

Abstract
The motor neuron disease spinal muscular atrophy (SMA) causes profound muscle weakness that most often leads to early death. At autopsy, SMA is characterized by loss of motor neurons and muscle atrophy, but the initial cellular events that precipitate motor unit dysfunction and loss remain poorly characterized. Here, we examined the function and corresponding structure of neuromuscular junction (NMJ) synapses in a mouse model of severe SMA (hSMN2/delta7SMN/mSmn−/−). Surprisingly, most SMA NMJs remained innervated even late in the disease course; however they showed abnormal synaptic transmission. There was a two-fold reduction in the amplitudes of the evoked endplate currents (EPCs), but normal spontaneous miniature EPC (MEPC) amplitudes. These features in combination indicate reduced quantal content. SMA NMJs also demonstrated increased facilitation suggesting a reduced probability of vesicle release. By electron microscopy, we found a decreased density of synaptic vesicles that is likely to contribute to the reduced release probability. In addition to presynaptic defects, there were postsynaptic abnormalities. EPC and MEPC decay time constants were prolonged because of a slowed switch from the fetal acetylcholine receptor (AChR) γ-subunit to the adult ε-subunit. There was also reduced size of AChR clusters and small myofibers, which expressed an immature pattern of myosin heavy chains. Together these results indicate that impaired synaptic vesicle release at NMJs in severe SMA is likely to contribute to failed postnatal maturation of motor units and muscle weakness.
Keywords: spinal muscular atrophy, motor neuron, neuromuscular junction, synapse, synaptic vesicle, acetylcholine receptor
Introduction
The motor neuron disease spinal muscular atrophy (SMA) is the leading inherited cause of infant mortality. The severe form of SMA manifests during the course of motor unit development and causes profound weakness, with proximal muscles more affected than distal muscles and with intercostal and paraspinal muscles more affected than the diaphragm (Dubowitz, 1995; Crawford, 2003).
SMA is caused by mutation of the survival motor neuron 1 (SMN1) gene and deficiency of the SMN protein (Lefebvre et al., 1995, 1997). Presently, the best characterized function of SMN is to regulate the assembly of small nuclear ribonuclear proteins (snRNPs), which are critical components of the spliceosome (Pellizzoni, 2007).
Despite advances in the understanding of the molecular functions of the SMN protein, little is known about the evolution of cellular events that causes weakness in SMA animals and humans. Pathological investigations of human disease tissue obtained at autopsy have revealed loss of anterior horn cells (AHCs) in the spinal cord, reduced numbers of large diameter axons in ventral roots, and atrophy of myofibers in muscle (Crawford and Pardo, 1996), but these studies at end-stage disease provide little insight into the earliest structural and functional abnormalities of the motor unit. Detailed investigation of animal models potentially provides this opportunity. Studies in mouse models of other motor neuron diseases including amyotrophic lateral sclerosis (ALS) suggest that axonal degeneration with disconnection of NMJ synapses occurs presymptomatically and before AHC death (Frey et al., 2000; Fischer et al., 2004; Gould et al., 2006). Furthermore, prevention of AHC death does not ameliorate distal pathology (Sagot et al., 1995; Gould et al., 2006). These studies have suggested that distal axonal degeneration may be an early contributor to muscle weakness in motor neuron diseases.
We and others have previously shown that SMA Δ7 mice (hSMN2/delta7SMN/mSmn−/−) are weak soon after birth and die at ∼2 weeks of life, recapitulating severe human SMA (Le et al., 2005; Avila et al., 2007). It has been reported that at postnatal day 4 (P4) no loss of AHCs is detectable (Le et al., 2005) and only modest loss is evident at the end-stage (Monani et al., 2000; Le et al., 2005; Avila et al., 2007; Kariya et al., 2008), indicating that AHC loss may be a late event in SMA disease pathogenesis. Recent studies suggest that the earliest detectable pathology in SMAΔ7 mice is at the neuromuscular junction (NMJ) synapse (Kariya et al., 2008; Murray et al., 2008). This pathology includes accumulation of neurofilaments (NFs) at presynaptic terminals and simplification of postsynaptic terminals. The functional consequences of these abnormalities remain unknown.
Here we examined the electrophysiology and corresponding structure of NMJs in SMAΔ7 mice. We show that NMJ synapses remain surprisingly well connected late in the disease course. However, we find reduced density and release of synaptic vesicles at SMA NMJs that is associated with impaired morphological and biochemical maturation of the motor unit. These findings may provide the basis of the muscle weakness in SMA.
Materials and Methods
Mice.
All experiments were performed in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals, and were approved by the Johns Hopkins University and Wright State University Animal Care Committees. Breeder pairs for SMAΔ7 mice on the FVB background (hSMN2/delta7SMN/mSmn−/−) were purchased from Jackson Laboratories. Mice were genotyped by PCR of tail DNA as previously reported (Avila et al., 2007) and control littermates (CL) were those that were heterozygous or wild type at the mSmn allele.
Histology.
Dissected muscle tissues were flash frozen in freezing isopentane. Cryostat sections were cut and stained with hematoxylin and eosin (H&E). Myofiber morphology and diameter was assessed using a Microcolor RGB-MS-C camera, OpenLab software, and a Zeiss Axiophot microscope. The diameter of myofibers was determined by measuring the longest aspect of the shortest axis using the OpenLab measurement tool.
Immunohistochemistry.
For whole-mount staining of the tibialis anterior (TA) and diaphragm muscle NMJs, freshly dissected muscles were fixed in 4% paraformaldehyde (PFA), connective tissues were removed, and muscles were incubated with Alexa Fluor 555-conjugated α-bungarotoxin (Invitrogen). After incubation with methanol at −20°C for 5 min, muscles were incubated with antibodies against phospho-nonspecific NF (SMI312, Covance, 1:1000) and synaptic vesicle protein 2 (SV2) [Developmental Studies Hybridoma Bank (DSHB), 1:100] antibodies. Secondary antibody was FITC goat-anti-mouse IgG1 (Jackson Immunology, 1:200).
NMJs were also examined in serial muscle sections. Mice were transcardially perfused with 4% PFA and muscles were isolated, postfixed in 4% PFA for 2 h, cryoprotected in 30% sucrose, and cut longitudinally on a cryostat at 50 μm thickness. Polyclonal rabbit-anti-human synaptophysin (DAKO, 1:250), SV2 (DSHB, 1:100), or monoclonal mouse anti-NF 160 (Millipore, 1:1000) were used followed by the appropriate Alexa Fluor-conjugated secondary antibodies (Invitrogen). Staining for phosphorylated and nonphosphorylated NF heavy chains was done with SMI31 and SMI32 antibodies (Covance). Antibodies specific for the acetylcholine receptor (AChR) γ-subunit were provided by Norihiro Yumoto (Yumoto et al., 2005). Images were obtained with an inverted Zeiss LSM 510 META confocal microscope. Z-stack projections were made from serial scanning every 0.5 μm to reconstruct the NMJ. A denervated NMJ was defined as α-bungarotoxin-labeled endplate without occupying synaptophysin-stained axons and partial denervation was defined as ≤50% occupancy. A presynaptic NF accumulation was defined as NF occupying ≥1/3 of the postsynaptic terminal area. Approximately 200 NMJs from each of 3 CL and 4 SMA mice were quantified. The NMJ size was determined by measuring the area of α-bungarotoxin staining using Stereo Investigator 7 software (MicroBrightField). Postsynaptic structural complexity was defined as the number of perforations per NMJ. ∼150 NMJs per group of 3 CL and 3 SMA mice were randomly selected to determine structure and size.
Electron microscopy.
Three P13 CL and SMA mice were transcardially perfused with 3% gluteraldehyde/4% PFA. Tissues were dissected and postfixed. NMJs were identified by staining for acetylcholine esterase using sodium sulfide (1.25%, pH 6) and tissues were trimmed to the endplate band. Tissues were rinsed with 0.1 m Sorensen's buffer, osmicated using 2% osmium, dehydrated with serial alcohol dilutions and propylene oxide, and embedded in plastic. Thick sections (1 μm) were cut on an ultramicrotome, stained with toluidine blue, and viewed by light microscopy. Thin sections were cut at 60–90 nm, placed on Formvar grids, and viewed using an H-600 electron microscope (Hitachi). Surface contours of total presynaptic terminal area and a subarea within 200 nm distance of the presynaptic membrane were manually traced from electron micrographs at 15, 000× or higher. Measurements of these areas were generated using the Stereo Investigator 7 software (MicroBrightField). The number of synaptic vesicles and mitochondria within this area from 22 to 27 CL and 30–33 SMA presynaptic terminals was counted in a blinded manner. Densities of vesicles and mitochondria in the total presynaptic terminal or within 200 nm of the presynaptic terminal were determined by the total number per unit area. The number of docked vesicles per micrometer was determined by measuring the total number of vesicles within 20 nm of the presynaptic membrane and dividing by the length of presynaptic membrane (Samigullin et al., 2004).
Quantitative reverse transcription PCR.
RNA was isolated from tissues using TRIzol reagent and converted to cDNA as previously described (Avila et al., 2007). Primers to amplify AChR γ- and ϵ-subunits, myosin heavy chain (MyHC) isoforms, and 18S RNA were purchased from Applied Biosystems (ABI). Reactions were run in triplicate using the ABI Prism 7900 Sequence Detector System as described previously (Avila et al., 2007).
Physiologic recordings from NMJs.
After sacrificing the mouse with carbon dioxide inhalation, the TA muscle was removed and placed in a chamber continuously perfused with Ringer solution containing (in millimoles per liter) 118 NaCl, 3.5 KCl, 2 CaCl2, 0.7 MgSO4, 26.2 NaHCO3, 1.7 NaH2PO4, 5.5 glucose, (pH 7.3–7.4, 20−22°C) equilibrated with 95% O2 and 5% CO2. After pinning, muscle was stained with 10 μm 4-(4-diethylaminostyryl)-N-methylpyridinium iodide (4-Di-2ASP) and imaged with an upright epifluorescence microscope (Leica DMR) as described previously (Wang et al., 2004, 2005). At this concentration, 4-Di-2ASP staining enables visualization of surface nerve terminals as well as individual surface muscle fibers. All NMJs were imaged and impaled within 100 μm. Muscle fibers were crushed away from the endplate band and voltage clamped to –45 mV to avoid movement after nerve stimulation (Glavinoviæ, 1979). Two-electrode voltage clamp was used to measure the amplitude of miniature endplate currents (MEPCs) and endplate currents (EPCs) that were evoked after nerve stimulation. Use of voltage clamp avoided issues introduced by differences in muscle fiber size (i.e., differences in capacitance and input resistance) between CL and SMA mice. Quantal content was determined directly by dividing EPC amplitude by the average MEPC amplitude for a given NMJ. Repetitive stimulation was given by applying a 50 Hz train of 10 pulses.
Statistics.
Morphological and biochemical data were analyzed using Excel and Statistica software package (1999). Statistical significance was determined using either Student's t tests or two-way ANOVA. Physiological data were analyzed using Student's t test or nested ANOVA using Systat (Cranes Software).
Results
Modest denervation of SMA NMJs is limited to particular muscle groups
In many diseases of peripheral nerve and motor neurons, muscle weakness is attributed to axonal degeneration and disruption of NMJ synapses. We examined lumbar ventral nerve roots (data not shown) and distal intramuscular nerves in the tibialis anterior (TA), paraspinal (PS), intercostal (IC), and diaphragm muscles at the light- and electron-microscopic level in SMA and control littermate (CL) mice at a late stage of disease, postnatal day 13 (P13) (Fig. 1 A). Surprisingly, myelin and axonal structures were well preserved and axons undergoing Wallerian-like degeneration could not be identified. To determine whether weakness in SMA mice was caused by loss of the distal presynaptic terminals of NMJs, we examined NMJs at P1, P5, P9, and P13 in TA, gastrocnemius, quadriceps, hamstring, PS, IC, and diaphragm muscles. The PS and IC muscles were examined because they are known to be severely involved in human SMA, whereas the diaphragm is relatively spared (Dubowitz, 1995; Crawford, 2003). Unlike NMJs in SOD1G93A ALS mice, which show abundant denervation of NMJs early in the disease course (Frey et al., 2000; Fischer et al., 2004; Gould et al., 2006), we saw minimal denervation in SMA mice at all ages examined (Fig. 1 B). We also did not observe extra-junctional AChR clusters or compensatory axon terminal sprouting, features often seen during denervation. We quantified the frequency of postsynaptic NMJ terminals lacking synaptophysin staining in the TA, PS, and IC muscles at P13, a time just before death caused by paralysis. The denervation percentages were as follows: IC, 6.8 ± 1.7% SMA and 0.1 ± 0.1% CL (p = 0.02); PS, 15.3 ± 3.4% SMA and 0.1 ± 0.1% CL (p = 0.01); and TA, 3.0 ± 0.9% SMA and 1.8 ± 1.2 CL (p = 0.44). Given this minimal increase in total denervation, we also quantified partial denervation. We found a 40% increase (p = 0.02) in SMA PS muscle NMJs, but again no statistically significant increase in the TA muscle NMJs (p = 0.5). Quantification of the number of multiply innervated NMJs in P13 TA muscle showed an increase in SMA mice (26%) compared with CLs (11%, p = 0.05). We also examined SMA muscles in cross-section for the presence of atrophied muscle fibers typically seen during denervation of muscle. Rare areas of very small myofibers were limited to the PS and IC muscles (supplemental Fig. 1, available at www.jneurosci.org as supplemental material). Together, these findings indicate that denervation of NMJs is not the sole cause of muscle weakness in SMA mice.
Figure 1.
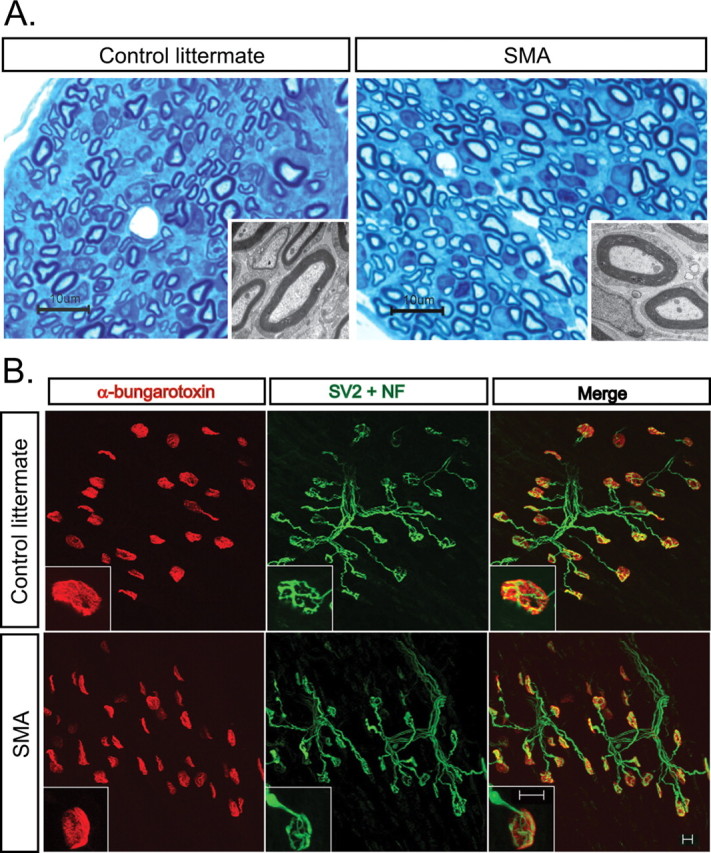
Limited denervation of SMA NMJs. A, Intramuscular axons examined by light microscopy and EM in the paraspinal muscle of P13 SMA and CL mice show normal axon and myelin sheath structure and no axonal degeneration. Scale bar, 10 μm and inset EM. B, IHC for NMJs in P13 TA muscle of SMA and CL mice showing that NMJs are well innervated. Scale bar, 10 μm.
Impaired synaptic vesicle release at SMA NMJs
Given that many NMJs were innervated in SMA mice at a stage when the mouse was profoundly weak, we postulated that there might be abnormal neuromuscular transmission at these structurally connected synapses. We therefore recorded endplate currents (EPCs) from the TA muscle isolated from P9 or P10 mice. Muscle fibers from the TA muscle were impaled with 2 electrodes, voltage clamped, and the nerve to the TA was stimulated as described previously (Wang et al., 2004). We compared the amplitude of EPCs between CL and SMA mice and found that the EPC amplitude was significantly decreased in SMA mice (p < 0.01, Fig. 2 A,B).
Figure 2.

Decreased EPC amplitude in SMA NMJs. A, Shown are representative EPCs from a CL and a SMA NMJ. The EPC from the SMA NMJ is less than ½ the amplitude of the CL. The stimulus artifact from nerve stimulation precedes the NMJ current by several milliseconds (ms). B, Shown is a bar graph of the average EPC amplitude from CL and SMA mice (*p < 0.01, n = 5 CL and 6 SMA mice).
The amplitude of the EPC is determined by both the number of synaptic vesicles released after nerve stimulation (quantal content) and the amplitude of the muscle response to the transmitter released from a single vesicle (quantal amplitude). We found that the amplitude of the miniature endplate current (MEPC), i.e., the postsynaptic response to transmitter released from an individual vesicle, was slightly reduced but did not reach statistical significance in SMA mice (Fig. 3 A,B). This suggested that both the amount of transmitter loaded into vesicles and the density of postsynaptic AChRs were normal. MEPC frequency was also similar in control littermates (1.65 ± 0.80 Hz) and SMA mice (1.71 ± 0.81, p = 0.97). High variability of MEPC frequency between individual NMJs was likely the result of the young age of the mice. To determine whether a reduction in number of vesicles released was the cause of the reduction in EPC amplitude, we calculated quantal content by dividing EPC amplitude by miniature endplate current (MEPC) amplitude and found a significant reduction (p < 0.01, Fig. 3 B).
Figure 3.

Reduction in quantal content underlies the reduction in EPC amplitude in SMA mice. A, Shown are representative average MEPCs from a CL and a SMA NMJ. There is no difference in amplitude. B, Shown are bar graphs of the average MEPC amplitude from CL and SMA pups (p = 0.38) and of quantal content (*p < 0.01).
Both the probability of vesicle release and the number of release sites determine quantal content. To determine whether reduced probability of vesicle release contributed to reduced quantal content in SMA mice, we inferred the probability of release by examining short-term synaptic plasticity during repetitive stimulation. Synapses with a high probability of release undergo depression, whereas those with a low probability of release undergo facilitation (Zucker and Regehr, 2002). We found that NMJs from SMA mice facilitated to a greater degree than CLs (Fig. 4 A,B). This suggests that reduced probability of release contributed to the reduction of NMJ current amplitude in SMA. Despite the increase in EPC amplitude during trains, EPCs from affected pups were still significantly decreased relative to control EPCs (19.2 ± 0.9 nA in control vs 10.8 ± 1.3 nA in SMA, p = 0.03). Unlike recently reported results in adult A2G mild SMA mice (Kariya et al., 2008), we did not observe synaptic failures during repetitive stimulation.
Figure 4.

Facilitation is increased in SMA NMJs. A, Shown are EPCs during a 50 Hz train of pulses for CL and SMA NMJs. In the CL NMJ there is a slight increase in EPC amplitude early in the train. By the end of the train, however, EPC amplitude has decreased back to the initial value. In the SMA NMJ, the initial EPC of the train is small, but there is an increase in EPC amplitude during the train. In both the CL and the SMA trace, stimulus artifacts were removed for clarity. B, The normalized change in average EPC amplitudes is plotted during trains of 50 Hz pulses for CL (n = 35 endplates from 3 muscles) and SMA NMJs (n = 17 endplates from 2 muscles). By the third pulse of the train, the SMA NMJs have facilitated to a greater degree that CL endplates. The facilitation is sustained throughout the train such that it is still statistically significant on the 10th pulse (p < 0.05).
It has been proposed that the morphological correlate of probability of release is the number of vesicles in the readily releasable pool (Regehr and Stevens, 2001). To examine the number of releasable synaptic vesicles in SMA presynaptic terminals, we examined NMJs in the TA muscle of P13 SMA and CL mice using electron microscopy (EM). In SMA mice, we observed abnormal accumulations of NFs in presynaptic terminals (Fig. 5 B). This correlated with accumulations of NFs that we observed by immunohistochemistry (IHC) (Fig. 5 A) and that have been reported by others (Kariya et al., 2008; Murray et al., 2008). IHC for SMI31, an antibody that recognizes phosphorylated NF, showed accumulations of NF in 84.8 ± 1.5% of SMA and in 1.0 ± 0.5% of CL P13 TA NMJs. In contrast, SMI32, an antibody specific for unphosphorylated NFs, showed accumulations in 34.4 ± 1.2% in SMA and 0.9 ± 0.4% in CL NMJs. This indicated that majority of accumulated NF was likely to be phosphorylated forms. We did not observe abundant accumulations of neurofilaments in more proximal intramuscular axons (data not shown). We also observed scattered branched tubulovesicular profiles in SMA presynaptic terminals. Mitochondrial density was reduced, although mitochondrial morphology was normal. Individual synaptic vesicles within presynaptic terminals had a normal diameter and morphology, but the overall density of synaptic vesicles within the total presynaptic terminal area was reduced by 56% (p < 0.001, Fig. 5 B,C).
Figure 5.
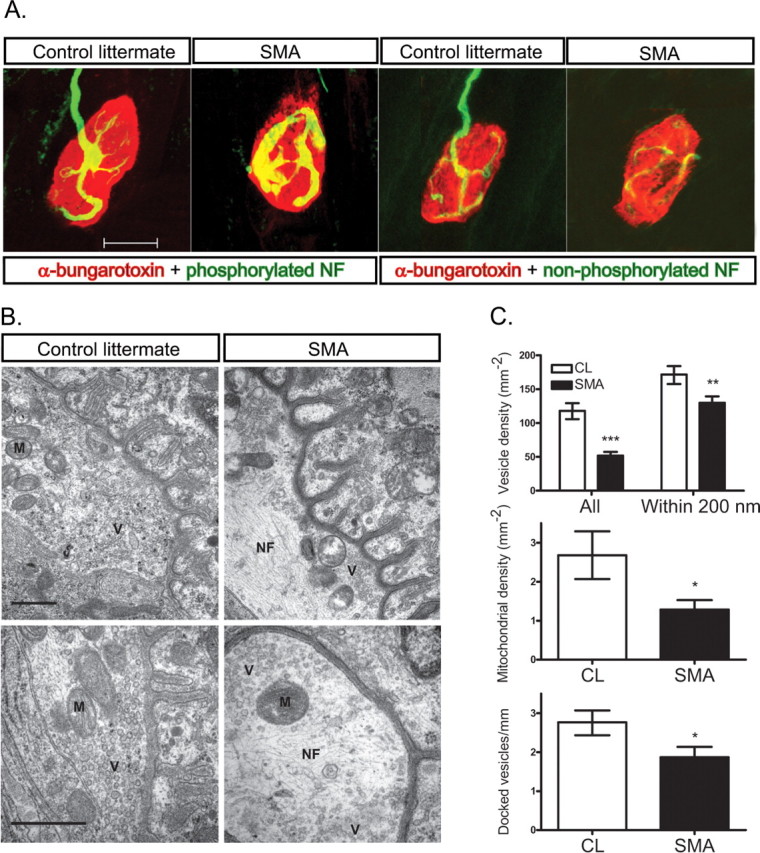
Decreased synaptic vesicle density at SMA NMJs. A, IHC for phosphorylated NFs and nonphosphorylated NFs shows accumulation of phosphorylated NFs within SMA presynaptic terminals. B, Electron microphotographs of NMJs in P13 TA muscle isolated from CL and SMA mice. Synaptic vesicles (V) are more homogeneously and densely distributed in CL compared with SMA mice. Accumulations of NF (N) are evident particularly at the center of SMA presynaptic terminals. Mitochondria (M) are reduced in number, but have normal morphology in SMA mice. Scale bar, 0.5 μm. C, Quantification of total synaptic vesicle density, synaptic density within 200 nm of the membrane, docked vesicle number, and mitochondria density (*p < 0.05, **p < 0.01, ***p < 0.001).
Synaptic vesicles were also abnormally distributed within presynaptic terminals in SMA mice. In control littermates, vesicles were diffusely and homogeneously scattered throughout the terminal, but in SMA mice they were shifted toward the membrane of the terminal and were patchy in distribution with some areas showing a high concentration of vesicles and other areas almost devoid of vesicles. Synaptic vesicle density was thus less severely reduced (24%) when considering just the area within 200 nm of the presynaptic terminal (Fig. 5 B,C). The number of docked vesicles, defined as those within 20 nm of the membrane, was reduced by 32% (p = 0.03) (Fig. 5 B,C). These studies indicate that reduced density of synaptic vesicles may contribute to reduced quantal content at SMA NMJs.
Immaturity of motor units in SMA mice
We next examined the decay times of the EPC and MEPC in SMA mice. Synaptic current decay time at the NMJ is determined by the activity of acetylcholine esterase, the duration of presynaptic vesicular release, and the intrinsic gating properties of the AChRs. The EPC decay time constant was significantly increased in SMA mice compared with control littermates (p = 0.02) (Fig. 6). Although the MEPC decay time constant was also prolonged, this difference did not reach statistical significance (p = 0.06) (Fig. 6). The AChR is made up of several subunits and during development there is a switch in the composition of the subunits from the γ-subunit, which defines the embryonic AChR, to the ε-subunit, which defines the adult AChR. Embryonic AChRs are known to have longer open times that adult AChRs (Sakmann and Brenner, 1978; Fischbach and Schuetze, 1980) and we have previously shown that the presence of embryonic AChRs prolongs the EPC more than the MEPC decay time (Wang et al., 2006). We therefore examined the expression of AChR subunits in SMA compared with CL mice. In rodents, the AChR subunit switch occurs between ∼P5 and P9 (Martinou and Merlie, 1991; Missias et al., 1996; Yumoto et al., 2005). In SMA hindlimb muscles, we observed abnormally increased gene expression of the γ-subunit and reduced expression of the ε-subunit by quantitative reverse transcription PCR (qRT-PCR) at the P5 and P9 time points (Fig. 7 A). IHC for the AChR γ confirmed persistent expression of the AChR γ protein in the TA, PS, and IC muscles of SMA mice at P9 (Fig. 7 B) and P13 (data not shown) when it was no longer detectable in CL mice (Fig. 3 B) (data not shown). This change in AChR γ protein expression appeared to lag behind and be more prominent than the change of gene expression as described previously (Yumoto et al., 2005).
Figure 6.

EPC and MEPC decay time constants are prolonged in NMJs from SMA mice. A, The CL and SMA MEPCs and EPCs from Figures 2 and 3 have been normalized and aligned by their peaks to allow for comparison of rates of decay. The MEPC and EPC from the SMA pups are gray and have slower rates of decay. B, Shown are bar graphs of the average EPC decay (*p = 0.02) and MEPC decay (p = 0.06) in CL and SMA NMJs.
Figure 7.
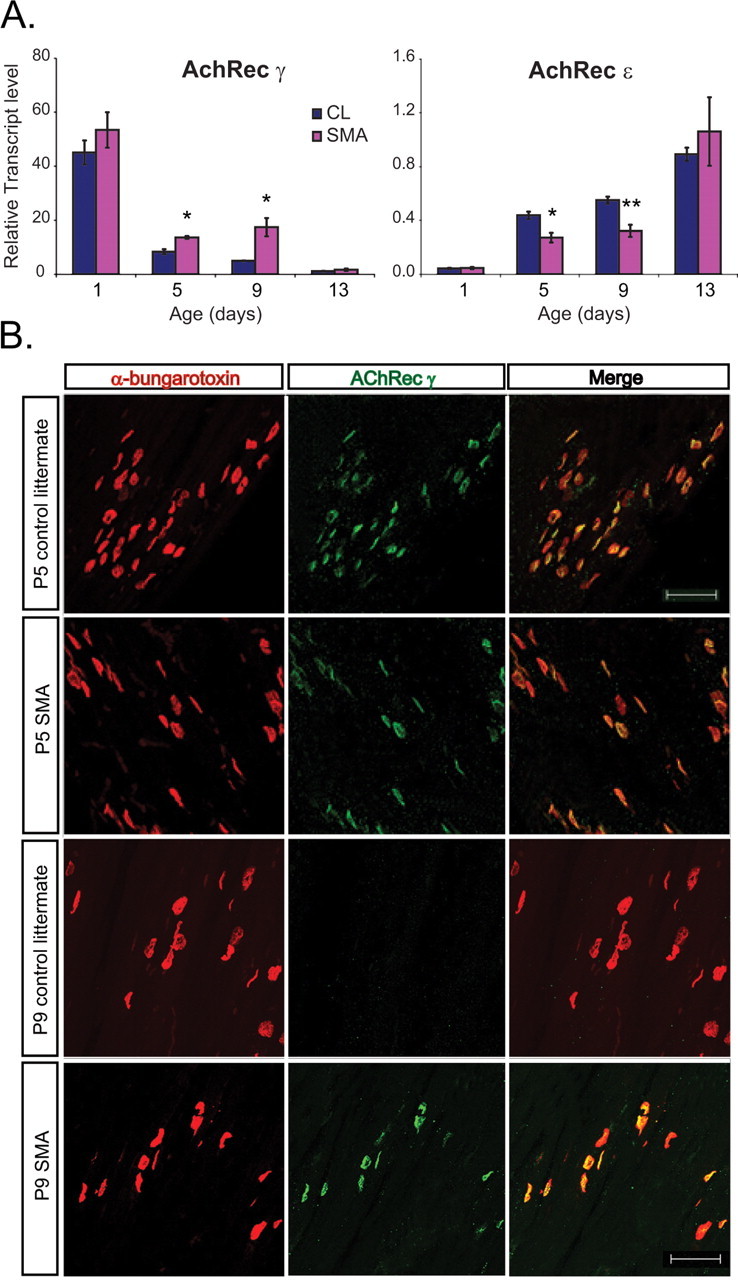
Delayed switch of AChR subunits in SMA NMJs. A, AChR γ and AChR ε transcript levels in all hindlimb muscle in SMA and CL mice at P1, P5, P9, and P13 show increased expression of AChR γ and reduced expression of AChR ε at P5 and P9 (*p < 0.05, **p < 0.005). B, IHC for AChR γ in the TA muscle indicates persistent expression at P9 SMA NMJs when expression is no longer evident in CL mice.
Comparison of AChR γ- and ϵ-subunit gene expression profiles in various muscle groups indicated muscle-specific differences in the rates of AChR maturation. NMJs in the IC and PS muscles, known to be severely affected in human SMA, showed the most significant increase in AChR γ expression, whereas the diaphragm, known to be relatively spared in SMA, showed less increase of AChR γ expression (supplemental Table 1, available at www.jneurosci.org as supplemental material). Gene expression profiles of other NMJ components including muscle specific kinase (MUSK), rapsyn, and acetylcholine esterase showed no differences between SMA and CL mice (data not shown).
In addition to AChR subunit switching, which changes the gating properties of the receptor, as NMJ postsynaptic terminals mature, they increase in size and sophistication evolving from a simple plaque-like appearance to a structure that contains multiple folds and perforations (Sanes and Lichtman, 2001). This elaboration is believed to amplify the action of Ach because of the high density of sodium channels in the depths of the folds and the high electrical resistance of the narrow sheets of cytoplasm. We observed that SMA NMJ postsynaptic terminals were small and simplified compared with CLs in all muscle groups examined (Fig. 8 A). Quantification of the area and number of perforations per NMJ in the TA muscle at P1, P5, P9, and P13 indicated that SMA NMJs were significantly smaller and less sophisticated beginning at P5 and showed little maturation thereafter (Fig. 8 B,C).
Figure 8.
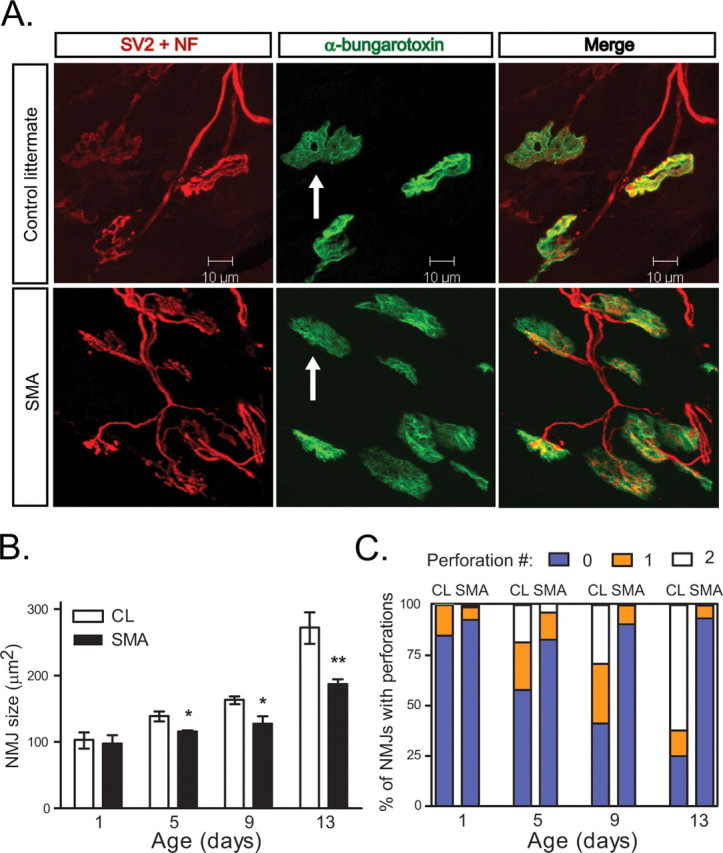
Simplified morphology of SMA NMJs. A, NMJs in paraspinal muscle of P13 SMA and CL mice showing simplification of the morphology of the postsynaptic terminals. Scale bar, 10 μm. B, NMJ area is decreased in the TA muscle of SMA mice at P5, P9, and P13 (*p < 0.05, **p < 0.005). C, SMA NMJs in the TA muscle have decreased numbers of perforations. The number of NMJs with no perforations is increased in SMA mice at P5, P9 (p < 0.05), and P13 (p < 0.005).
We further postulated that this delay of NMJ maturation might cause a failure of normal myofiber growth. Beginning at P9, SMA mice showed a reduced average diameter of myofibers and a reduced total number of myofibers in the TA muscle compared with CLs (Fig. 9 A–C). This was associated with an immature pattern of expression of MyHCs, major components of the contractile apparatus that are encoded by a developmentally regulated, multigene family (Shi and Garry, 2006). Specifically, SMA muscle showed an over-abundance of the perinatal form of MyHC and a deficiency of adult MyHC isoforms, particularly MyHC type IIb (Fig. 9 D). Examination of individual muscles showed that those muscles that showed the highest expression of the γ receptor, i.e., the PS and IC muscles, also showed the most profound deficit of MyHC IIb expression. In contrast, the diaphragm again showed minimal change (supplemental Table 1, available at www.jneurosci.org as supplemental material). This correlated with a reduced diameter of myofibers in the PS muscles, but little change in the diaphragm (supplemental Fig. 2, available at www.jneurosci.org as supplemental material). There was no significant change in the expression levels of muscle regulatory factors including MyoD, myogenin, MRF4, Mef2C, and Pax7 at P1, P5, and P9 (data not shown).
Figure 9.
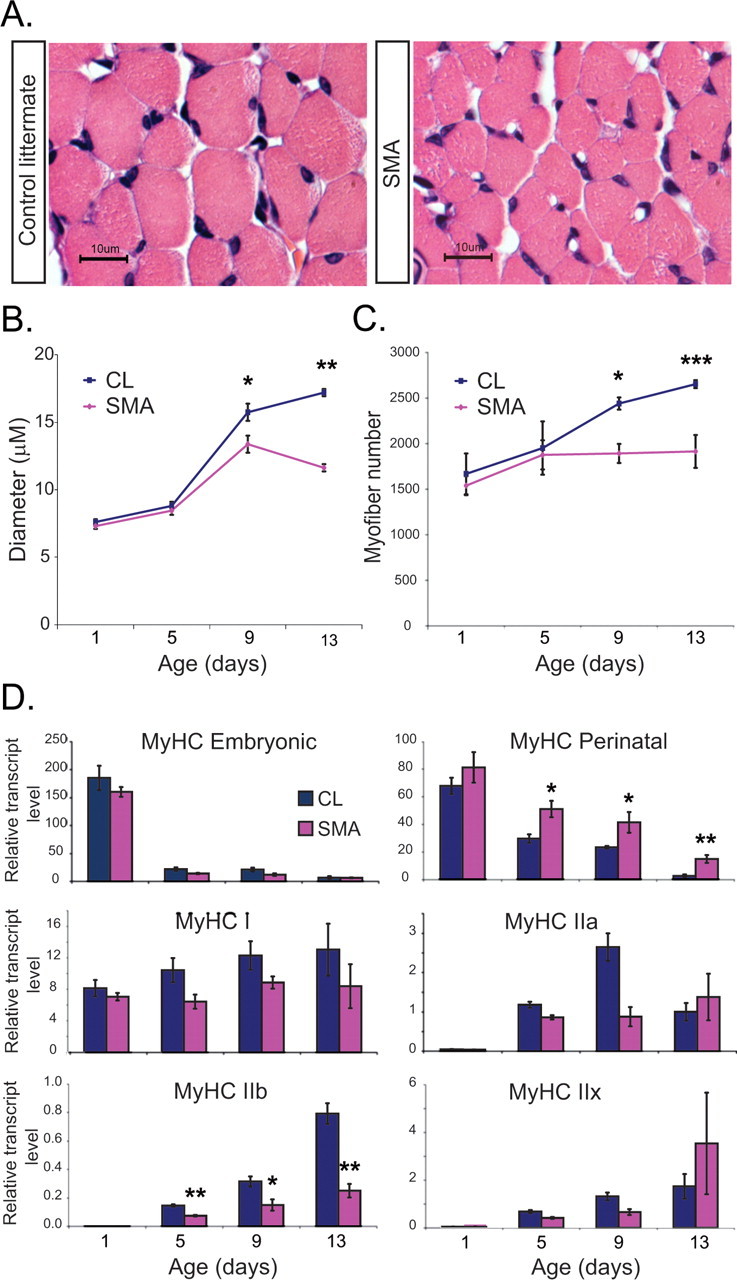
Impaired SMA neonatal muscle growth. A, Cross-sections of TA muscle stained with H&E from P13 CL and SMA mice showing reduced myofiber diameter in SMA mice. B, Quantification of average myofiber diameter in the TA muscle at P1, P5, P9, and P13 (n = 3 CL and 3 SMA mice per time point). C, Quantification of total myofiber number in TA muscle at P1, P5, P9, and P13 (n = 3 CL and 3 SMA mice per time point). D, Embryonic MyHC, perinatal MyHC, MyHC type I, MyHC type IIa, MyHC IIb, and MyHC type IIx transcript levels were quantified in all hind-limb muscle in SMA and CL mice at P1, P5, P9, and P13. Perinatal MyHC levels are increased and adult MyHC isoforms are reduced in SMA mice compared with CLs starting at P5 (n = 4–6 CL and SMA mice per time point, *p < 0.05, **p < 0.01, ***p < 0.005).
Discussion
Recent studies in different SMA mouse models have emphasized early morphological abnormalities of NMJ synapses including presynaptic terminal NF accumulation and simplified postsynaptic terminals (summarized in supplemental Table 2, available at www.jneurosci.org as supplemental material). In this study, we have extended these findings to show that there is NMJ synaptic dysfunction that precedes axonal degeneration and/or AHC death in severe SMA mice. This abnormality of neurotransmission is characterized by reduced quantal content that can be attributed to a decrease in the density of synaptic vesicles at motor nerve terminals. This abnormality of presynaptic vesicle release is accompanied by retarded maturation of postsynaptic NMJ terminals and myofibers, which may account for the profound muscle weakness and early death of severe SMA mice.
Degeneration of axon terminals has been proposed to be a critical early step in the pathogenesis of many neurodegenerative diseases, including motor neuron diseases. Motor neurons and muscle are dependent on one another for electrical, chemical, and trophic signals and early denervation of muscle has been observed in several motor neuron disease models (Fischer and Glass, 2007; Gould and Oppenheim, 2007). We were therefore surprised that even at late stages of disease in SMA mice when animals have fatal muscle weakness and nearly universally small myofibers, distal axonal degeneration was not evident and terminal denervation of NMJs was modest and limited only to those specific muscles groups known to be particularly affected in human SMA such as the intercostal and paraspinal muscles. Our results are in agreement with others (supplemental Table 2, available at www.jneurosci.org as supplemental material). These data suggest that axonal degeneration is not the initial cause of motor unit dysfunction in severe forms of SMA. This conclusion is consistent with a recent study showing that the Wallerian degeneration slow (Wld s) gene, which delays axonal degeneration and disease onset in some models of motor neuron disease, provides no benefit to severe SMA mice (Rose et al., 2008).
Rather than marked denervation of NMJs in severe SMA, we observed abnormal neurotransmission at connected synapses. Several human diseases characterized by muscle weakness are caused by either presynaptic or postsynaptic dysfunction of the NMJ including myasthenia gravis, Lambert-Eaton myasthenic syndrome (LEMS), and botulism. Abnormal NMJ function before denervation has also been shown in the motor neuron disease, canine SMA (Pinter et al., 1995; Balice-Gordon et al., 2000). Although the causative gene abnormality has not been identified for this dominantly inherited genetic disease, canine SMA causes severe weakness of proximal more than distal muscles phenocopying human and mouse SMA. Like SMA mice, SMA dogs demonstrate reduced quantal content at NMJs before degeneration of nerve terminals (Rich et al., 2002). In the case of canine SMA, reduced quantal content is associated with depression during repetitive stimulation rather than facilitation.
Facilitation of SMA NMJs during repetitive stimulation indicates that reduced probability of synaptic vesicle release contributes to the decrease of quantal content. Release probability is decreased when there are less vesicles available for immediate exocytosis; i.e., readily releasable vesicles. Readily releasable vesicles are thought to be those already docked at the presynaptic membrane, whereas those vesicles located farther from the membrane make up the reserve pool (Regehr and Stevens, 2001). In SMA NMJs, we observed a 56% decrease of overall synaptic vesicle density, but only a 32% reduction in the number of vesicles within 20 nm of the presynaptic membrane. This raises the question of whether reduced synaptic vesicle density fully accounts for the observed abnormality of SMA NMJ neurotransmission. Short term synaptic facilitation can be an indicator of reduced Ca2+ entry at presynaptic terminals (Zucker and Regehr, 2002). Facilitation is a prominent feature of LEMS in which autoantibodies target presynaptic P/Q channels reducing Ca2+ influx (Flink and Atchison, 2002). Cultured primary motor neuron isolated from SMA mice show impaired clustering of Cav2.2 channels at the distal terminal and abnormal calcium transients (Jablonka et al., 2007) suggesting that a functional abnormality of Ca2+ channels could contribute to the reduced probability of vesicle release seen in SMA mice.
At the time of NMJ dysfunction, most SMA presynaptic terminals also contained abnormal accumulations of NFs. Presynaptic terminal NF accumulations have been previously described in multiple SMA mouse models and increase with SMA disease progression (Cifuentes-Diaz et al., 2002; Kariya et al., 2008; Murray et al., 2008). NF accumulations have been observed in the soma and proximal axon of several neurodegenerative disorders including ALS; however, accumulation specifically at the motor nerve terminal is not seen in ALS mice and may be a specific feature of SMA (Cifuentes-Diaz et al., 2002; Kariya et al., 2008; Murray et al., 2008). NFs are synthesized in the cell body and transported anterogradely and retrogradely in the axon via the microtubule based motors, kinesin and dynein (for review, see Perrot et al., 2008). Carboxy-terminal phosphorylation of NFs may increase their affinity for dynein and promote retrograde transport. In the nerve terminal, degradation of NFs by calcium activated proteases is facilitated by their dephosphorylation. It is possible that NF accumulation resulting from altered phosphorylation or impaired degradation is itself a primary disease mechanism in SMA that results in structural distortion of the terminal, displacement of the normal organelles, including vesicles, and impaired neurotransmission. Another alternative is that accumulated NFs are a marker of a more generally disrupted neuronal cytoskeleton that has resulted in abnormal axonal transport and/or impaired vesicle trafficking within nerve terminals. The predominant cytoskeletal constituent of presynaptic terminals is actin, which together with synapsins likely plays important roles in regulating vesicle mobility and release at presynaptic terminals (Cingolani and Goda, 2008). The SMN protein has been shown to colocalize with β-actin mRNA at neuronal growth cones (Rossoll et al., 2003) and it has been postulated that SMN may be critical for the localization and subsequent processing of β-actin mRNA at nerve terminals. Recently, plastin 3, which regulates actin polymerization, was shown to modify the SMA disease phenotype in humans and SMA animal models (Oprea et al., 2008).
In addition to impaired presynaptic function, we also documented prolonged decay times of EPCs and MEPCs consistent with the abnormally persistent expression of embryonic type AChRs at SMA NMJs. This is associated with small and immature postsynaptic terminals and myofibers as has also been observed by others (supplemental Table 2, available at www.jneurosci.org as supplemental material). Because the rapid postnatal maturation of NMJs and growth of myofibers are known to be dependent on excitation from motor nerve terminals (Misgeld et al., 2002), we postulate that retarded motor unit maturation is a direct and expected compensation for impaired presynaptic terminal function during development. However, SMN deficiency has been proposed to cause intrinsic abnormalities of muscle development (Braun et al., 1995; Rajendra et al., 2007) and we cannot exclude the possibility that abnormalities of postsynaptic terminals retrogradely modulate presynaptic terminal function. Retrograde bone morphogenic protein (BMP) signaling regulates NMJ synaptic development and plasticity in Drosophila (Marqués and Zhang, 2006) and this pathway has recently been shown to modify disease in SMA flies (Chang et al., 2008). Regardless of the sequence of events, the end result is SMA myofibers that are small and have decreased levels of adult MyHC, particularly MyHC IIb. Myofiber contractile strength is determined by diameter (Lieber, 1992) and composition of MyHCs (Gokhin et al., 2008), and it is therefore likely that these deficits contribute to the profound muscle weakness observed in SMA mice. Importantly, the severity of these abnormalities varies with different muscle groups with the axial paraspinal and intercostal muscles being the most severely affected. This mirrors the pattern of muscle weakness seen in human SMA patients. The preferential involvement of these muscles suggests that there may be more severe synaptic dysfunction of medial compared with lateral motor column motor neurons. Future studies are needed to investigate whether there is also dysfunction of synaptic inputs to AHCs in SMA.
We have shown that impaired synaptic vesicle release at SMA NMJs may be an early step in SMA pathogenesis that precedes synaptic disconnection, axonal degeneration, or AHC death. These findings provide hope that therapeutic interventions that improve neurotransmission during the critical period of motor unit development could provide lasting benefit to motor unit function. Our recent results showing that SMA mice treated with the combination of a histone deacetylase inhibitor, trichostatin A, and improved nutrition have significantly increased survival, stable motor function, and well innervated NMJs (Narver et al., 2008) suggest that this may be possible. Dissection of the specific molecular mechanisms that cause synaptic dysfunction in SMA will likely lead to new therapeutic approaches for this devastating disease.
Footnotes
L.K. was supported by funding from the Robert Packard Center for ALS Research. C.J.S. was supported by a National Institute of Neurological Disorders and Stroke (NINDS) Career Transition Award (K22-NS0048199-01), Families of Spinal Muscular Atrophy Grant, and Howard Hughes Medical Institute Physician Scientist Award. M.M.R. was supported by NINDS Grant P01NS057228. B.G.B. was supported by an NINDS Competitive Fellowship Award. We thank Carol Rubright for EM support and Norihiro Yumoto for providing the AChR γ antibody.
References
- Avila et al., 2007.Avila AM, Burnett BG, Taye AA, Gabanella F, Knight MA, Hartenstein P, Cizman Z, Di Prospero NA, Pellizzoni L, Fischbeck KH, Sumner CJ. Trichostatin A increases SMN expression and survival in a mouse model of spinal muscular atrophy. J Clin Invest. 2007;117:659–671. doi: 10.1172/JCI29562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balice-Gordon et al., 2000.Balice-Gordon RJ, Smith DB, Goldman J, Cork LC, Shirley A, Cope TC, Pinter MJ. Functional motor unit failure precedes neuromuscular degeneration in canine motor neuron disease. Ann Neurol. 2000;47:596–605. [PubMed] [Google Scholar]
- Braun et al., 1995.Braun S, Croizat B, Lagrange MC, Warter JM, Poindron P. Constitutive muscular abnormalities in culture in spinal muscular atrophy. Lancet. 1995;345:694–695. doi: 10.1016/s0140-6736(95)90869-2. [DOI] [PubMed] [Google Scholar]
- Chang et al., 2008.Chang HC, Dimlich DN, Yokokura T, Mukherjee A, Kankel MW, Sen A, Sridhar V, Fulga TA, Hart AC, Van Vactor D, Artavanis-Tsakonas S. Modeling spinal muscular atrophy in Drosophila. PLoS ONE. 2008;3:e3209. doi: 10.1371/journal.pone.0003209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cifuentes-Diaz et al., 2002.Cifuentes-Diaz C, Nicole S, Velasco ME, Borra-Cebrian C, Panozzo C, Frugier T, Millet G, Roblot N, Joshi V, Melki J. Neurofilament accumulation at the motor endplate and lack of axonal sprouting in a spinal muscular atrophy mouse model. Hum Mol Genet. 2002;11:1439–1447. doi: 10.1093/hmg/11.12.1439. [DOI] [PubMed] [Google Scholar]
- Cingolani and Goda, 2008.Cingolani LA, Goda Y. Actin in action: the interplay between the actin cytoskeleton and synaptic efficacy. Nat Rev Neurosci. 2008;9:344–356. doi: 10.1038/nrn2373. [DOI] [PubMed] [Google Scholar]
- Crawford, 2003.Crawford TO. Spinal muscular atrophies. In: Jones H, Vivo DD, Darras B, editors. Neuromuscular disorders of infancy, childhood and adolescence: a clinician's approach. Philadelphia: Butterworth Heinemann; 2003. pp. 145–166. [Google Scholar]
- Crawford and Pardo, 1996.Crawford TO, Pardo CA. The neurobiology of childhood spinal muscular atrophy. Neurobiol Dis. 1996;3:97–110. doi: 10.1006/nbdi.1996.0010. [DOI] [PubMed] [Google Scholar]
- Dubowitz, 1995.Dubowitz V. Muscle disorders in childhood. Ed 2. Philadelphia: WB Saunders; 1995. [Google Scholar]
- Fischbach and Schuetze, 1980.Fischbach GD, Schuetze SM. A post-natal decrease in acetylcholine channel open time at rat end-plates. J Physiol. 1980;303:125–137. doi: 10.1113/jphysiol.1980.sp013275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischer and Glass, 2007.Fischer LR, Glass JD. Axonal degeneration in motor neuron disease. Neurodegener Dis. 2007;4:431–442. doi: 10.1159/000107704. [DOI] [PubMed] [Google Scholar]
- Fischer et al., 2004.Fischer LR, Culver DG, Tennant P, Davis AA, Wang M, Castellano-Sanchez A, Khan J, Polak MA, Glass JD. Amyotrophic lateral sclerosis is a distal axonopathy: evidence in mice and man. Exp Neurol. 2004;185:232–240. doi: 10.1016/j.expneurol.2003.10.004. [DOI] [PubMed] [Google Scholar]
- Flink and Atchison, 2002.Flink MT, Atchison WD. Passive transfer of Lambert-Eaton syndrome to mice induces dihydropyridine sensitivity of neuromuscular transmission. J Physiol. 2002;543:567–576. doi: 10.1113/jphysiol.2002.021048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frey et al., 2000.Frey D, Schneider C, Xu L, Borg J, Spooren W, Caroni P. Early and selective loss of neuromuscular synapse subtypes with low sprouting competence in motoneuron diseases. J Neurosci. 2000;20:2534–2542. doi: 10.1523/JNEUROSCI.20-07-02534.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glavinoviæ, 1979.Glavinoviæ MI. Voltage clamping of unparalysed cut rat diaphragm for study of transmitter release. J Physiol. 1979;290:467–480. doi: 10.1113/jphysiol.1979.sp012784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gokhin et al., 2008.Gokhin DS, Ward SR, Bremner SN, Lieber RL. Quantitative analysis of neonatal skeletal muscle functional improvement in the mouse. J Exp Biol. 2008;211:837–843. doi: 10.1242/jeb.014340. [DOI] [PubMed] [Google Scholar]
- Gould and Oppenheim, 2007.Gould TW, Oppenheim RW. Synaptic dysfunction in disease and after injury in the developing and adult nervous system: caveats in the choice of therapeutic intervention. Neurosci Biobehav Rev. 2007;31:1073–1087. doi: 10.1016/j.neubiorev.2007.04.015. [DOI] [PubMed] [Google Scholar]
- Gould et al., 2006.Gould TW, Buss RR, Vinsant S, Prevette D, Sun W, Knudson CM, Milligan CE, Oppenheim RW. Complete dissociation of motor neuron death from motor dysfunction by Bax deletion in a mouse model of ALS. J Neurosci. 2006;26:8774–8786. doi: 10.1523/JNEUROSCI.2315-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jablonka et al., 2007.Jablonka S, Beck M, Lechner BD, Mayer C, Sendtner M. Defective Ca2+ channel clustering in axon terminals disturbs excitability in motoneurons in spinal muscular atrophy. J Cell Biol. 2007;179:139–149. doi: 10.1083/jcb.200703187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kariya et al., 2008.Kariya S, Park GH, Maeno-Hikichi Y, Leykekhman O, Lutz C, Arkovitz MS, Landmesser LT, Monani UR. Reduced SMN protein impairs maturation of the neuromuscular junctions in mouse models of spinal muscular atrophy. Hum Mol Genet. 2008;17:2552–2569. doi: 10.1093/hmg/ddn156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le et al., 2005.Le TT, Pham LT, Butchbach ME, Zhang HL, Monani UR, Coovert DD, Gavrilina TO, Xing L, Bassell GJ, Burghes AH. SMNDelta7, the major product of the centromeric survival motor neuron (SMN2) gene, extends survival in mice with spinal muscular atrophy and associates with full-length SMN. Hum Mol Genet. 2005;14:845–857. doi: 10.1093/hmg/ddi078. [DOI] [PubMed] [Google Scholar]
- Lefebvre et al., 1995.Lefebvre S, Bürglen L, Reboullet S, Clermont O, Burlet P, Viollet L, Benichou B, Cruaud C, Millasseau P, Zeviani M, LePaslier D, Frézal J, Cohen D, Weissenbach J, Munnich A, Melki J. Identification and characterization of a spinal muscular atrophy-determining gene. Cell. 1995;80:155–165. doi: 10.1016/0092-8674(95)90460-3. [DOI] [PubMed] [Google Scholar]
- Lefebvre et al., 1997.Lefebvre S, Burlet P, Liu Q, Bertrandy S, Clermont O, Munnich A, Dreyfuss G, Melki J. Correlation between severity and SMN protein level in spinal muscular atrophy. Nat Genet. 1997;16:265–269. doi: 10.1038/ng0797-265. [DOI] [PubMed] [Google Scholar]
- Lieber, 1992.Lieber RL. Skeletal muscle structure and function. Baltimore: Williams & Wilkins; 1992. [Google Scholar]
- Marqués and Zhang, 2006.Marqués G, Zhang B. Retrograde signaling that regulates synaptic development and function at the Drosophila neuromuscular junction. Int Rev Neurobiol. 2006;75:267–285. doi: 10.1016/S0074-7742(06)75012-7. [DOI] [PubMed] [Google Scholar]
- Martinou and Merlie, 1991.Martinou JC, Merlie JP. Nerve-dependent modulation of acetylcholine receptor epsilon-subunit gene expression. J Neurosci. 1991;11:1291–1299. doi: 10.1523/JNEUROSCI.11-05-01291.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Misgeld et al., 2002.Misgeld T, Burgess RW, Lewis RM, Cunningham JM, Lichtman JW, Sanes JR. Roles of neurotransmitter in synapse formation: development of neuromuscular junctions lacking choline acetyltransferase. Neuron. 2002;36:635–648. doi: 10.1016/s0896-6273(02)01020-6. [DOI] [PubMed] [Google Scholar]
- Missias et al., 1996.Missias AC, Chu GC, Klocke BJ, Sanes JR, Merlie JP. Maturation of the acetylcholine receptor in skeletal muscle: regulation of the AChR gamma-to-epsilon switch. Dev Biol. 1996;179:223–238. doi: 10.1006/dbio.1996.0253. [DOI] [PubMed] [Google Scholar]
- Monani et al., 2000.Monani UR, Sendtner M, Coovert DD, Parsons DW, Andreassi C, Le TT, Jablonka S, Schrank B, Rossoll W, Prior TW, Morris GE, Burghes AH. The human centromeric survival motor neuron gene (SMN2) rescues embryonic lethality in Smn(−/−) mice and results in a mouse with spinal muscular atrophy. Hum Mol Genet. 2000;9:333–339. doi: 10.1093/hmg/9.3.333. [DOI] [PubMed] [Google Scholar]
- Murray et al., 2008.Murray LM, Comley LH, Thomson D, Parkinson N, Talbot K, Gillingwater TH. Selective vulnerability of motor neurons and dissociation of pre and post-synaptic pathology at the neuromuscular junction in mouse models of spinal muscular atrophy. Hum Mol Genet. 2008;17:949–962. doi: 10.1093/hmg/ddm367. [DOI] [PubMed] [Google Scholar]
- Narver et al., 2008.Narver HL, Kong L, Burnett BG, Choe DW, Bosch-Marcé M, Taye AA, Eckhaus MA, Sumner CJ. Sustained improvement of spinal muscular atrophy mice treated with trichostatin a plus nutrition. Ann Neurol. 2008;64:465–470. doi: 10.1002/ana.21449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oprea et al., 2008.Oprea GE, Kröber S, McWhorter ML, Rossoll W, Müller S, Krawczak M, Bassell GJ, Beattie CE, Wirth B. Plastin 3 is a protective modifier of autosomal recessive spinal muscular atrophy. Science. 2008;320:524–527. doi: 10.1126/science.1155085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pellizzoni, 2007.Pellizzoni L. Chaperoning ribonucleoprotein biogenesis in health and disease. EMBO Rep. 2007;8:340–345. doi: 10.1038/sj.embor.7400941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrot et al., 2008.Perrot R, Berges R, Bocquet A, Eyer J. Review of the multiple aspects of neurofilament functions, and their possible contribution to neurodegeneration. Mol Neurobiol. 2008;38:27–65. doi: 10.1007/s12035-008-8033-0. [DOI] [PubMed] [Google Scholar]
- Pinter et al., 1995.Pinter MJ, Waldeck RF, Wallace N, Cork LC. Motor unit behavior in canine motor neuron disease. J Neurosci. 1995;15:3447–3457. doi: 10.1523/JNEUROSCI.15-05-03447.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajendra et al., 2007.Rajendra TK, Gonsalvez GB, Walker MP, Shpargel KB, Salz HK, Matera AG. A Drosophila melanogaster model of spinal muscular atrophy reveals a function for SMN in striated muscle. J Cell Biol. 2007;176:831–841. doi: 10.1083/jcb.200610053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Regehr and Stevens, 2001.Regehr WG, Stevens CF. Physiology of synaptic transmission and short-term plasticity. Baltimore: Johns Hopkins UP; 2001. [Google Scholar]
- Rich et al., 2002.Rich MM, Wang X, Cope TC, Pinter MJ. Reduced neuromuscular quantal content with normal synaptic release time course and depression in canine motor neuron disease. J Neurophysiol. 2002;88:3305–3314. doi: 10.1152/jn.00271.2002. [DOI] [PubMed] [Google Scholar]
- Rose et al., 2008.Rose FF, Jr, Meehan PW, Coady TH, Garcia VB, Garcia ML, Lorson CL. The Wallerian degeneration slow (Wld. (s)) gene does not attenuate disease in a mouse model of spinal muscular atrophy. Biochem Biophys Res Commun. 2008;375:119–123. doi: 10.1016/j.bbrc.2008.07.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rossoll et al., 2003.Rossoll W, Jablonka S, Andreassi C, Kröning AK, Karle K, Monani UR, Sendtner M. Smn, the spinal muscular atrophy-determining gene product, modulates axon growth and localization of beta-actin mRNA in growth cones of motoneurons. J Cell Biol. 2003;163:801–812. doi: 10.1083/jcb.200304128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sagot et al., 1995.Sagot Y, Dubois-Dauphin M, Tan SA, de Bilbao F, Aebischer P, Martinou JC, Kato AC. Bcl-2 overexpression prevents motoneuron cell body loss but not axonal degeneration in a mouse model of a neurodegenerative disease. J Neurosci. 1995;15:7727–7733. doi: 10.1523/JNEUROSCI.15-11-07727.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakmann and Brenner, 1978.Sakmann B, Brenner HR. Change in synaptic channel gating during neuromuscular development. Nature. 1978;276:401–402. doi: 10.1038/276401a0. [DOI] [PubMed] [Google Scholar]
- Samigullin et al., 2004.Samigullin D, Bill CA, Coleman WL, Bykhovskaia M. Regulation of transmitter release by synapsin II in mouse motor terminals. J Physiol. 2004;561:149–158. doi: 10.1113/jphysiol.2004.073494. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanes and Lichtman, 2001.Sanes JR, Lichtman JW. Induction, assembly, maturation and maintenance of a postsynaptic apparatus. Nat Rev Neurosci. 2001;2:791–805. doi: 10.1038/35097557. [DOI] [PubMed] [Google Scholar]
- Shi and Garry, 2006.Shi X, Garry DJ. Muscle stem cells in development, regeneration, and disease. Genes Dev. 2006;20:1692–1708. doi: 10.1101/gad.1419406. [DOI] [PubMed] [Google Scholar]
- Wang et al., 2004.Wang X, Engisch KL, Li Y, Pinter MJ, Cope TC, Rich MM. Decreased synaptic activity shifts the calcium dependence of release at the mammalian neuromuscular junction in vivo . J Neurosci. 2004;24:10687–10692. doi: 10.1523/JNEUROSCI.2755-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang et al., 2005.Wang X, Li Y, Engisch KL, Nakanishi ST, Dodson SE, Miller GW, Cope TC, Pinter MJ, Rich MM. Activity-dependent presynaptic regulation of quantal size at the mammalian neuromuscular junction in vivo . J Neurosci. 2005;25:343–351. doi: 10.1523/JNEUROSCI.3252-04.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang et al., 2006.Wang X, Engisch KL, Teichert RW, Olivera BM, Pinter MJ, Rich MM. Prolongation of evoked and spontaneous synaptic currents at the neuromuscular junction after activity blockade is caused by the upregulation of fetal acetylcholine receptors. J Neurosci. 2006;26:8983–8987. doi: 10.1523/JNEUROSCI.2493-06.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yumoto et al., 2005.Yumoto N, Wakatsuki S, Sehara-Fujisawa A. The acetylcholine receptor gamma-to-epsilon switch occurs in individual endplates. Biochem Biophys Res Commun. 2005;331:1522–1527. doi: 10.1016/j.bbrc.2005.04.081. [DOI] [PubMed] [Google Scholar]
- Zucker and Regehr, 2002.Zucker RS, Regehr WG. Short-term synaptic plasticity. Annu Rev Physiol. 2002;64:355–405. doi: 10.1146/annurev.physiol.64.092501.114547. [DOI] [PubMed] [Google Scholar]