vitro intrinsic clearance (CLint,in vitro), predicted in vivo intrinsic clearance value (CLint), and predicted in vivo hepatic extraction ratio (EH) as previously described, 32 and assuming predominantly hepatic cytochrome P450-mediated clearance in vivo. The scaling parameters used in these calculations included a human hepatic blood flow of 20.7 mL/min/kg, liver mass of 25.7 g liver/kg body weight, and microsomal content of 45 mg microsomal protein/g liver. 33, 34
Mouse pharmacokinetics
All animal studies were performed in accordance with the Australian and New Zealand Council for the Care of Animals in Research and Training Guidelines and the study protocol was approved by the Monash University (Victorian College of Pharmacy) Animal Experimentation Ethics Committee. The pharmacokinetics of 2, 3 and 21 were studied individually in non-fasted male Swiss outbred mice weighing 23 – 33 g. Mice had access to food and water ad libitum throughout the pre- and post-dose sampling period. Compounds were administered orally by gavage (0.1 mL dose volume per mouse) in a suspension of aqueous vehicle (0.5% w/v sodium caboxymethylcellulose, 0.5% v/v benzyl alcohol and 0.4% v/v Tween 80). A single blood sample was collected from each mouse via cardiac puncture at each indicated time point and transferred to heparinised tubes. Blood samples were centrifuged immediately, supernatant plasma was removed and stored at −20 °C until analysis (within 1–2 weeks).
Plasma samples were assayed by LC-MS following protein precipitation with acetonitrile. Chromatography was conducted using a Waters Acquity UPLC. The analysis of 2 was conducted on a Phenomenex Luna C8(2) column (50 × 1 mm) and a Phenomenex Synergi Polar RP column (50 × 1 mm) was used for 3 and 21. Each column was maintained at 40 °C, and a mobile phase consisting of acetonitrile/water containing 0.05% formic acid was delivered via a linear gradient at a flow rate of 0.15 mL/min. Mass spectrometry was conducted using a Waters/Micromass Quattro Premier triple quadrupole mass spectrometer with positive mode electrospray ionisation and MS-MS conditions with a capillary voltage of 3.2 kV, detector multiplier gain of 600 V and source block and desolvation temperature of 90 °C and 350 °C, respectively. Analytes were quantified using multiple reaction mode with the following conditions: 2 (MS-MS transition 276.3>109.0, cone voltage 35 V, collision energy 30 V), 3 (MS-MS transition 326.3>109.0, cone voltage 35 V, collision energy 30 V), 21 (MS-MS transition 294.1>108.7, cone voltage 35 V, collision energy 30 V) and the internal standard (IS) diazepam (MS-MS transition 285.1>154.1, cone voltage 40 V, collision energy 30 V). Plasma concentrations were determined by comparison of the peak area to a calibration curve prepared in plasma and analysed along with the study samples. Pharmacokinetic parameters were calculated on the basis of plasma concentration-time profiles that used the mean plasma concentration at each sample time.
P. berghei mouse in vivo efficacy testing
All animal care and experimental procedures were approved by the Institutional Animal Care and Use Committee at the University of Washington. Eight to ten week old female BALB/c mice were obtained from Jackson Laboratories (Bar Harbor, ME, USA) and maintained in a temperature/humidity controlled SPF facility with a 12 hour light/dark cycle. Plasmodium berghei isolate NK65 was obtained from MR4 (Malaria research and Reference Resource Center, Monassas, VA, USA). Erythrocytic stages of P. berghei were harvested from infected donor mice via cardiac puncture and diluted in RPMI 1640 media (ATCC, Monassas, VA, USA) supplemented with 20% heat inactivated FBS (Altanta Biologicals, Atlanta, GA, USA). Recipient mice were infected with 2×107 infected erythrocytes, 200 µL IP injection, 24 hours prior to receiving the first drug dose. Tail vein bleeds and weighing were performed daily for the duration of the experiment. All infected mice were screened prior to receiving drug or vehicle to verify an established minimum infection of 0.1% parasitemia.
Parasitemia was determined from thin blood smears stained with Wright-Giemsa stain (Fisher Scientific, Houston, TX, USA). A minimum of 10 fields (1000x oil immersion) of >100 erythrocytes per field were counted by light microscopy.
2, 3 or 21 were dissolved in vehicle composed of 0.5% w/v carboxymethylcellulose, 0.4% v/v Tween 80, and 0.5% v/v benzyl alcohol in deionized water (all Sigma, St. Louis, MO, USA). All doses were administered to infected mice in weight-matched groups via oral gavage and calibrated at 50 or 100 mg drug/kg body weight or 200 uL of vehicle only. All animals were euthanized when the no-drug control group exhibited a loss of 20% pre-infected body weight (in compliance with institutional guidelines). For 2 and 3, mice were dosed orally with vehicle or with 100 mg/kg/dose every 4 hours × 10 doses, then every 6 hours × 6 doses (n = 6 mice for each group). For 21 dosing was 50 mg/kg or vehicle 1x day for 4 consecutive days (n=3 mice for each group), or 50 mg/kg or vehicle 2x daily (n = 5 mice for each group) for 4 consecutive days.
Molecular Modeling
The active site structure of P. falciparum bound to 2-cyano-3-hydroxy-N-[4-(trifluoromethyl)phenyl]-2-butenamide 1 was displayed using PyMol (DeLano Scientific LLC, San Carlos, CA) from the file 1TV5.pdb. 16
General Chemistry
Unless otherwise noted, reagents and solvents were obtained from commercial suppliers and were used without further purification. The reaction progress was monitored by thin layer chromatography (TLC) using silica gel 60 F-254 (0.25 mm) plates. Visualization was achieved with UV light and iodine vapor. Flash chromatography was carried out with silica gel (32–63 µm). Compounds 8 – 47 were synthesized as described in our previous paper for 2 – 4, 22 and as illustrated by scheme 1. Briefly, condensation of 3-amino-[1,2,4]triazole 5 with ethyl acetoacetate in acetic acid to form the 7-hydroxy-[1,2,4]triazolo[1,5-a]pyrimidine 6. Chlorination with phosphorous oxychloride gave the corresponding 7-chloro-[1,2,4]triazolo[1,5-a]pyrimidine 7, which upon treatment with substituted aryl amines in ethanol resulted in the desired products 8 – 47.
Analysis
1H NMR spectra were recorded on dilute solutions in CDCl3 or DMSO-d6 at 300 MHz. Chemical shifts are reported in parts per million (δ) downfield from tetramethylsilane (TMS). Coupling constants (J) are reported in Hz. Spin multiplicities are described as s (singlet), brs (broad singlet), d (doublet), t (triplet), q (quartet) and m (multiplet). Electrospray ionization mass spectra were acquired on a Bruker Esquire Liquid Chromatograph-Ion Trap Mass Spectrometer. FABHRMS obtained on JEOL HX-110 Mass Spectrometer. Melting points (Pyrex capillary) were determined on a Mel-Temp apparatus and are uncorrected. As a test of purity, compounds were subjected to HPLC analysis on an automated Varian Prep star system using a gradient of 20% MeOH to 100% MeOH (with 0.1% trifluoroacetic acid) over 30 min, using a YMC S5 ODS column (20 × 100 mm, Waters, Inc.). Compounds eluted as a single peak and the activity of the peak fraction was confirmed by demonstrating PfDHODH inhibitory activity. Representative 1HNMR, Mass and HPLC peaks for the most active compound 21, is provided as Supplemental data.
Physical properties
(2-Fluoro-phenyl)-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-amine (8)
Mp 180 °C. 1H NMR (300 MHz, CDCl3): δ 8.49 (s, 1H), 7.58 (m, 1H), 7.41 (m, 1H), 7.35–7.30 (m, 2H), 6.33 (s, 1H), 2.64 (s, 3H). MS m/z 244 [M + H]+.
(2-Chloro-phenyl)-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-amine (9)
Mp 174 °C (lit.35 174 °C). 1H NMR (300 MHz, DMSO-d6): δ 10.14 (brs, NH, exchangeable), 8.49 (s, 1H), 7.68 (m, 1H), 7.62-7.39 (m, 3H), 5.82 (s, 1H), 2.36 (s, 3H). MS m/z 260.2 [M + H]+.
(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-o-tolyl-amine (10)
Mp 183 °C (lit.35 179–180 °C). 1H NMR (300 MHz, DMSO-d6): δ 9.92 (brs, NH, exchangeable), 8.48 (s, 1H), 7.47-7.26 (m, 4H), 5.73 (s, 1H), 2.33 (s, 3H), 2.21 (s, 3H). MS m/z 240.1 [M + H]+.
(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-(2-trifluoromethyl-phenyl)-amine (11)
Mp 223 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.15 (brs, NH, exchangeable), 8.52 (s, 1H), 8.05-7.56 (m, 4H), 5.85 (s, 1H), 2.39 (s, 3H). MS m/z 294.1 [M + H]+.
2-(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ylamino)-benzonitrile (12)
Mp 246 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.54 (brs, NH, exchangeable), 8.51 (s, 1H), 7.98 (m, 1H), 7.82 (m, 1H), 7.55 (m, 2H), 6.08 (s, 1H), 2.38 (s, 3H). MS m/z 251 [M + H]+.
(3-Fluoro-phenyl)-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-amine (13)
Mp 223 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.35 (brs, NH, exchangeable), 8.54 (s, 1H), 7.51 (m, 1H), 7.35 (m, 2H), 7.11 (m, 1H), 6.58 (s, 1H), 2.47 (s, 3H). MS m/z 244 [M + H]+.
(3-Chloro-phenyl)-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-amine (14)
Mp 227 °C (lit.36 228 °C). 1H NMR (300 MHz, DMSO-d6): δ 10.31 (brs, NH, exchangeable), 8.52 (s, 1H), 7.54 (s, 1H), 7.50-7.48 (m, 2H), 7.36 (m, 1H), 6.49 (s, 1H), 2.44 (s, 3H). MS m/z 260.1 [M + H]+.
(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-m-tolyl-amine (15)
Mp 194 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.10 (brs, NH, exchangeable), 8.48 (s, 1H), 7.35 (m, 1H), 7.25 (m, 2H), 7.12 (m, 1H), 6.34 (s, 1H), 2.40 (s, 3H), 2.33 (s, 3H). MS m/z 240 [M + H]+.
(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-(3-trifluoromethyl-phenyl)-amine (16)
Mp 200 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.43 (brs, NH, exchangeable), 8.55 (s, 1H), 7.82 (m, 2H), 7.72 (m, 1H), 7.65 (m, 1H), 6.53 (s, 1H), 2.46 (s, 3H). MS m/z 294.1 [M + H]+.
(4-Fluoro-phenyl)-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-amine (17)
Mp 224 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.12 (brs, NH, exchangeable), 8.49 (s, 1H), 7.51-7.47 (m, 2H), 7.35-7.29 (m, 2H), 6.29 (s, 1H), 2.41 (s, 3H). MS m/z 244.1 [M + H]+.
(4-Chloro-phenyl)-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-amine (18)
Mp 267 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.22 (brs, NH, exchangeable), 8.47 (s, 1H), 7.55-7.45 (m, 4H), 6.42 (s, 1H), 2.42 (s, 3H). MS m/z 260.1 [M + H]+.
(4-Bromo-phenyl)-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-amine (19)
Mp 268 °C (lit.36 265 °C). 1H NMR (300 MHz, DMSO-d6): δ 10.24 (brs, NH, exchangeable), 8.50 (s, 1H), 7.65 (d, J = 8.7 Hz, 2H), 7.45 (d, J = 8.7 Hz, 2H), 6.46 (s, 1H), 2.43 (s, 3H). MS m/z 305.9 [M + H]+.
(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-p-tolyl-amine (20)
Mp 206 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.09 (brs, NH, exchangeable), 8.48 (s, 1H), 7.35-7.27 (m, 4H), 6.29 (s, 1H), 2.40 (s, 3H), 2.35 (s, 3H). MS m/z 240 [M + H]+.
(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-(4-trifluoromethyl-phenyl)-amine (21)
Mp 244 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.51 (brs, NH, exchangeable), 8.52 (s, 1H), 7.81 (m, 2H), 7.71 (m, 2H), 6.69 (s, 1H), 2.46 (s, 3H). MS m/z 294.1 [M + H]+; FABHRMS calcd for [C13H10N5F3 + H]+ 294.09669, determined 294.09716.
(4-Methoxy-phenyl)(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-amine (22)
Mp 212 °C (lit.36 211 °C). 1H NMR (300 MHz, DMSO-d6): δ 10.01 (brs, NH, exchangeable), 8.46 (s, 1H), 7.35 (d, J = 8.4 Hz, 2H), 7.05 (d, J = 8.4 Hz, 2H), 6.16 (s, 1H), 3.80 (s, 3H), 2.38 (s, 3H). MS m/z 256.1 [M + H]+.
(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-(4-trifluoromethoxy-phenyl)-amine (23)
Mp 214 °C. 1H NMR (300 MHz, CDCl3): δ 8.37 (s, 1H), 7.95 (s, 1H), 7.44-7.39 (m, 3H), 6.37 (s, 1H), 2.58 (s, 3H). MS m/z 310.1 [M + H]+.
(4-Difluoromethoxy-phenyl)-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-amine (24)
Mp 200 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.23 (brs, NH, exchangeable), 8.50 (s, 1H), 7.52-7.49 (m, 2H), 7.30-7.27 (m, 3H), 6.34 (s, 1H), 2.41 (s, 3H). MS m/z 292.1 [M + H]+.
(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-(4-nitro-phenyl)-amine (25)
Mp 338 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.55 (s, 1H), 8.30 (d, J = 9 Hz, 2H), 7.71 (m, 2H), 6.85 (s, 1H), 2.49 (s, 3H). MS m/z 271 [M + H]+.
Biphenyl-4-yl-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-amine (26)
Mp 114 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.31 (brs, NH, exchangeable), 8.52 (s, 1H), 7.78-7.69 (m, 4H), 7.56-7.37 (m, 4H), 7.21 (m, 1H), 6.49 (s, 1H), 2.43 (s, 3H). MS m/z 302.1 [M + H]+.
(4-Benzyl-phenyl)-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-amine (27)
Mp 190 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.11 (brs, NH, exchangeable), 8.48 (s, 1H), 7.35-7.21 (m, 9H), 6.34 (s, 1H), 3.97 (s, 2H), 2.40 (s, 3H). MS m/z 316.2 [M + H]+.
(2,3-Difluoro-phenyl)-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-amine (28)
Mp 231 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.76 (s, 1H), 7.57-7.48 (m, 1H), 7.41-7.33 (m, 2H), 6.33 (s, 1H), 2.46 (s, 3H). MS m/z 262.1 [M + H]+.
(2,5-Difluoro-phenyl)-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-amine (29)
Mp 227 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.21 (brs, NH, exchangeable), 8.53 (s, 1H), 7.54-7.44 (m, 2H), 7.35-7.28 (m, 1H), 6.12 (s, 1H), 2.43 (s, 3H). MS m/z 262.1 [M + H]+.
(2,6-Difluoro-phenyl)-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-amine (30)
Mp 231 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.76 (s, 1H), 7.62-7.53 (m, 1H), 7.39-7.34 (m, 2H), 6.17 (s, 1H), 2.45 (s, 3H). MS m/z 262 [M + H]+.
(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-(2,3,4-trifluoro-phenyl)-amine (31)
Mp 246 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.73 (s, 1H), 7.55-7.37 (m, 2H), 6.34 (s, 1H), 2.45 (s, 3H). MS m/z 280.2 [M + H]+.
(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-(2,4,5-trifluoro-phenyl)-amine (32)
Mp 272 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.67 (s, 1H), 7.89-7.72 (m, 2H), 6.25 (s, 1H), 2.44 (s, 3H). MS m/z 280.1 [M + H]+.
(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-(2,4,6-trifluoro-phenyl)-amine (33)
Mp 178 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.02 (brs, NH, exchangeable), 8.54 (s, 1H), 7.51-7.46 (m, 2H), 6.08 (s, 1H), 2.41 (s, 3H). MS m/z 280.1 [M + H]+.
(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-(2,3,5,6-tetrafluoro-phenyl)-amine (34)
Mp 211 °C. 1H NMR (300 MHz, DMSO-d6): δ 13.25 (brs, NH, exchangeable), 8.53 (s, 1H), 8.18 (s, 1H), 5.83 (s, 1H), 2.32 (s, 3H). MS m/z 298.1 [M + H]+.
(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-(2,3,4,5,6-pentafluorophenyl)-amine (35)
Mp 278 °C. 1H NMR (300 MHz, DMSO-d6): δ 13.20 (brs, NH, exchangeable), 8.18 (s, 1H), 5.83 (s, 1H), 2.32 (s, 3H). MS m/z 316.2 [M + H]+.
(2-Fluoro-4-methyl-phenyl)-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-amine (36)
Mp 208 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.05 (brs, NH, exchangeable), 8.50 (s, 1H), 7.38-7.12 (m, 3H), 5.95 (s, 1H), 2.35 (s, 3H). MS m/z 258.1 [M + H]+.
(3,4-Difluoro-phenyl)-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-amine (37)
Mp 265 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.54 (s, 1H), 7.62-7.51 (m, 2H), 7.38-7.31 (m, 1H), 6.47 (s, 1H), 2.44 (s, 3H). MS m/z 262.1 [M + H]+.
(3,5-Difluoro-phenyl)-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-amine (38)
Mp 256 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.41 (brs, NH, exchangeable), 8.54 (s, 1H), 7.25 (m, 2H), 7.15 (m, 1H), 6.73 (s, 1H), 2.49 (s, 3H). MS m/z 262.1 [M + H]+.
(3,5-Bis-trifluoromethyl-phenyl)-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-amine (39)
Mp 257 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.57 (brs, NH, exchangeable), 8.53 (s, 1H), 8.13 (m, 2H), 7.96 (m, 1H), 6.64 (s, 1H), 2.45 (s, 3H). MS m/z 362.2 [M + H]+.
(4-Fluoro-3-methyl-phenyl)-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-amine (40)
Mp 231 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.09 (brs, NH, exchangeable), 8.48 (s, 1H), 7.36-7.23 (m, 3H), 6.25 (s, 1H), 2.40 (s, 3H), 2.26 (s, 3H). MS m/z 258.1 [M + H]+.
(4-Chloro-3-trifluoromethyl-phenyl)-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-amine (41)
Mp 233 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.42 (brs, NH, exchangeable), 8.52 (s, 1H), 7.93 (s, 1H), 7.80 (m, 2H), 6.62 (s, 1H), 2.45 (s, 3H). MS m/z 328.2 [M + H]+.
(4-Bromo-3-trifluoromethyl-phenyl)-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-amine (42)
Mp 243 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.41 (brs, NH, exchangeable), 8.52 (s, 1H), 7.92 (s, 1H), 7.68 (m, 2H), 6.61 (s, 1H), 2.40 (s, 3H). MS m/z 373.9 [M + H]+.
(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-(4-methyl-3-trifluoromethyl-phenyl)-amine (43)
Mp 200 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.28 (brs, NH, exchangeable), 8.50 (s, 1H), 7.74 (s, 1H), 7.68 (d, J = 8.1 Hz, 1H), 7.55 (d, J = 8.4 Hz , 1H), 6.41 (s, 1H), 2.47 (s, 3H), 2.43 (s, 3H). MS m/z 308.2 [M + H]+.
(3,4-Dimethyl-phenyl)-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-amine (44)
Mp 224 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.01 (brs, NH, exchangeable), 8.44 (s, 1H), 7.46-7.06 (m, 3H), 6.20 (s, 1H), 2.37 (s, 3H), 2.23 (s, 6H). MS m/z 254.2 [M + H]+.
4-(5-Methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-ylamino)-2-trifluoromethyl-benzonitrile (45)
Mp 264 °C. 1H NMR (300 MHz, DMSO-d6): δ 8.55 (s, 1H), 8.20 (m, 1H), 8.05 (s, 1H), 7.95 (m, 1H), 6.90 (s, 1H), 2.48 (s, 3H). MS m/z 319.1 [M + H]+.
(3-Fluoro-4-methyl-phenyl)-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-amine (46)
Mp 251 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.20 (brs, NH, exchangeable), 8.48 (s, 1H), 7.36-7.23 (m, 3H), 6.43 (s, 1H), 2.41 (s, 3H), 2.25 (s, 3H). MS m/z 258.1 [M + H]+.
(3-Fluoro-4-trifluoromethyl-phenyl)-(5-methyl-[1,2,4]triazolo[1,5-a]pyrimidin-7-yl)-amine (47)
Mp 247 °C. 1H NMR (300 MHz, DMSO-d6): δ 10.65 (brs, NH, exchangeable), 8.56 (s, 1H), 7.85-7.78 (m, 1H), 7.68-7.60 (m, 1H), 7.55-7.45 (m, 1H), 6.88 (s, 1H), 2.50 (s, 3H). MS m/z 312.2 [M + H]+.
Supplementary Material
1HNMR, Mass and HPLC traces for compound 21 are provided.
Acknowledgements
The authors would like to thank David Floyd, Kip Guy, David Matthews and Carl Craft of the Medicines for Malaria Venture (MMV) Expert Scientific Advisory Committee for helpful discussions, Rajan K Paranji and Martin Sadilek for analytical support. This work was supported by the United States National Institutes of Health grants, U01AI075594 (to MAP, PKR and WNC; Ian Bathurst (PI); Malaria Medicines Venture), AI053680 (to MAP and PKR), AI60360 (to PKR), and by a grant from Malaria Medicines Venture (to MAP, PKR and WNC). MAP also acknowledges the support of the Welch Foundation (I-1257) and PKR also acknowledges support from a Senior Scholar Award in Global Infectious Diseases from the Ellison Medical Foundation and from the UW Keck Center for Microbial Pathogens.
Abbreviations
- PfDHODH
Plasmodium falciparum dihydroorotate dehydrogenase
- PbDHODH
Plasmodium berghei dihydroorotate dehydrogenase
- hDHODH
human dihydroorotate dehydrogenase
- CoQ
ubiquinone
- FMN
flavin mononucleotide
- DSM
Compound Code names, Dallas, Seattle, Melbourne
References
- 1.Greenwood BM, Fidock DA, Kyle DE, Kappe SHI, Alonso PL, Collins FH, Duffy PE. Malaria: progress, perils, and prospects for eradication. J. Clin. Invest. 2008;118:1266–1276. doi: 10.1172/JCI33996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Guerra CA, Gikandi PW, Tatem AJ, Noor AM, Smith DL, Hay SI, Snow RW. The limits and intensity of Plasmodium falciparum transmission: implications for malaria control and elimination worldwide. PLoS Med. 2008;5:e38. doi: 10.1371/journal.pmed.0050038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.White NJ. Antimalarial drug resistance. J Clin Invest. 2004;113:1084–1092. doi: 10.1172/JCI21682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rosenthal PJ. Artesunate for the treatment of severe falciparum malaria. N Engl J Med. 2008;358:1829–1836. doi: 10.1056/NEJMct0709050. [DOI] [PubMed] [Google Scholar]
- 5.Gardner MJ, Hall N, Fung E, White O, Berriman M, Hyman RW, Carlton JM, Pain A, Nelson KE, Bowman S, Paulsen IT, James K, Eisen JA, Rutherford K, Salzberg SL, Craig A, Kyes S, Chan MS, Nene V, Shallom SJ, Suh B, Peterson J, Angiuoli S, Pertea M, Allen J, Selengut J, Haft D, Mather MW, Vaidya AB, Martin DMA, Fairlamb AH, Fraunholz MJ, Roos DS, Ralph SA, McFadden GI, Cummings LM, Subramanian GM, Mungall C, Venter JC, Carucci DJ, Hoffman SL, Newbold C, Davis RW, Fraser CM, Barrell B. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature. 2002;419:498–511. doi: 10.1038/nature01097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pink R, Hudson A, Mouries MA, Bendig M. Opportunities and challenges in antiparasitic drug discovery. Nat Rev Drug Discov. 2005;4:727–740. doi: 10.1038/nrd1824. [DOI] [PubMed] [Google Scholar]
- 7.Srivastava IK, Morrisey JM, Darrouzet E, Daldal F, Vaidya AB. Resistance mutations reveal the atovaquone-binding domain of cytochrome b in malaria parasites. Mol Microbio. 1999;33:704–711. doi: 10.1046/j.1365-2958.1999.01515.x. [DOI] [PubMed] [Google Scholar]
- 8.Fry M, Pudney M. Site of action of the antimalarial hydroxynaphthoquinone, 2-[trans-4-(4'-chlorophenyl) cyclohexyl]-3-hydroxy-1,4-naphthoquinone (566C80) Biochem Pharmacol. 1992;43:1545–1553. doi: 10.1016/0006-2952(92)90213-3. [DOI] [PubMed] [Google Scholar]
- 9.Gutteridge WE, Trigg PI. Incorporatin of radioactive precursors into DNA and RNA of Plasmodium knowlesi in vitro. J. Protozool. 1970;17:89–96. doi: 10.1111/j.1550-7408.1970.tb05163.x. [DOI] [PubMed] [Google Scholar]
- 10.Reyes P, Rathod PK, Sanchez DJ, Mrema JEK, Rieckmann K, Heidrich H. Enzymes of purine and pyrimidine metabolism from the human malaria parasite, Plasmodium falciparum. Mol. Biochem. Parasitol. 1982;5:275–290. doi: 10.1016/0166-6851(82)90035-4. [DOI] [PubMed] [Google Scholar]
- 11.Jones M. Pyrimidine nucleotide biosynthesis in animals: genes, enzymes, and regulation of UMP biosynthesis. Annu. Rev. Biochem. 1980;49:253–279. doi: 10.1146/annurev.bi.49.070180.001345. [DOI] [PubMed] [Google Scholar]
- 12.Nagy M, Lacroute F, Thomas D. Divergent evolution of pyrimidine biosynthesis between anaerobic and aerobic yeasts. Proc. Natl. Acad. Sci. USA. 1992;89:8966–8970. doi: 10.1073/pnas.89.19.8966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Painter HJ, Morrisey JM, Mather MW, Vaidya AB. Specific role of mitochondrial electron transport in blood-stage Plasmodium falciparum. Nature. 2007;446:88–91. doi: 10.1038/nature05572. [DOI] [PubMed] [Google Scholar]
- 14.Goldenberg MM. Leflunomide, a novel immunomodulator for the treatment of active rheumatoid arthritis. Clinical therapeutics. 1999;21:1837–1852. doi: 10.1016/S0149-2918(00)86732-6. [DOI] [PubMed] [Google Scholar]
- 15.Hansen M, Le Nours J, Johansson E, Antal T, Ullrich A, Loeffler M, Larsen S. Inhibitor binding in a class 2 dihydroorotate dehydrogenase causes variations in the membrane-associated N-terminal domain. Protein Science. 2004;13:1031–1042. doi: 10.1110/ps.03533004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hurt DE, Widom J, Clardy J. Structure of Plasmodium falciparum dihydroorotate dehydrogenase with a bound inhibitor. Acta Crystallogr D Biol Crystallogr. 2006;62:312–323. doi: 10.1107/S0907444905042642. [DOI] [PubMed] [Google Scholar]
- 17.Liu S, Neidhardt E, Grossman T, Ocain T, Clardy J. Structures of human dihydroorotate dehydrogenase in complex with antiproliferative agents. Structure. 1999;8:25–33. doi: 10.1016/s0969-2126(00)00077-0. [DOI] [PubMed] [Google Scholar]
- 18.Baldwin J, Farajallah AM, Malmquist NA, Rathod PK, Phillips MA. Malarial dihydroorotate dehydrogenase: substrate and inhibitor specificity. J. Biol. Chem. 2002;277:41827–41834. doi: 10.1074/jbc.M206854200. [DOI] [PubMed] [Google Scholar]
- 19.Baldwin J, Michnoff CH, Malmquist NA, White J, Roth MG, Rathod PK, Phillips MA. High-throughput screening for potent and selective inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. J Biol Chem. 2005;280:21847–21853. doi: 10.1074/jbc.M501100200. [DOI] [PubMed] [Google Scholar]
- 20.Copeland RA, Marcinkeviciene J, Haque T, Kopcho LM, Jiang W, Wang K, Ecret LD, Sizemore C, Amsler KA, Foster L, Tadesse S, Combs AP, Stern AM, Trainor GL, Slee A, Rogers MJ, Hobbs F. Helicobacter pylori-selective antibacterials based on inhibition of pyrimidine biosynthesis. J. Biol. Chem. 2000;275:33373–33378. doi: 10.1074/jbc.M004451200. [DOI] [PubMed] [Google Scholar]
- 21.Malmquist NA, Gujjar R, Rathod PK, Phillips MA. Analysis of Flavin Oxidation and Electron-Transfer Inhibition in Plasmodium falciparum Dihydroorotate Dehydrogenase. Biochemistry. 2008;47:2466–2475. doi: 10.1021/bi702218c. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Phillips MA, Gujjar R, Malmquist NA, White J, El Mazouni F, Baldwin J, Rathod PK. Triazolopyrimidine-based dihydroorotate dehydrogenase inhibitors with potent and selective activity against the malaria parasite, Plasmodium falciparum. J. Med. Chem. 2008;51:3649–3653. doi: 10.1021/jm8001026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lin JH. CYP induction-mediated drug interactions: in vitro assessment and clinical implications. Pharm Res. 2006;23:1089–1116. doi: 10.1007/s11095-006-0277-7. [DOI] [PubMed] [Google Scholar]
- 24.Topliss JG. Utilization of operational schemes for analog synthesis in drug design. J Med Chem. 1972;15:1006–1011. doi: 10.1021/jm00280a002. [DOI] [PubMed] [Google Scholar]
- 25.Topliss JG. A manual method for applying the Hansch approach to drug design. J Med Chem. 1977;20:463–469. doi: 10.1021/jm00214a001. [DOI] [PubMed] [Google Scholar]
- 26.Boa AN, Cnanavan SP, Hirst PR, Ramsey C, Stead AMW, McConkey GA. Synthesis of brequinar analogue inhibitors of malaria parasite dihydroorotate dehydrogenase. Bioorganic and Medicinal Chemistry. 2005;13:1945–1967. doi: 10.1016/j.bmc.2005.01.017. [DOI] [PubMed] [Google Scholar]
- 27.Heikkilae T, Thirumalairajan S, Davies M, Parsons MR, McConkey AG, Fishwick CWG, Johnson AP. The first de novo designed inhibitors of Plasmodium falciparum dihydroorotate dehydrogenase. Bioorg Med Chem Lett. 2006;16:88–92. doi: 10.1016/j.bmcl.2005.09.045. [DOI] [PubMed] [Google Scholar]
- 28.Malmquist NA, Baldwin J, Phillips MA. Detergent-dependent kinetics of truncated Plasmodium falciparum dihydroorotate dehydrogenase. J Biol Chem. 2007;282:12678–12686. doi: 10.1074/jbc.M609893200. [DOI] [PubMed] [Google Scholar]
- 29.Copeland RA. A guide for medicinal chemists and pharmacologists. Hoboken, New Jersey: John Wiley and Sons, Inc; 2005. Evaluation of enzyme inhibitors in drug discovery; pp. 185–192. [PubMed] [Google Scholar]
- 30.Desjardins RE, Canfield CJ, Haynes JD, Chulay JD. Quantitative assessment of antimalarial activity in vitro by a semiautomated microdilution technique. Antimicrob Agents Chemother. 1979;16:710–718. doi: 10.1128/aac.16.6.710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang L, Lee PC, White J, Rathod PK. Potent and selective activity of a combination of thymidine and 1843U89, a folate-based thymidylate synthase inhibitor, against Plasmodium falciparum. Antimicrob. Agents and Chemotherapy. 2000;44:1047–1050. doi: 10.1128/aac.44.4.1047-1050.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Obach RS. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: An examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab Dispos. 1999;27:1350–1359. [PubMed] [Google Scholar]
- 33.Davies B. Physiological parameters in laboratory animal and humans. Pharm Res. 1993;10:1093–1095. doi: 10.1023/a:1018943613122. [DOI] [PubMed] [Google Scholar]
- 34.Houston JB. Utility of in vitro drug metabolism data in predicting in vivo metabolic clearance. Biochem Pharmacol. 1994;47:1469–1479. doi: 10.1016/0006-2952(94)90520-7. [DOI] [PubMed] [Google Scholar]
- 35.Levin YA, Sergeeva EM, Kukhtin VA. Condensed heterocycles V. Reaction of 4-chloro-6-methyl-1,2,4-triazolo[2,3-α]pyrimidine with some nitrogenous bases. Zhurnal Obshchei Khimii. 1964;34:205–209. [Google Scholar]
- 36.Reynolds GA, VanAllan JA. Structure of certain polyazaindenes. VII. 4-Amino-6-methyl-1,3,3a,7-tetraazaindene and its derivatives. J. Org. Chem. 1961;26:115–117. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
1HNMR, Mass and HPLC traces for compound 21 are provided.