

Endothelial cells constitute a selective barrier that controls the passage of plasma proteins and circulating cells from the blood to tissues. This is achieved by the caveolar-vesicular system and by the dynamic opening and closing of intercellular junctions.1 Inter-endothelial cell junctions are almost absent in the postcapillary venules, where cellular extravasation and exchange of plasma constituents occur, but are well-organized in the large vessels to ensure strict control of vascular permeability. Cadherins are Ca2+- and protease-sensitive molecules that mediate homotypic cell-to-cell adhesion. Endothelial cells express N-, P- and VE-cadherin. N-cadherin is diffusely spread on the cell surface, while P-cadherin is present in trace amounts. VE-cadherin is a single-chain transmembrane glycoprotein localized at specialized inter-endothelial cell contact regions referred to as adherens junctions.2
Vascular inflammation has been implicated in the development and progress of vascular diseases such as hypertension and atherosclerosis.3 The inflammatory response is initiated by injury of the endothelium inflicted by factors such as oxidized low-density lipoprotein, reactive oxygen species and viruses. Endothelial cell injury prompts the recruitment of circulating leukocytes to the injury site, and the disruption of the endothelial cell barrier allows leukocyte infiltration of the vessel wall (Figure). Leukocyte recruitment and infiltration of the vascular wall is a complex process encompassing a series of adhesion and deadhesion events and distinct adhesion molecules on the activated endothelium and leukocytes.
Figure.
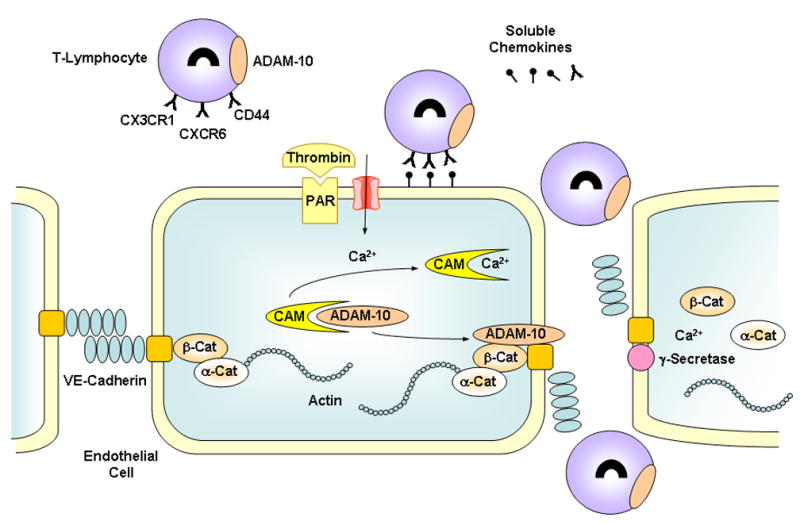
Role of ADAM-10 in leukocyte recruitment and transmigration and endothelial cell permeability. In activated endothelium, upregulated cell adhesion molecules such as fractalkine, CXCL16 and hyaluronan ( ) bind their respective receptors CX3CR1, CXCR6 and CD44 (Y) resulting in initial capture of inflammatory cells. Inflammatory mediators such as thrombin (acting via protease-activated receptor, PAR), and leukocyte adhesion increase endothelial cell [Ca2+]i, causing the release of pro-ADAM-10 from calmodulin (CAM) and activation of ADAM-10. Activated ADAM-10 cleaves the adhesion molecules resulting in leukocyte deadhesion, initial rolling and subsequent migration to inter-endothelial junction. The released ectodomains act as soluble chemokines to further recruit inflammatory cells to the endothelium. Activated ADAM-10 also cleaves VE-cadherin ectodomain resulting in endothelial cell gap formation and inflammatory cell transmigration. The remaining membrane bound stub of VE-cadherin is cleaved by Ca2+-sependent γ-secretase leading to the release of β-catenin, α-catenin and actin from the plasma membrane and alterations in cell morphology and nuclear signaling.
Cadherin Shedding and Leukocyte Rolling
An important mechanism regulating leukocyte recruitment to the vascular wall involves the proteolytic cleavage of adhesion molecules by cell-associated metalloproteases, a process called “shedding”. The shedding process can be exemplified in the adhesion protein selectin. Leukocyte L-selectin acts as a homing receptor for the targeting and initial interaction between leukocytes and activated endothelium, a process called “rolling”. Endothelial E-selectin and platelet P-selectin are stored intracellulary, but when the cells are activated they translocate to the cell surface, where they participate in the initial rolling of leukocytes over the activated endothelium. Focal shedding of endothelial E-selectin is thought to promote leukocyte infiltration and lesion formation.4 Also, blood levels of soluble vascular cell and intracellular adhesion molecule-1 (VCAM-1 and ICAM-1) are increased in atherosclerosis, suggesting their shedding during lesion development. Additionally, inflammatory mediators such as thrombin cause proteolysis of VE-cadherin, thereby disrupting the organization of endothelial adherens junctions and increasing vascular permeability and edema.5 The role of VE-cadherin shedding in endothelial cell permeability is further supported by reports that induction of apoptosis in human umbilical vein endothelial cells (HUVEC) is associated with dissolution of adherens junctions, loss of cell-cell contacts, and shedding of VE-cadherin.6 While factors associated with the expression of transmembrane adhesion molecules and their dysregulation in atherosclerosis have been studied,7 the mechanisms involved in the release of these adhesion molecules from the cell surface by controlled cleavage or shedding are not well-understood.
Role of ADAMs as Adhesion Molecule Sheddases
Disintegrin and metalloproteases (ADAMs) are membrane-anchored glycoproteins and regulatory enzymes that have been implicated in cell adhesion as well as the proteolytic conversion or shedding of membrane-bound proteins to soluble forms.8-10 ADAMs, like matrix metalloproteinases (MMPs), are members of the metzincin zinc-dependent metalloprotease superfamily. At least thirty members of the ADAMs protein family share a common structure consisting of a prodomain, a metalloprotease domain, a disintegrin domain, a cysteine-rich domain, an epidermal growth factor (EGF)-like domain, a transmembrane domain, and a cytoplasmic domain.11,12 Adams are involved in diverse biological functions including fertilization, neurogenesis and angiogenesis as well as disease states such cancer.13,14
ADAM-17 (TNF-converting enzyme or TACE) was the first member of the ADAMs family with a defined role as a sheddase that releases TNF-α and its receptors from neutrophils and macrophages during inflammation.15 ADAM-1, -12, -15 and -17 have been identified in vascular smooth muscle cells, and ADAM-10, -15 and -17 may have potential roles in the regulation of endothelial function via their metalloprotease and proteolytic properties. Also, the adhesive disintegrin domain of ADAMs allows them to interact with integrins. For example, ADAM-15 may be involved in endothelial-leukocyte or endothelial-tumor adhesion by binding to the classic RGD-binding integrins α5β1 and αvβ3. The binding of ADAM-12 and -15 disintegrin domains to non-RGD-type integrin α9β1 may also mediate cell-cell interaction.16
ADAM-10 and -17 are upregulated in activated endothelium and play a role in ectodomain shedding of adhesion molecules during leukocyte recruitment (Figure).17 Also, fractalkine (CX3CL1) and CXCL16 are adhesion molecules that are upregulated in activated endothelium and macrophages, and take part in the initial capture of inflammatory cells. ADAM-10 or -17 cleaves these adhesion molecules to soluble chemoattractant cytokines or chemokines to attract additional inflammatory cells expressing chemokine receptors CX3CR1 and CXCR6.18,19 Also, CD44, another adhesion molecule in inflammatory cells, cross-links to endothelial cell hyaluronan and initiates intracellular signaling cascade, leading to activation of ADAM-10 or -17 and cleavage of the soluble ectodomain of CD44. Soluble CD44 fragments in turn compete for uncleaved hyaluronan and promote leukocyte deadhesion and rolling.20 Thus, ADAMs-induced release of the soluble ectodomain from the adhesion molecules L-selectin, VCAM, ICAM, fractalkine, CXCL16 and CD44 contribute to leukocyte deadhesion and rolling on activated endothelial cells and their migration to the inter-endothelial junction.4,21,22
ADAMs and Vascular Permeability
ADAMs-mediated shedding of cell surface molecules is emerging as a critical pathway not only in the regulation of leukocyte recruitment but also in the control of vascular and non-vascular cell-cell interactions. ADAM-15 is upregulated in atherosclerotic lesions suggesting that ADAMs participate in lesion formation. Also, ADAM-10 is localized in the membrane of epithelial cells of benign glands suggesting a role in cell-cell, cell-matrix, and cell-basement membrane interactions. Although ADAM-10 does not interact directly with integrins, it may indirectly influence integrin-related adhesion activity by cleaving the L1 membrane adhesion molecule, a type 1 membrane glycoprotein implicated in the migration of neural and tumor cells.23 Also, in resting cells, calmodulin (CaM) constitutively associates with the pro-ADAM-10 inactive form. An increase in endothelial cell [Ca2+]i in response to activators such as thrombin or subsequent to leukocyte adhesion would promote the dissociation of CaM and the activation of ADAM-10 (Figure).20,24 However, the molecular mechanisms via which activated ADAM-10 could affect endothelial cell permeability and leukocyte infiltration are not clear.
In this issue of Circulation Research Schulz and colleagues 25 describe the effects of ADAM-10 on permeability of HUVECs and T cell transmigration. They found that ADAM-10 cleaves VE-cadherin ectodomain into a soluble fragment, and the remaining carboxyterminal membrane bound stub is further cleaved by γ-secretase. ADAM-10-mediated cleavage of VE-cadherin is induced by thrombin activation of endothelial cells, Ca2+ influx as well as induction of apoptosis by staurosporine treatment. Inhibition of ADAM-10 by GI254023X decreased endothelial cell permeability and transmigration of T cells. Also, transfecting T cells with ADAM-10 siRNA caused a decrease in the rate of transmigration of activated T cells. These elegant studies highlight the importance of ADAM-10 in VE-cadherin removal from endothelial cell surface by controlled cleavage or shedding, a potential regulatory pathway of vascular permeability and the inflammation process associated with atherosclerosis.26
Extracellular Proteases and Intracellular Kinases
The identification of the role of ADAM-10 in VE-cadherin shedding poses several challenging questions. An important question is whether the observed effects on endothelial adhesion molecules and cell permeability are unique to ADAM-10 or involve cooperative interactions with other ADAMs. For instance, shedding of VCAM-1 can be mediated by ADAM-17.21 Also, binding of ADAM-28 to P-selectin glycoprotein ligand-1 enhances P-selectin-mediated leukocyte adhesion to endothelial cells.27 Additionally, ADAM-15 is an adherens junction molecule whose surface expression can be driven by VE-cadherin.28
Because ADAMs have multidomain structure, they are potentially multifunctional with multiple roles depending on their cellular localization. Also, ADAMs may function in concert with other metallorotease superfamilies such as ADAMTS and MMPs. ADAMTS (a disintegrin-like and metalloproteinase with thrombospondin type 1 motifs) include at least 19 members evolved as non-integral membrane proteins associated with the cell surface and ECM through specific protein domains. Interestingly, thrombin, an activator of ADAM-10, as well as plasmin promote proteolytic inactivation of ADAMTS-13.29 Also, ADAMs may have substrate overlap with MMPs and thereby influence the degradation of ECM components. This is supported by reports that purified bovine ADAM-10 cleaves native type IV collagen, a major component of the basement membrane and ECM.30 MMPs are also known to activate each other.31 Therefore, the potential interactions between ADAMs and MMPs on endothelial cell permeability need to be examined. In this regard, it is important to investigate the effects of inhibitors of ADAMs and MMPs on leukocyte infiltration. Tissue inhibitors of MMPs (TIMPs) are being considered to target specific MMPs in localized vascular diseases such as abdominal aortic aneurysm. However, the isolated N-terminal domains of TIMP-1 and TIMP-3 may not be sufficient for ADAM-10 inhibition,32 and specific siRNA may provide a more specific approach.
ADAMs-induced proteolysis can also change the activity of remnant surface molecular complexes which in turn affect signaling pathways inside the cell. While the effects of thrombin can be related to increased Ca2+ influx in endothelial cells, the role of localized Ca2+ gradients and the relation between the intracellular Ca2+ stores and other Ca2+ regulatory pathways in the surface membrane pumps and exchangers need to be further characterized. Also, while an increase in endothelial cell [Ca2+]i could activate ADAM-10, the proeolytic fragments generated from this reaction may affect the activity of the same or other membrane channels. For instance, studies have suggested an effect of MMPs on membrane Ca2+ and/or K+ channel activity, possibly through an interaction with membrane αvβ3 integrin.33-36
An important question also relates to the cellular remnants of cadherin shedding. β-catenin links the cytoplasmic domain of VE-cadherin to the actin cytoskeleton via α-catenin and therefore contributes to establishing VE-cadherin-mediated cell-cell adhesion (Figure). Ca2+ influx and ADAMs activation not only induce the proteolysis of extracellular VE-cadherin, and separation of cell-cell adhesion, but also facilitate the degradation of cytoplasmic domain of VE-cadherin by γ-secretase resulting in translocation of β-catenin from the plasma membrane to the cytoplasm where it may alter cell morphology, motility and proliferation.37
In addition to Ca2+, the role of Rho kinase and protein kinase C (PKC) in proteolytic dissolution and shedding of adhesion molecules should be considered. Activation of PKC-α may increase endothelial cell permeability by disassembly of VE-cadherin junctions.38 Also, vascular endothelial growth factor upregulates the expression of ADAMTS1 through PKC signaling.39 While ADAM-10 is a weak substrate of PKC,40 phorbol esters activating PKC and the small GTPase Rac can activate ADAM17-dependent shedding of a variety of substrates.21
Thus the discovery of the role of specific ADAMs in VE-cadherin shedding paves the way for further investigations to identify the potential interactions of ADAMs with other adhesion molecules, transmembrane and extracellular metalloproteases, as well as intracellular ion and protein kinase-dependent regulatory pathways. The identification of the mechanisms of cleavage of adhesion molecules by various metalloproteases during vascular lesion formation provides novel leads for the development of therapeutic interventions in vascular diseases.
Acknowledgments
Sources of Funding: The authors acknowledge the support of grants from the National Heart, Lung & Blood Institute (HL65998, HL70659).
Footnotes
Disclosures: None
References
- 1.Rippe B, Haraldsson B. Transport of macromolecules across microvascular walls: the two-pore theory. Physiol Rev. 1994;74:163–219. doi: 10.1152/physrev.1994.74.1.163. [DOI] [PubMed] [Google Scholar]
- 2.Lampugnani MG, Resnati M, Raiteri M, Pigott R, Pisacane A, Houen G, Ruco LP, Dejana E. A novel endothelial-specific membrane protein is a marker of cell-cell contacts. J Cell Biol. 1992;118(6):1511–22. doi: 10.1083/jcb.118.6.1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Packard RR, Libby P. Inflammation in atherosclerosis: from vascular biology to biomarker discovery and risk prediction. Clin Chem. 2008;54(1):24–38. doi: 10.1373/clinchem.2007.097360. [DOI] [PubMed] [Google Scholar]
- 4.Hafezi-Moghadam A, Thomas KL, Prorock AJ, Huo Y, Ley K. L-selectin shedding regulates leukocyte recruitment. J Exp Med. 2001;193(7):863–72. doi: 10.1084/jem.193.7.863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rabiet MJ, Plantier JL, Rival Y, Genoux Y, Lampugnani MG, Dejana E. Thrombin-induced increase in endothelial permeability is associated with changes in cell-to-cell junction organization. Arterioscler Thromb Vasc Biol. 1996;16(3):488–96. doi: 10.1161/01.atv.16.3.488. [DOI] [PubMed] [Google Scholar]
- 6.Herren B, Levkau B, Raines EW, Ross R. Cleavage of beta-catenin and plakoglobin and shedding of VE-cadherin during endothelial apoptosis: evidence for a role for caspases and metalloproteinases. Mol Biol Cell. 1998;9(6):1589–601. doi: 10.1091/mbc.9.6.1589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Blankenberg S, Barbaux S, Tiret L. Adhesion molecules and atherosclerosis. Atherosclerosis. 2003;170(2):191–203. doi: 10.1016/s0021-9150(03)00097-2. [DOI] [PubMed] [Google Scholar]
- 8.Herren B. ADAM-mediated shedding and adhesion: a vascular perspective. News Physiol Sci. 2002;17:73–6. doi: 10.1152/nips.01373.2001. [DOI] [PubMed] [Google Scholar]
- 9.Moss ML, Lambert MH. Shedding of membrane proteins by ADAM family proteases. Essays Biochem. 2002;38:141–53. doi: 10.1042/bse0380141. [DOI] [PubMed] [Google Scholar]
- 10.Becherer JD, Blobel CP. Biochemical properties and functions of membrane-anchored metalloprotease-disintegrin proteins (ADAMs) Curr Top Dev Biol. 2003;54:101–23. doi: 10.1016/s0070-2153(03)54006-6. [DOI] [PubMed] [Google Scholar]
- 11.Seals DF, Courtneidge SA. The ADAMs family of metalloproteases: multidomain proteins with multiple functions. Genes Dev. 2003;17(1):7–30. doi: 10.1101/gad.1039703. [DOI] [PubMed] [Google Scholar]
- 12.Duffy MJ, Lynn DJ, Lloyd AT, O'Shea CM. The ADAMs family of proteins: from basic studies to potential clinical applications. Thromb Haemost. 2003;89(4):622–31. [PubMed] [Google Scholar]
- 13.Trochon V, Li H, Vasse M, Frankenne F, Thomaidis A, Soria J, Lu H, Gardner C, Soria C. Endothelial metalloprotease-disintegrin protein (ADAM) is implicated in angiogenesis in vitro. Angiogenesis. 1998;2(3):277–85. doi: 10.1023/A:1009206817829. [DOI] [PubMed] [Google Scholar]
- 14.Rocks N, Paulissen G, El Hour M, Quesada F, Crahay C, Gueders M, Foidart JM, Noel A, Cataldo D. Emerging roles of ADAM and ADAMTS metalloproteinases in cancer. Biochimie. 2008;90(2):369–79. doi: 10.1016/j.biochi.2007.08.008. [DOI] [PubMed] [Google Scholar]
- 15.Bell JH, Herrera AH, Li Y, Walcheck B. Role of ADAM17 in the ectodomain shedding of TNF-α and its receptors by neutrophils and macrophages. J Leukoc Biol. 2007;82:173–6. doi: 10.1189/jlb.0307193. [DOI] [PubMed] [Google Scholar]
- 16.Eto K, Puzon-McLaughlin W, Sheppard D, Sehara-Fujisawa A, Zhang XP, Takada Y. RGD-independent binding of integrin α9β1 to the ADAM-12 and -15 disintegrin domains mediates cell-cell interaction. J Biol Chem. 2000;275(45):34922–30. doi: 10.1074/jbc.M001953200. [DOI] [PubMed] [Google Scholar]
- 17.Boulday G, Coupel S, Coulon F, Soulillou JP, Charreau B. Antigraft antibody-mediated expression of metalloproteinases on endothelial cells. Differential expression of TIMP-1 and ADAM-10 depends on antibody specificity and isotype. Circ Res. 2001;88(4):430–7. doi: 10.1161/01.res.88.4.430. [DOI] [PubMed] [Google Scholar]
- 18.Hundhausen C, Misztela D, Berkhout TA, Broadway N, Saftig P, Reiss K, Hartmann D, Fahrenholz F, Postina R, Matthews V, Kallen KJ, Rose-John S, Ludwig A. The disintegrin-like metalloproteinase ADAM10 is involved in constitutive cleavage of CX3CL1 (fractalkine) and regulates CX3CL1-mediated cell-cell adhesion. Blood. 2003;102:1186–95. doi: 10.1182/blood-2002-12-3775. [DOI] [PubMed] [Google Scholar]
- 19.Gough PJ, Garton KJ, Wille PT, Rychlewski M, Dempsey PJ, Raines EW. A disintegrin and metalloproteinase 10-mediated cleavage and shedding regulates the cell surface expression of CXC chemokine ligand 16. J Immunol. 2004;172(6):3678–85. doi: 10.4049/jimmunol.172.6.3678. [DOI] [PubMed] [Google Scholar]
- 20.Nagano O, Murakami D, Hartmann D, De Strooper B, Saftig P, Iwatsubo T, Nakajima M, Shinohara M, Saya H. Cell-matrix interaction via CD44 is independently regulated by different metalloproteinases activated in response to extracellular Ca2+ influx and PKC activation. J Cell Biol. 2004;165(6):893–902. doi: 10.1083/jcb.200310024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garton KJ, Gough PJ, Philalay J, Wille PT, Blobel CP, Whitehead RH, Dempsey PJ, Raines EW. Stimulated shedding of vascular cell adhesion molecule 1 (VCAM-1) is mediated by tumor necrosis factor-alpha-converting enzyme (ADAM 17) J Biol Chem. 2003;278(39):37459–64. doi: 10.1074/jbc.M305877200. [DOI] [PubMed] [Google Scholar]
- 22.Tsakadze NL, Sithu SD, Sen U, English WR, Murphy G, D'Souza SE. Tumor necrosis factor-alpha-converting enzyme (TACE/ADAM-17) mediates the ectodomain cleavage of intercellular adhesion molecule-1 (ICAM-1) J Biol Chem. 2006;281(6):3157–64. doi: 10.1074/jbc.M510797200. [DOI] [PubMed] [Google Scholar]
- 23.Mechtersheimer S, Gutwein P, Agmon-Levin N, Stoeck A, Oleszewski M, Riedle S, Postina R, Fahrenholz F, Fogel M, Lemmon V, Altevogt P. Ectodomain shedding of L1 adhesion molecule promotes cell migration by autocrine binding to integrins. J Cell Biol. 2001;155(4):661–73. doi: 10.1083/jcb.200101099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huang AJ, Manning JE, Bandak TM, Ratau MC, Hanser KR, Silverstein SC. Endothelial cell cytosolic free calcium regulates neutrophil migration across monolayers of endothelial cells. J Cell Biol. 1993;120(6):1371–80. doi: 10.1083/jcb.120.6.1371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Schulz B, Pruessmeyer J, Maretzky T, Ludwig A, Blobel CP, Saftig P, Reiss K. Disintegrin Metalloprotease (ADAM) 10 Regulates Endothelial Permeability and T Cell Transmigration by Proteolysis of Vascular Endothelial Cadherin. Circ Res. 2008;102:XXX–XXX. doi: 10.1161/CIRCRESAHA.107.169805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Moss ML, Bartsch JW. Therapeutic benefits from targeting of ADAM family members. Biochemistry. 2004;43(23):7227–35. doi: 10.1021/bi049677f. [DOI] [PubMed] [Google Scholar]
- 27.Shimoda M, Hashimoto G, Mochizuki S, Ikeda E, Nagai N, Ishida S, Okada Y. Binding of ADAM28 to P-selectin glycoprotein ligand-1 enhances P-selectin-mediated leukocyte adhesion to endothelial cells. J Biol Chem. 2007;282(35):25864–74. doi: 10.1074/jbc.M702414200. [DOI] [PubMed] [Google Scholar]
- 28.Ham C, Levkau B, Raines EW, Herren B. ADAM15 is an adherens junction molecule whose surface expression can be driven by VE-cadherin. Exp Cell Res. 2002;279:239–47. doi: 10.1006/excr.2002.5606. [DOI] [PubMed] [Google Scholar]
- 29.Crawley JT, Lam JK, Rance JB, Mollica LR, O'Donnell JS, Lane DA. Proteolytic inactivation of ADAMTS13 by thrombin and plasmin. Blood. 2005;105(3):1085–93. doi: 10.1182/blood-2004-03-1101. [DOI] [PubMed] [Google Scholar]
- 30.Millichip MI, Dallas DJ, Wu E, Dale S, McKie N. The metallo-disintegrin ADAM10 (MADM) from bovine kidney has type IV collagenase activity in vitro. Biochem Biophys Res Commun. 1998;245(2):594–8. doi: 10.1006/bbrc.1998.8485. [DOI] [PubMed] [Google Scholar]
- 31.Raffetto JD, Khalil RA. Matrix metalloproteinases and their inhibitors in vascular remodeling and vascular disease. Biochem Pharmacol. 2008;75(2):346–59. doi: 10.1016/j.bcp.2007.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rapti M, Atkinson SJ, Lee MH, Trim A, Moss M, Murphy G. The isolated N-terminal domains of TIMP-1 and TIMP-3 are insufficient for ADAM10 inhibition. Biochem J. 2008;411(2):433–9. doi: 10.1042/BJ20071430. [DOI] [PubMed] [Google Scholar]
- 33.Chew DKW, Conte MS, Khalil RA. Matrix metalloproteinase-specific inhibition of Ca2+ entry mechanisms of vascular contraction. J Vasc Surg. 2004;40:1001–10. doi: 10.1016/j.jvs.2004.08.035. [DOI] [PubMed] [Google Scholar]
- 34.Raffetto JD, Ross RL, Khalil RA. Matrix metalloproteinase-2 induced venous dilation via hyperpolarization and activation of K+ channels: Relevance to varicose vein formation. J Vasc Surg. 2007;45:373–80. doi: 10.1016/j.jvs.2006.10.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.D'Angelo G, Mogford JE, Davis GE, Davis MJ, Meininger GA. Integrin-mediated reduction in vascular smooth muscle [Ca2+]i induced by RGD-containing peptide. Am J Physiol. 1997;272(4 Pt 2):H2065–70. doi: 10.1152/ajpheart.1997.272.4.H2065. [DOI] [PubMed] [Google Scholar]
- 36.Wu X, Mogford JE, Platts SH, Davis GE, Meininger GA, Davis MJ. Modulation of calcium current in arteriolar smooth muscle by αvβ3 and α5β1 integrin ligands. J Cell Biol. 1998;143:241–52. doi: 10.1083/jcb.143.1.241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ito K, Okamoto I, Araki N, Kawano Y, Nakao M, Fujiyama S, Tomita K, Mimori T, Saya H. Calcium influx triggers the sequential proteolysis of extracellular and cytoplasmic domains of E-cadherin, leading to loss of beta-catenin from cell-cell contacts. Oncogene. 1999;18(50):7080–90. doi: 10.1038/sj.onc.1203191. [DOI] [PubMed] [Google Scholar]
- 38.Sandoval R, Malik AB, Minshall RD, Kouklis P, Ellis CA, Tiruppathi C. Ca2+ signalling and PKCalpha activate increased endothelial permeability by disassembly of VE-cadherin junctions. J Physiol. 2001;533(Pt 2):433–45. doi: 10.1111/j.1469-7793.2001.0433a.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xu Z, Yu Y, Duh EJ. Vascular endothelial growth factor upregulates expression of ADAMTS1 in endothelial cells through protein kinase C signaling. Invest Ophthalmol Vis Sci. 2006;47(9):4059–66. doi: 10.1167/iovs.05-1528. [DOI] [PubMed] [Google Scholar]
- 40.Sanderson MP, Erickson SN, Gough PJ, Garton KJ, Wille PT, Raines EW, Dunbar AJ, Dempsey PJ. ADAM10 mediates ectodomain shedding of the betacellulin precursor activated by p-aminophenylmercuric acetate and extracellular calcium influx. J Biol Chem. 2005;280(3):1826–37. doi: 10.1074/jbc.M408804200. [DOI] [PubMed] [Google Scholar]