

Abstract
The essential metals iron, zinc and copper deposit near the Aβ (amyloid β-peptide) plaques in the brain cortex of AD (Alzheimer’s disease) patients. Plaque-associated iron and zinc are in neurotoxic excess at 1 mM concentrations. APP (amyloid precursor protein) is a single transmembrane metalloprotein cleaved to generate the 40-42-amino-acid Aβs, which exhibit metal-catalysed neurotoxicity. In health, ubiquitous APP is cleaved in a non-amyloidogenic pathway within its Aβ domain to release the neuroprotective APP ectodomain, APP(s). To adapt and counteract metal-catalysed oxidative stress, as during reperfusion from stroke, iron and cytokines induce the translation of both APP and ferritin (an iron storage protein) by similar mechanisms. We reported that APP was regulated at the translational level by active IL (interleukin)-1 (IL-1-responsive acute box) and IRE (iron-responsive element) RNA stem-loops in the 5′ untranslated region of APP mRNA. The APP IRE is homologous with the canonical IRE RNA stem-loop that binds the iron regulatory proteins (IRP1 and IRP2) to control intracellular iron homoeostasis by modulating ferritin mRNA translation and transferrin receptor mRNA stability. The APP IRE interacts with IRP1 (cytoplasmic cis-aconitase), whereas the canonical ferritin-H IRE RNA stem-loop binds to IRP2 in neural cell lines, and in human brain cortex tissue and in human blood lysates. The same constellation of RNA-binding proteins [IRP1/IRP2/poly(C) binding protein] control ferritin and APP translation with implications for the biology of metals in AD.
Keywords: amyloid precursor protein (APP), copper, ferritin, iron, oxidative stress, zinc
Iron accelerates amyloid/tau pathology, contributing to AD (Alzheimer’s disease)
Pathology of iron and AD
Several neurodegenerative disorders have been linked to brain neuronal loss owing to perturbations in iron metabolism, including AD [1], PD (Parkinson’s disease) [2], neurodegeneration with brain iron accumulation Type 1, and, more recently, prion-related disease [3] and ALS (amyotrophic lateral sclerosis) [4]. Disrupted iron metabolism in PD was detected by histological Perl’s staining of the SN (substantia nigra) region of brain sections, whereas, more recently, specific wavelength dispersive electron probe X-ray microanalysis coupled with cathodoluminescence spectroscopy showed iron levels to be increased almost 2-fold in single SN neurons in PD (mean neuronal iron 2838 compared with 1611, P<0.0001) [2].
Iron, in excess, is linked to neuron loss in the AD brain, wherein neurotoxic damage has been often associated with oxidative damage in affected brain cortex specimens [5], as demonstrated by enhanced membrane peroxidation by TBARS (thiobarbituric acid-reacting substances) and induced 8-oxoguanine accumulation in the DNA [6]. Haem binds Aβ (amyloid β-peptide) and has altered metabolism in the AD brain [7], where iron in the reduced Fe2+ state may accelerate cell damage by catalysing toxic hydroxyl radical formation. Here, iron imbalance may well have caused protracted cellular oxidative stress by iron catalysis of O2· [and superoxide (O2-)] to generate toxic hydroxyl radicals as a result of Fenton chemistry (see [8] for a review).
Iron and copper significantly enhanced the toxicity of Aβ in cultured neural cells, providing a direct link between excessive iron and loss of neuronal function seen in AD patients [9,10].
Metals provide one of the ultrastructural requirements needed for polymerization of Aβ in addition to pathological chaperones such as ACT (α1-antichymotrypsin) or ApoE (apolipoprotein E) [11]. Elemental profiles (sulfur, iron, copper and zinc) were observed microscopically to physically associate with amyloid plaques using synchrotron-scanning μ-XRF (X-ray fluorescence microscopy) [12]. This integral role iron plays in the aetiology of AD pathology was supported from MRI (magnetic resonance imaging) studies that revealed disease-associated elevated levels of ferritin iron, particularly in the neurons of the basal ganglia [13]. Spectroscopy confirmed that amyloid plaques harbour an increased burden of iron, copper and zinc [14], in which iron and zinc levels were at concentrations as high as the 1 mM level in the vicinity of amyloid plaques. Neurofibrillary tangles that are the second major hallmark of AD are negatively charged and capable of binding iron, which promotes further neurotoxicity during disease progression [15].
The intracellular iron chelator DFO (desferrioxamine) was at first thought to provide clinical benefit to AD patient by removing aluminium neurotoxic effect [16], but its therapeutic capacity now appears in line with the action of DFO chelator to remove excess iron as Fe3+ and thus decelerate disease progression [17].
Genetic links between iron and AD
Genetic evidence implicates synergy between the C282Y allele of the HFE (haemochromatosis) gene and the C2 allele of transferrin (blood iron carrier protein) as risk factors for developing AD [18]. Consistent with this, overexpression of the common HFE H63D and C282Y mutations in SH-SY5Y neuroblastoma cells lines enhanced expression of oxidative genes and mitochondrial gene expression patterns to lower mitochondrial membrane potential, lipid and protein peroxidative stress similarly to events observed in the AD brain [5,19]. Transgenically expressed H-ferritin (heavy ferritin)-null mutants were found to be lethal in the homozygous state, but heterozygotes exhibited reduced H-ferritin expression and an altered mouse brain gene expression profile that mimicked iron management seen in AD and PD [20]. Also brain metallothionein-III, a known potent copper/zinc-binding antioxidant protein, was measured to be lowered in the AD brain compared with control subjects [21]. Significant to the regulation for ferritin being associated with disease progression, mutation in the IRE (iron-responsive element) RNA stem-loop of the H-ferritin transcript caused autosomal dominant iron overload [22], and mice with the IRP2 (iron-regulatory protein 2) gene knocked out express a neurodegenerative phenotype reminiscent of PD [23] (see Figure 3).
Figure 3. Translational control of ferritin mRNA: model for APP mRNA translation.
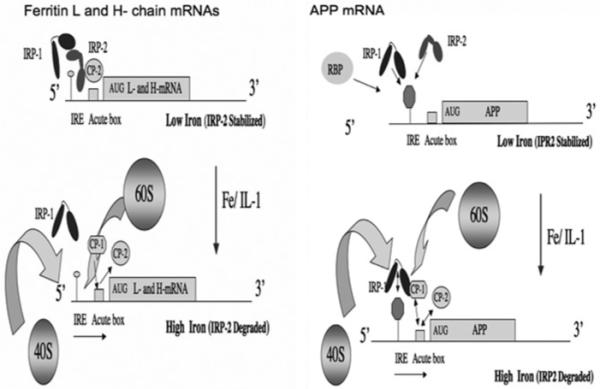
Left-hand panel: under conditions of low intracellular iron, IRP2 is stabilized and, with IRP1, interacts with the 5′-cap-specific IRE RNA stem-loops in the L- and H-ferritin 5′-UTR to repress their translation. Iron influx releases both IRP1 and IRP2 from binding to relieve suppression of L- and H-subunit translation [IRP1 and IRP2 comprise domains 1-3 separated from domain 4 by a hinge region (line)]. CP-1 and CP-2 mediate H-ferritin mRNA translation via the GC-rich IL-1-responsive acute box domain which is immediately in front of the H-ferritin start codon. IL-1 activates the 60S ribosome joining in presence of iron after removal of both CP-1 and CP-2 from the acute box, an event that occurs in the presence of iron where CP-1 binding to Aβ is replaced by CP-2. Right-hand panel: under investigation is the model for the iron-induced change of IRP1 interaction with the APP IRE to activate either 5′ cap translation or internal 40S ribosome entry and the onset of APP protein synthesis. Under conditions of low intracellular iron levels (i.e. iron chelation with DFO), IRP1 may bind at a higher affinity to the APP IRE to repress precursor translation. A role for CP1 and CP2 has yet to be determined for IL-1 activation of 60S ribosome subunit joining to the APP acute box in the presence of normal levels of intracellular iron.
APP (amyloid precursor protein) and ferritin expression is cytoprotective from iron-catalysed oxidative stress
APP is expressed in most cell types, both brain-derived and other tissues, including blood cells. In the amyloidogenic pathway, APP is a transmembrane metalloprotein [24] that is cleaved by β-secretase, also known as BACE (β-site amyloid precursor protein-cleaving enzyme), and γ-secretase (presenilin-associated complex) to generate the neurotoxic 40-42-amino-acid prefibrillar amyloid [25]. During the non-amyloidogenic path of APP metabolism, α-secretase is the zinc-metalloprotease that cleaves APP in the Aβ-peptide domain to release the 90 kDa ectodomain of APP [APP(s)]. In fact, APP(s) is at the highest physiological abundance when secreted from the α granules of platelets in response to thrombin where secreted APP can act as an anti-coagulant by binding to Factor IXa, leading to the production of fibrinogen from fibrin [26].
In health, the natural function of ferritin is for iron storage, and intrinsic ferroxidase activity imparted by its H-subunit protects endothelium from haem-aggravated oxidative stress as occurs during stroke and related diseases [27]. Likewise the secreted ectodomain of the membrane-associated APP, APP(s), was shown to protect neurons during conditions of haem release during haemorrhagic stroke [28,29], and this neurorescue activity was imparted via with the RERMS domain of immediately downstream of the KPI (Kunitz protease inhibitor) domain of APP(s) [30]. We reported that secretion of the APP is increased after IL (interleukin)-1-mediated stress responses by translational up-regulation of APP mRNA translation at the same time by as induction of α-secretase activity to generate the 90 kDa APP(s) [31]. These finding are consistent with existing published data that APP provides protection against catalysed oxidative stress, e.g. from mini-haemorrhagic lesions during the pathogenesis of AD or from ischaemic lesion and reperfusion after stroke [29].
APP translational control by iron and IL-1 (ferritin model)
The 3 kb APP transcript binds to many RNA-binding proteins, including proteins that bind to the AU-rich stability element in the 3′-UTR (untranslated region) of the transcript [32]. Most recently, the polyG quartet of coding region of the APP transcript was demonstrated to bind to the FMRP (fragile X mental retardation protein), indicating that post-synaptic FMRP binds to and regulates the translation of APP mRNA through metabotropic glutamate receptor activation [33]. An IRE-like RNA stem-loop sequence was hypothesized for the coding region of APP mRNA near the Aβ domain (Figure 1) [6]. However, as discussed below, the only bone fide and fully functional IRE RNA stem-loop that controls iron-dependent APP expression is present in the APP 5′-UTR [17,34]. Overall, the APP transcript can be seen as a ribonucleoprotein complex in which both IRP1 and IRP2 bind at its 5′-UTR site, FMRP binds to its coding region, whereas the U-rich AUF [ARE (AU-rich element)/poly(U)-binding/degradation factor] protein binds to the APP 3′-UTR to control mRNA stability according to serum status (Figure 1). In terms of AD pathology, we note that the APP IRE RNA secondary structure may be disrupted in the presence of an adjacent 5′-UTR-specific SNP (single nucleotide polymorphism) that has been genetically linked to increased risk for spontaneous AD [35].
Figure 1. RNA regulatory domains in the APP transcript.
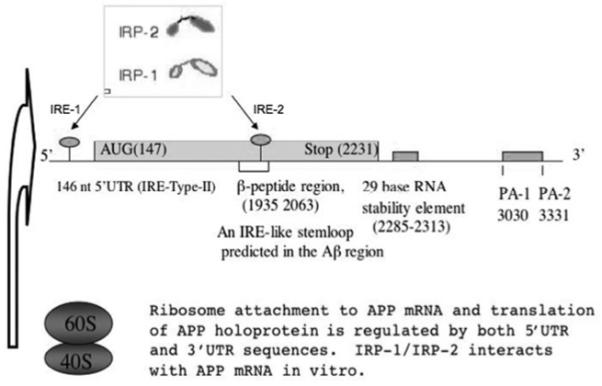
The 3 kb APP transcript is controlled at the level of mRNA translation by the action of 5′-UTR regulatory domains that are responsive to IL-1 and iron. The 3′-UTR is alternatively polyadenylated, and the longer APP transcript is translated more efficiently than the shorter transcript. A 29 nt RNA destabilizing element was mapped to the 3′-UTR of APP mRNA. A second IRE-like RNA sequence is depicted in the Aβ domain of the APP coding region, and FMRP is a cytoplasmic mRNA-binding protein that binds to the coding region of APP mRNA at a G-rich, G-quartet-like sequence. IL-1 co-induces APP mRNA translation and α-secretase cleavage in the APP Aβ domain where this primary inflammatory cytokine induces the non-amyloidogenic secretion of APP(s) from astrocytes.
IRPs control the translation of ferritin and APP mRNAs
Ferritin is the universal iron storage protein composed of a mixture of 24 subunits of light (L) and heavy (H) subunits. TfR (transferrin receptor) is responsible for transferrin-mediated iron transport from the blood into cells, and the regulation of iron homoeostasis is controlled by IRP binding to IREs in the UTRs of both of these key transcripts.
We found that intracellular levels of APP (and hence brain Aβ), like ferritin, is closely regulated at the translational level by an active IRE RNA stem-loop in the 146 nt 5′-UTR of APP mRNA [31]. Mutation in the H-ferritin IREs has been shown to cause human genetic diseases associated with perturbed iron metabolism (iron overload) [22], whereas a well-described SNP associated with AD risk exists in the APP 5′-UTR [35].
IRP1 and IRP2 control ferritin mRNA translation and TfR mRNA stability [8] (Figure 2 and 3). IRP1 and IRP2 bind at high affinity (Kd 40-100 pM) to the highly conserved IRE RNA stem-loops at the 5′ cap sites of the L- and H-ferritin mRNAs and the 3′-UTRs of TfR mRNA {other iron-specific transcripts also interact with IRP1 and IRP2 [36], including eALAS (erythroid isoform of aminolevulinate synthase) (haem biosynthesis)} [37]. Iron influx increases ferritin mRNA translation by releasing IRP1-IRP2 binding to the 5′ cap site IRE stem-loop, and the same IRP1-IRP2 interactions control TfR mRNA stability by 3′-UTR-specific IREs [36]. Relative to IRP2, IRP1 appears to be less critical as the iron-dependent mediator of the post-transcriptional regulation of intracellular ferritin and TfR levels, since only the IRP2-knockout mice exhibited a disruption to iron metabolism and caused an ataxia associated with neurodegeneration [23]. IRP1 exhibits a dual role as an RNA repressor protein in the absence of iron (binding tightly to IREs and preventing ribosome access to the 5′ cap sites of ferritin mRNAs), whereas iron promotes the formation of an [4Fe-4S] cluster in IRP1, which then exhibits enzymatic activity as a cytoplasmic cis-aconitase and no longer binds to IREs [6]. IRP2 is destabilized under conditions of iron influx, whereas IRP2 avidly binds the canonical IREs in ferritin and TfR mRNAs when intracellular iron is chelated [6].
Figure 2. Intracellular iron homoeostasis: the impact of inflammation on iron-associated gene expression.
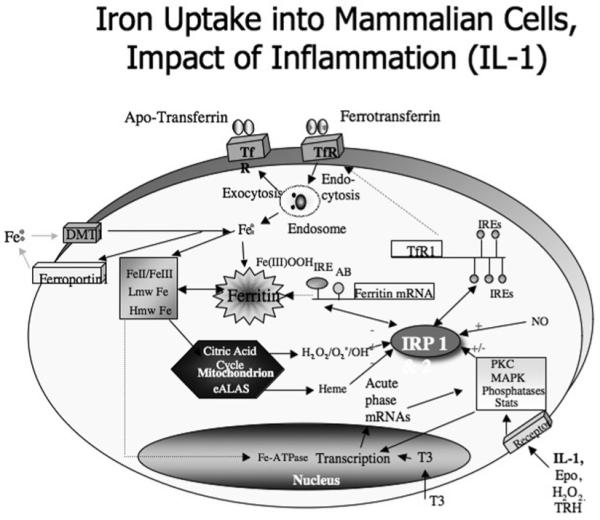
Iron transit across the cell-surface membrane is mediated by (i) ferrotransferrin internalization by the TfR, (ii) DMT1 (divalent metal transporter 1), (iii) ferroportin-mediated iron efflux from the duodenum into the blood. Ferritin mRNA translation is regulated by the modulated interaction between the IRPs and the IREs in the 5′-UTR of ferritin mRNA. MAPK signalling events influence ferritin translation and TfR activity and expression. AB, Aβ; Epo, erythropoietin; Hmw, high-molecular mass; Lmw, low-molecular-mass; PKC, protein kinase C; Stats, signal transducers and activators of transcription; TRH, thyrotropin-releasing hormone.
We reported that APP gene expression is controlled in response to cellular iron levels by related, but distinct, pathways to those governing ferritin translation. We established the presence of high-affinity binding of IRP1, and potentially IRP2, to radiolabelled probes for the precursor 5′-UTR (using both SH-SY5Y cells and recombinant IRP1) [17]. Work in progress is testing the relative strength of the IRP1-IRP2 interaction to APP mRNA in human brain (AD compared with control subjects). Figure 1 shows that the 3 kb APP mRNA sequence encodes two RNA stem-loops, encoding two 35 nt stretches related to the canonical IREs RNA stem-loop in ferritin mRNA, but transfection experiments validated the presence of the fully functional IRE (APP IRE-1) to APP 5′-UTR sequences [17,38]. It will be critical to rank whether IRP1 or IRP2 binds more selectively to the APP 5′-UTR, and determine whether this interaction drives iron-responsive APP mRNA translation and even mediates DFO-dependent therapeutic repression of neural APP (and amyloid) expression in vivo in human blood and brain tissue.
IL-1, APP and ferritin translational regulation: the role of poly(C)-binding proteins in their 5′-UTR acute box sequences
IL-1-positive microglial cells cluster around amyloid plaques, a phenomenon that also occurs after brain trauma, but illustrates that inflammation is a major component of AD pathophysiology. The primary inflammatory cytokine IL-1 is present at enhanced levels in the brains of patients with AD and Down’s syndrome [39] Meta-analysis has supported a significant association between the T/T genotype of the IL-1α gene and AD in which carriers of the IL-1α promoter-specific T/T genotype experience an onset of AD 9 years earlier than do carriers of the IL-lα C/C genotype [40].
IL-1, in addition to iron, regulates the translational control of both the L and H subunits of ferritin via distinct acute box domain sequences downstream of the IREs in their 5′-UTRs [41]. We demonstrated that the 5′-UTR of APP mRNA [42] is also IL-1-responsive through a related acute box domain [41] (Figures 2 and 3). In the case of ferritin, IL-1 stimulates the H-subunit gene expression at the level of mRNA translation as mediated by pyrimidine tract RNA poly(C)-binding proteins (CP-1 and CP-2), operating through the GC-rich IL-1-responsive acute box domain in the H-ferritin mRNA 5′-UTR which is 105 nt downstream from the 5′-cap-specific IRE in H-ferritin mRNA [41]. The APP mRNA acute box domain, like H-ferritin, is located immediately upstream of the start codon and may well also interact with CP-1 and CP-2 (Figure 3).
Like IRP1 and IRP2, the poly(C)-binding proteins (CP-1, CP-2, CP-3 and CP-4) are important in the post-transcriptional regulation of genes involved in iron metabolism (among other functions), including the translational control of 15-lipoxygenase (red blood cell nuclear membrane during erythropoeisis) [43], and in controlling α-globin mRNA stability during erythropoiesis [44]. Most interestingly, levels of the poly(C)-binding proteins in brain are increased after hypoxic assault (as in ischaemic stroke) [45], suggesting a link whereby CP-1 and CP-2 may themselves drive the translational activation of brain ferritin and APP generate neuroprotection after stroke. After the stoppage of blood flow to the brain during ischaemic stroke, the abundance of APP(s) levels are dramatically increased in region of the rat brain in the penumbra of an ischaemic lesion caused by mid artery occlusion [29].
Using astrocytes, we reported that short-term IL-1 (6-24 h) induced an increase in APP gene expression at the level of mRNA translation [3], while co-inducing α-secretase activity [ADAM (a disintegrin and metalloproteinase)-10 and ADAM-17] [31] expression, as a non-amyloidogenic pathway of increased neuroprotective APP(s) production. We concluded that an astrocytic p38 MAPK (mitogen-activated protein kinase) mediates a short-term IL-1-dependent burst of secretion of neuroprotective APP(s) in this novel non-amyloidogenic pathway [31]. Therefore, via the translational control pathway shown in Figures 2 and 3, IL-1 may induce astrocytes as key generators of increased production of neuroprotective APP(s), which is released to stressed neurons during an acute-phase response, such as occurs during stroke. In contrast, in the long term, this prolonged IL-1-activation of astrocytes and neurons in the AD brain may be corrupted towards pro-amyloidogenic events of increase APP translation with the development of amyloid protofibrils and neurodegenerative events.
The metalloprotein-attenuating agent, clioquinol, representing a low-affinity metal-sequestering agent [46], dissolved amyloid plaque formation and prevented cognitive decline in mice [46] and in human clinical trials. However, therapeutics based on iron chelation are consistent with earlier reports of a decreased rate of decline of AD in patients treated with intramuscular DFO (Kd for binding Fe3+ of 10-31 M) [16].
Abbreviations used
- Aβ
amyloid β-peptide
- AD
Alzheimer’s disease
- ADAM
a disintegrin and metalloproteinase
- APP
amyloid precursor protein
- APP(s)
secreted APP ectodomain
- CP
poly(C)-binding protein
- DFO
desferrioxamine
- FMRP
fragile X mental retardation protein
- H-
heavy
- HFE
haemochromatosis
- IL
interleukin
- IRE
iron-responsive element
- IRP
iron regulatory protein
- L-
light
- MAPK
mitogen-activated protein kinase
- PD
Parkinson’s disease
- SN
substantia nigra
- SNP
single nucleotide polymorphism
- TfR
transferrin receptor
- UTR
untranslated region
References
- 1.Zecca L, Youdim MB, Riederer P, Connor JR, Crichton RR. Iron, brain ageing and neurodegenerative disorders. Nat. Rev. Neurosci. 2004;5:863–873. doi: 10.1038/nrn1537. [DOI] [PubMed] [Google Scholar]
- 2.Oakley AE, Collingwood JF, Dobson J, Love G, Perrott HR, Edwardson JA, Elstner M, Morris CM. Individual dopaminergic neurons show raised iron levels in Parkinson disease. Neurology. 2007;68:1820–1825. doi: 10.1212/01.wnl.0000262033.01945.9a. [DOI] [PubMed] [Google Scholar]
- 3.Fernaeus S, Reis K, Bedecs K, Land T. Increased susceptibility to oxidative stress in scrapie-infected neuroblastoma cells is associated with intracellular iron status. Neurosci. Lett. 2005;389:133–136. doi: 10.1016/j.neulet.2005.07.032. [DOI] [PubMed] [Google Scholar]
- 4.Olsen MK, Roberds SL, Ellerbrock BR, Fleck TJ, McKinley DK, Gurney ME. Disease mechanisms revealed by transcription profiling in SOD1-G93A transgenic mouse spinal cord. Ann. Neurol. 2001;50:730–740. doi: 10.1002/ana.1252. [DOI] [PubMed] [Google Scholar]
- 5.Smith CD, Carney JM, Starke-Reed PE, Oliver CN, Stadtman ER, Floyd RA, Markesbery WR. Excess brain protein oxidation and enzyme dysfunction in normal aging and in Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 1991;88:10540–10543. doi: 10.1073/pnas.88.23.10540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Rogers JT. In: Sessler JL, Doctrow SR, McMurry TJ, Lippard SJ, editors. Amyloid precursor protein and ferritin translation: implications for metals and Alzheimer’s disease therapeutics; Medicinal Inorganic Chemistry (ACS Symposium Series 903); 2005; ???-??? [Google Scholar]
- 7.Atamna H, Frey WH., 2nd A role for heme in Alzheimer’s disease: heme binds amyloid β and has altered metabolism. Proc. Natl. Acad. Sci. U.S.A. 2004;101:11153–11158. doi: 10.1073/pnas.0404349101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rogers JT, Lahiri DK. Metal and inflammatory targets for Alzheimer’s disease. Curr. Drug Targets. 2004;5:535–551. doi: 10.2174/1389450043345272. [DOI] [PubMed] [Google Scholar]
- 9.Schubert D, Chevion M. The role of iron in β amyloid toxicity. Biochem. Biophys. Res. Commun. 1995;216:702–707. doi: 10.1006/bbrc.1995.2678. [DOI] [PubMed] [Google Scholar]
- 10.Huang X, Cuajungco MP, Atwood CS, Hartshorn MA, Tyndall JD, Hanson GR, Stokes KC, Leopold M, Multhaup G, Goldstein LE, et al. Cu(II) potentiation of Alzheimer Aβ neurotoxicity: correlation with cell-free hydrogen peroxide production and metal reduction. J. Biol. Chem. 1999;274:37111–37116. doi: 10.1074/jbc.274.52.37111. [DOI] [PubMed] [Google Scholar]
- 11.Nilsson LN, Bales KR, DiCarlo G, Gordon MN, Morgan D, Paul SM, Potter H. α-1-Antichymotrypsin promotes β-sheet amyloid plaque deposition in a transgenic mouse model of Alzheimer’s disease. J. Neurosci. 2001;21:1444–1451. doi: 10.1523/JNEUROSCI.21-05-01444.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu G, Huang W, Moir RD, Vanderburg CR, Lai B, Peng Z, Tanzi RE, Rogers JT, Huang X. Metal exposure and Alzheimer’s pathogenesis. J. Struct. Biol. 2006;155:45–51. doi: 10.1016/j.jsb.2005.12.011. [DOI] [PubMed] [Google Scholar]
- 13.Bartzokis G, Sultzer D, Mintz J, Holt LE, Marx P, Phelan CK, Marder SR. In vivo evaluation of brain iron in Alzheimer’s disease and normal subjects using MRI. Biol. Psychiatry. 1994;35:480–487. doi: 10.1016/0006-3223(94)90047-7. [DOI] [PubMed] [Google Scholar]
- 14.Lovell MA, Robertson JD, Teesdale WJ, Campbell JL, Markesbery WR. Copper, iron and zinc in Alzheimer’s disease senile plaques. J. Neurol. Sci. 1998;158:47–52. doi: 10.1016/s0022-510x(98)00092-6. [DOI] [PubMed] [Google Scholar]
- 15.Shin RW, Kruck TP, Murayama H, Kitamoto T. A novel trivalent cation chelator Feralex dissociates binding of aluminum and iron associated with hyperphosphorylated tau of Alzheimer’s disease. Brain Res. 2003;961:139–146. doi: 10.1016/s0006-8993(02)03893-3. [DOI] [PubMed] [Google Scholar]
- 16.Crapper McLachlan DR, Dalton AJ, Kruck TPA, Bell MY, Smith WL, Kalow W, Andrews DF. Intramuscular desferrioxamine in patients with Alzheimer’s disease. Lancet. 1991;337:1304–1308. doi: 10.1016/0140-6736(91)92978-b. [DOI] [PubMed] [Google Scholar]
- 17.Rogers JT, Randall JD, Cahill CM, Eder PS, Huang X, Gunshin H, Leiter L, McPhee J, Sarang SS, Utsuki T, et al. An iron-responsive element type II in the 5′-untranslated region of the Alzheimer’s amyloid precursor protein transcript. J. Biol. Chem. 2002;277:45518–45528. doi: 10.1074/jbc.M207435200. [DOI] [PubMed] [Google Scholar]
- 18.Robson KJ, Lehmann DJ, Wimhurst VL, Livesey KJ, Combrinck M, Merryweather-Clarke AT, Warden DR, Smith AD. Synergy between the C2 allele of transferrin and the C282Y allele of the haemochromatosis gene (HFE) as risk factors for developing Alzheimer’s disease. J. Med. Genet. 2004;41:261–265. doi: 10.1136/jmg.2003.015552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lee SY, Patton SM, Henderson RJ, Connor JR. Consequences of expressing mutants of the hemochromatosis gene (HFE) into a human neuronal cell line lacking endogenous HFE. FASEB J. 2007;21:564–576. doi: 10.1096/fj.06-6397com. [DOI] [PubMed] [Google Scholar]
- 20.Thompson K, Menzies S, Muckenthaler M, Torti FM, Wood T, Torti SV, Hentze MW, Beard J, Connor J. Mouse brains deficient in H-ferritin have normal iron concentration but a protein profile of iron deficiency and increased evidence of oxidative stress. J. Neurosci. Res. 2003;71:46–63. doi: 10.1002/jnr.10463. [DOI] [PubMed] [Google Scholar]
- 21.Yu WH, Lui W, Bergeron C, Niznik HB, Fraser P. Metallothionein III is reduced in Alzheimer’s disease. Brain Res. 2001;894:37–45. doi: 10.1016/s0006-8993(00)03196-6. [DOI] [PubMed] [Google Scholar]
- 22.Kato J, Fujikawa K, Kanda M, Fukuda N, Sasaki K, Takayama T, Kobune M, Takada K, Takimoto R, Hamada H, et al. A mutation, in the iron-responsive element of H ferritin mRNA, causing autosomal dominant iron overload. Am. J. Hum. Genet. 2001;69:191–197. doi: 10.1086/321261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.LaVaute T, Smith S, Cooperman S, Iwai K, Land W, Meyron-Holtz E, Drake SK, Miller G, Abu-Asab M, Tsokos M, et al. Targeted deletion of the gene encoding iron regulatory protein-2 causes misregulation of iron metabolism and neurodegenerative disease in mice. Nat. Genet. 2001;27:209–214. doi: 10.1038/84859. [DOI] [PubMed] [Google Scholar]
- 24.Multhaup G, Schlicksupp A, Hesse L, Beher D, Ruppert T, Masters CL, Beyreuther K. The amyloid precursor protein of Alzheimer’s disease in the reduction of copper(II) to copper(I) Science. 1996;271:1406–1409. doi: 10.1126/science.271.5254.1406. [DOI] [PubMed] [Google Scholar]
- 25.Walsh DM, Klyubin I, Fadeeva JV, Cullen WK, Anwyl R, Wolfe MS, Rowan MJ, Selkoe DJ. Naturally secreted oligomers of amyloid β protein potently inhibit hippocampal long-term potentiation in vivo. Nature. 2002;416:535–539. doi: 10.1038/416535a. [DOI] [PubMed] [Google Scholar]
- 26.Van Nostrand WE, Schmaier AH, Farrow JS, Cunningham DD. Protease nexin-II (amyloid β-protein precursor): a platelet α-granule protein. Science. 1990;248:745–748. doi: 10.1126/science.2110384. [DOI] [PubMed] [Google Scholar]
- 27.Balla G, Jacob HS, Balla J, Rosenberg M, Nath K, Apple F, Eaton JW, Vercellotti GM. Ferritin: a cytoprotective antioxidant strategem of endothelium. J. Biol. Chem. 1992;267:18148–18153. [PubMed] [Google Scholar]
- 28.Komori N, Kittel A, Kang D, Shackelford D, Masliah E, Zivin JA, Saitoh T. Reversible ischemia increases levels of Alzheimer amyloid protein precursor without increasing levels of mRNA in the rabbit spinal cord. Mol. Brain Res. 1997;49:103–112. doi: 10.1016/s0169-328x(97)00133-2. [DOI] [PubMed] [Google Scholar]
- 29.Fujioka M, Taoka T, Matsuo Y, Mishima K, Ogoshi K, Kondo Y, Tsuda M, Fujiwara M, Asano T, Sakaki T, et al. Magnetic resonance imaging shows delayed ischemic striatal neurodegeneration. Ann. Neurol. 2003;54:732–747. doi: 10.1002/ana.10751. [DOI] [PubMed] [Google Scholar]
- 30.Roch JM, Masliah E, Roch-Levecq AC, Sundsmo MP, Otero DA, Veinbergs I, Saitoh T. Increase of synaptic density and memory retention by a peptide representing the trophic domain of the amyloid β/A4 protein precursor. Proc. Natl. Acad. Sci. U.S.A. 1994;91:7450–7454. doi: 10.1073/pnas.91.16.7450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Bandyopadhyay S, Hartley DM, Cahill CM, Lahiri DK, Chattopadhyay N, Rogers JT. Interleukin-1α stimulates non-amyloidogenic pathway by α-secretase (ADAM-10 and ADAM-17) cleavage of APP in human astrocytic cells involving p38 MAP kinase. J. Neurosci. Res. 2006;84:106–118. doi: 10.1002/jnr.20864. [DOI] [PubMed] [Google Scholar]
- 32.Zaidi SH, Malter JS. Amyloid precursor protein mRNA stability is controlled by a 29-base element in the 3′-untranslated region. J. Biol. Chem. 1994;269:24007–24013. [PubMed] [Google Scholar]
- 33.Westmark CJ, Malter JS. FMRP mediates mGluR5-dependent translation of amyloid precursor protein. PLoS Biol. 2007;5:e52. doi: 10.1371/journal.pbio.0050052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bandyopadhyay S, Huang X, Cho H, Greig NH, Youdim MB, Rogers JT. Metal specificity of an iron-responsive element in Alzheimer’s APP mRNA 5′ untranslated region, tolerance of SH-SY5Y and H4 neural cells to desferrioxamine, clioquinol, VK-28, and a piperazine chelator. J. Neural Transm, Suppl. 2006:237–247. doi: 10.1007/978-3-211-33328-0_25. [DOI] [PubMed] [Google Scholar]
- 35.Athan ES, Lee JH, Arriaga A, Mayeux RP, Tycko B. Polymorphisms in the promoter of the human APP gene: functional evaluation and allele frequencies in Alzheimer disease. Arch. Neurol. 2002;59:1793–1799. doi: 10.1001/archneur.59.11.1793. [DOI] [PubMed] [Google Scholar]
- 36.Thomson AM, Rogers JT, Leedman PJ. Iron-regulatory proteins, iron-responsive elements and ferritin mRNA translation. Int. J. Biochem. Cell Biol. 1999;31:1139–1152. doi: 10.1016/s1357-2725(99)00080-1. [DOI] [PubMed] [Google Scholar]
- 37.Piccinelli P, Samuelsson T. Evolution of the iron-responsive element. RNA. 2007;13:952–966. doi: 10.1261/rna.464807. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Venti A, Giordano T, Eder P, Lahiri DK, Greig NH, Rogers JT. The integrated role of desferrioxamine and phenserine targeted to an iron-responsive element in the APP-mRNA 5′-untranslated region. Ann. N.Y. Acad. Sci. 2004;1035:34–48. doi: 10.1196/annals.1332.003. [DOI] [PubMed] [Google Scholar]
- 39.Griffin WS, Stanley LC, Ling C, White L, MacLeod V, Perrot LJ, White CL, 3rd, Araoz C. Brain interleukin 1 and S-100 immunoreactivity are elevated in Down syndrome and Alzheimer disease. Proc. Natl. Acad. Sci. U.S.A. 1989;86:7611–7615. doi: 10.1073/pnas.86.19.7611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Grimaldi LM, Casadei VM, Ferri C, Veglia F, Licastro F, Annoni G, Biunno I, De Bellis G, Sorbi S, Mariani C. Association of early-onset Alzheimer’s disease with an interleukin-1α gene polymorphism. Ann. Neurol. 2000;47:361–365. [PubMed] [Google Scholar]
- 41.Thomson AM, Cahill CM, Cho HH, Kassachau KD, Epis MR, Bridges KR, Leedman PJ, Rogers JT. The acute box cis-element in human heavy ferritin mRNA 5′-untranslated region is a unique translation enhancer that binds poly(C)-binding proteins. J. Biol. Chem. 2005;280:30032–30045. doi: 10.1074/jbc.M502951200. [DOI] [PubMed] [Google Scholar]
- 42.Rogers JT, Leiter LM, McPhee J, Cahill CM, Zhan SS, Potter H, Nilsson LN. Translation of the Alzheimer amyloid precursor protein mRNA is up-regulated by interleukin-1 through 5′-untranslated region sequences. J. Biol. Chem. 1999;274:6421–6431. doi: 10.1074/jbc.274.10.6421. [DOI] [PubMed] [Google Scholar]
- 43.Ostareck DH, Ostareck-Lederer A, Shatsky IN, Hentze MW. Lipoxygenase mRNA silencing in erythroid differentiation: the 3′UTR regulatory complex controls 60S ribosome subunit joining. Cell. 2001;104:281–290. doi: 10.1016/s0092-8674(01)00212-4. [DOI] [PubMed] [Google Scholar]
- 44.Chkheidze AN, Lyakhov DL, Makeyev AV, Moraqles J, Kong J, Liebhaber SA. Assembly of the α-globin mRNA stability complex reflects binary interaction between the pyrimidine-rich 3′untranslated region determinant and poly(C) binding protein αCP. Mol. Cell. Biol. 1999;19:4572–4681. doi: 10.1128/mcb.19.7.4572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Zhu Y, Sun Y, Mao XO, Jin KL, Greenberg DA. Expression of poly(C)-binding proteins is differentially regulated by hypoxia and ischemia in cortical neurons. Neuroscience. 2002;110:191–198. doi: 10.1016/s0306-4522(01)00522-x. [DOI] [PubMed] [Google Scholar]
- 46.Cherny RA, Atwood CS, Xilinas ME, Gray DN, Jones WD, McLean CA, Barnham KJ, Volitakis I, Fraser FW, Kim Y. Treatment with a copper-zinc chelator markedly and rapidly inhibits β-amyloid accumulation in Alzheimer’s disease transgenic mice. Neuron. 2001;30:665–676. doi: 10.1016/s0896-6273(01)00317-8. [DOI] [PubMed] [Google Scholar]