

Abstract
Development of axonal tracts requires interactions between growth cones and the environment. Tracts such as the anterior commissure and internal capsule are defective in mice with null mutation of Celsr3. We generated a conditional Celsr3 allele, allowing regional inactivation. Inactivation in telencephalon, ventral forebrain, or cortex demonstrated essential roles for Celsr3 in neurons that project axons to the anterior commissure and subcerebral targets, as well as in cells that guide axons through the internal capsule. When Celsr3 was inactivated in cortex, subcerebral projections failed to grow, yet corticothalamic axons developed normally, indicating that besides guidepost cells, additional Celsr3-independent cues can assist their progression. These observations provide in vivo evidence that Celsr3-mediated interactions between axons and guidepost cells govern axonal tract formation in mammals.
Formation of axonal tracts is essential for brain wiring, and several cues, such as extracellular molecules, guidepost cells, and fiber-to-fiber interactions, guide growing axons to their targets (1). We showed previously that the anterior commissure (AC) and the internal capsule (IC) are defective in constitutive Celsr3 mutant mice (2). Celsr1, Celsr2, Celsr3 are homologous to Drosophila flamingo/starry night (Fmi/stan) (3, 4), which collaborates with Frizzled and Van Gogh to regulate planar cell polarity (PCP) and neurite development. Celsr proteins are seven-pass transmembrane cadherins and are thought to mediate cell adhesion via homophilic interactions. Celsr3 is expressed in postmigratory neurons in cortex, ventral telencephalon, olfactory structures, and thalamus during development and progressively down-regulated during maturation (5).
To probe forebrain wiring, we generated a conditional mutant allele that allows inactivation of Celsr3 by crosses with mice that express the Cre recombinase in region-specific manners (6). This allele was produced by flanking exons 19 to 27, deletion of which generates a null allele (2), with loxP sites (“floxed” allele, Celsr3f ). Mice with regional inactivation were produced by crossing double heterozygous males (Celsr3+/−;Cre/+) with homozygous Celsr3f/f females. Celsr3 inactivation requires Cre-mediated modification of one floxed allele only, thereby increasing efficiency. To facilitate reading, we use the shorthand “|”: for example, Celsr3|Foxg1 is short for Celsr3f/−;Foxg1-Cre/+. Cre-expressing strains were crossed with ROSA26R mice (7) to verify that Cre activation proceeded as described. Inactivation was further confirmed by in situ hybridization with a probe complementary to exons 19 to 27 that allows detection of intact Celsr3 mRNA only and by Western blot (fig. S1). Control animals were double heterozygotes, e.g., Celsr3f/+;Foxg1-Cre/+.
We first examined the role of Celsr3 during formation of the AC. In Celsr3|Foxg1 mice in which Celsr3 is inactivated in the telencephalon (8), the AC was absent (Fig. 1, A, B, D, and E). It was also drastically affected in Celsr3|Emx1 mice that express Cre in olfactory structures and neocortex (9). Diminutive bundles originating from olfactory nuclei ran caudally and turned, but never crossed the midline, as confirmed by injecting 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (DiI) in the olfactory bulb (Fig. 1, C and F). This phenotype could reflect the kinetics of Cre activation: Axonal growth from olfactory nuclei may be initiated before a full inactivation of Celsr3 is achieved. In Celsr3|Emx1 mice, Celsr3 mRNA is absent from the olfactory and the temporal neurons of origin of the AC but present in the cells located along the pathway. Thus, normal Celsr3 activity is likely required cell-autonomously in the neurons of origin of the AC. In all other crosses, namely Celsr3|Gsh2, Celsr3|Nkx2.1, double Celsr3|(Gsh2 and Nkx2.1), and Celsr|Dlx5/6, that express Cre in large sectors of basal telencephalon but not in olfactory nuclei nor temporal cortex (10, 11), the AC developed normally. This suggests that Celsr3 expression may not be required in cells along the AC pathway. Alternatively, functionally relevant cells in intermediate regions may have escaped Celsr3 inactivation, because Gsh2-, Nkx2.1-, and Dlx5/6-Cre mice do not express Cre strongly in the medial region where AC axons cross the mid-line. We favor the latter interpretation because it fits with the observations on the IC described below.
Fig. 1.

Celsr3 expression is required intrinsically in neurons of origin of the AC. (A) Schematic representation of the AC, composed of a rostral component (R) that contains commissural axons connecting olfactory nuclei and a caudal component (C) made of axons that connect temporal lobes. OB indicates olfactory bulb. (B and C) Montages of coronal sections at the level of the AC in newborn animals. The control phenotype is shown on the left side, and the right side shows Celsr3| Foxg1 (B) and Celsr3|Emx1 (C) mice. Note the rudiment of AC in Celsr3| Emx1 mice (arrow in C). (D to F) Horizontal sections at birth day (P0) after DiI injection in control (D), Celsr3| Foxg1 (E), and Celsr3|Emx1 mice (F). Arrows in (C) and (F) point to rudiments of AC that never cross the midline.
We next investigated the function of Celsr3 during development of the three components of the IC, namely corticothalamic axons (CTA), thalamocortical axons (TCA), and subcerebral projections [terminology of Molyneaux et al. (12)]. In Celsr3|Foxg1 mice, despite normal Celsr3 expression in dorsal thalamus, all three components were defective (Fig. 2, A and D). No thalamic fibers turned toward the striatum, and no thalamic neurons were labeled after DiI injection in cortex. Reciprocally, injection of DiI in thalamus did not label cortical neurons but stained thalamic axons that ran into the basal forebrain or crossed the midline ventrally, like in constitutive Celsr3 and Fzd3 mutants (2, 13) (fig. S2). With use of focal DiI injections in the basal forebrain at embryonic day 13.5 (E13.5), we found that early thalamic fibers reached the medial ganglionic eminence in normal but not in Celsr3−/− mice. In contrast, in both genotypes, corticofugal fibers crossed the pallial subpallial boundary and reached the lateral ganglionic eminence at E13.5 (fig. S3). Thus, Celsr3 is required for the early growth of TCA, but not CTA, toward ventral telencephalon.
Fig. 2.

Region-specific Celsr3 inactivation affects development of the IC in different manners. (A to F) Montages of P0 sections stained with Cresyl violet [(A) to (C)] or neurofilament antibody (NF) [(D) to (F)]. The IC is fully defective in Celsr3|Foxg1 mice in which some thalamic axons cross to the contralateral diencephalon [arrow in (D)]. In Celsr3| Dlx5/6 mice, the IC does not form, but cortical axons stall and form a mass at the level of the striatum [asterisks in (B) and (E)], whereas thalamic fibers are mis-routed to the amygdala [arrow in (E)]. In Celsr3| Emx1 mice, the IC is present and thalamocortical connections are similar to that in normal mice [(C) and (F)]. (G) Schematic summary of the IC phenotypes in the various mice used, in relation to areas of Celsr3 inactivation (gray) and expression of markers (Dlx5/6, Gsh2, Nkx2.1, and Rora). dTh and vTh, dorsal and ventral thalamus; HT, hypothalamus; VP, ventral pallidum; NCx, neocortex; PSPB, pallial subpallial boundary; and DTB, diencephalon telencephalon border.
The three components of the IC were also defective in Celsr3|Dlx5/6 mice that express Cre in the ventral forebrain (11) (Fig. 2, B and E). A subset of TCA managed an incomplete turning at the diencephalon-telencephalon border but then ran aberrantly through the pallidum and amygdala. Corticofugal fibers crossed the pallial-subpallial boundary and entered the lateral part of the basal forebrain. However, they failed to progress and spiraled in a disorderly manner, forming an abnormal mass that filled most of the dorsal striatum and protruded in the lateral ventricle (Fig. 2E). After DiI injections in cortex and thalamus, no labeling of thalamic or cortical cells was observed, confirming that both TCA and CTA were defective (fig. S2). Similarly, no sub-cerebral projections formed, as shown by absence of corticospinal tract (CST) (fig. S4). Thus, Celsr3 expression by Dlx5/6-positive cells is required for progression of TCA, CTA, and subcerebral projections through the ventral telencephalon. Partial progression of CTA through the pallial subpial boundary in Celsr3|Dlx5/6 but not in Celsr3|Foxg1 mice may reflect Celsr3-mediated fiber-to-fiber interactions among CTA, in which Celsr3 is expressed, or interactions between CTA and Celsr3-positive, Dlx5/6-negative cells. Similarly, the partial turning of TCA in Celsr3|Dlx5/6 mice may reflect interactions with Celsr3-positive, Dlx5/6-negative cells.
To define which subset of Dlx5/6-positive cells could qualify as guidepost cells, we used Gsh2-Cre and Nkx2.1-Cre mice that express Cre in the lateral and medial ganglionic eminences, respectively. In Celsr3|Gsh2, Celsr3|Nkx2.1, and double Celsr3|Gsh2-Nkx2.1 mice, all components of the IC developed normally (fig. S5). This showed that a sufficient number of Celsr3-expressing guidepost cells along the IC originate from Nkx2.1- and Gsh2-negative precursors. We compared the distribution of Celsr3 (in situ hybridization), Nkx2.1 and Gsh2 (LacZ histochemistry and immunohistochemistry), Dlx5/6 [enhanced green fluorescent protein (EGFP) reporter], and Islet1 (immunohistochemistry) at E12.5, before any fiber growth in the ventral telencephalon, and at E14.5, when the IC begins to form. At E12.5 (Fig. 3), Celsr3 expression was very similar to that of Dlx5/6, with maximal signal along the ganglionic eminences where Islet1 was also present (compare Fig. 3, D and E, to A). Expression of LacZ in ROSA26R| Nkx2.1 and ROSA26R|Gsh2 mice was strong in large sectors of the ganglionic eminences and in the striatal mantle but low in the intermediate region where expression of Celsr3 and Dlx5/6 was maximal. At E14.5 (fig. S6), zones of Gsh2 and Nkx2.1 expression flanked axonal bundles of the incipient IC, whereas Islet1-expressing cells were in close contact with axonal tracts, in the region of highest Dlx5/6 signal and minimal Gsh2 and Nkx2.1 signal. These observations suggest that Celsr3 is required in basal forebrain guide-post cells that are positive for Dlx5/6 and possibly Islet1 and that are derived from Nkx2.1-Cre–and Gsh2-Cre–negative precursors.
Fig. 3.

The IC corridor expresses Celsr3, Dlx5/6, and Islet1 and is flanked with expression of Nkx2.1 and Gsh2. Coronal sections at rostral (A to E) and caudal levels (F to J) of the forebrain at E12.5, showing expression of Celsr3 [(A) and (F), in situ hybridization], activation of the LacZ reporter in ROSA26R| Nkx2.1 mice [(B) and (G)] and ROSA26R| Gsh2 mice [(C) and (H)], expression of Dlx5/6 [(D) and (I), EGFP in the transgene], and expression of Islet1 [(E) and (J), immunohistochemistry]. A central corridor is characterized by high levels of Celsr3, Dlx5/6, and Islet1, and low levels of LacZ. Arrows in (F) and (I) indicate a Celsr3-positive, Dlx5/6-negative zone that could explain partial growth of TCA in Celsr3|Dlx5/6 mice. CGE, LGE, and MGE, caudal, lateral, and medial ganglionic eminences.
To test whether Celsr3 is required intrinsically for progression of corticofugal axons, we studied Celsr3|Emx1 mice, in which Celsr3 is inactivated early in the cortical anlage (9) (fig. S1). In those mice, subcerebral projections such as CST were defective. In crosses with Thy1–yellow fluorescent protein (YFP) transgenic mice (14), the CST was clearly defined in control mice but absent in Celsr3|Emx1 mice in which the number of cortical layer 5 neurons was dramatically reduced (Fig. 4). After injections of Fluoro-Gold (Biotum, Incorporated, Haywood, CA) in the spinal cord, cells were labeled in the hindbrain, red nucleus, and layer 5 in normal mice but only in the hind-brain and red nucleus, and not in layer 5, in Celsr3|Emx1 mice (fig. S7). Thus, Celsr3 is required, presumably cell autonomously, in the neurons of origin of subcerebral axons, like in those of the AC.
Fig. 4.
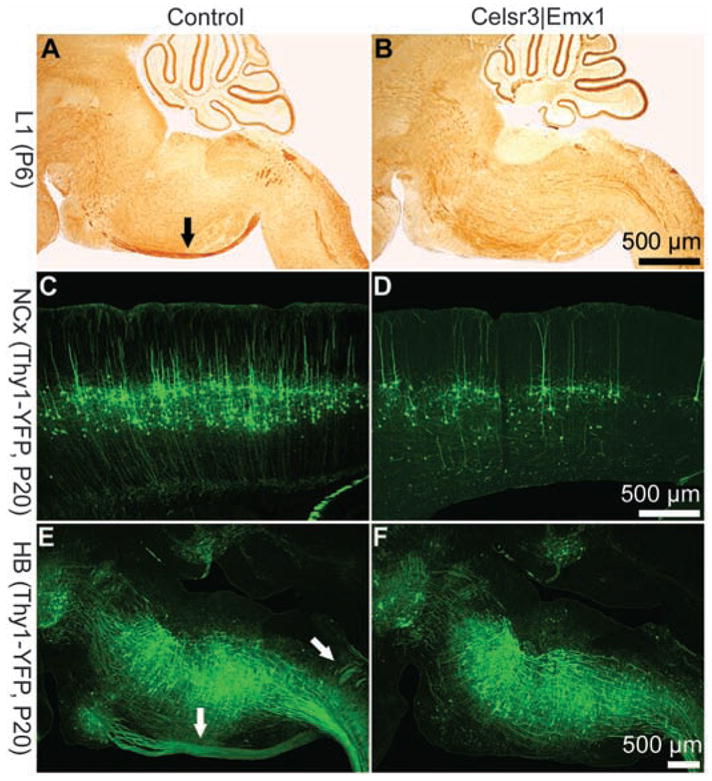
The corticospinal tract is defective in Celsr3|Emx1 mice. Comparison of control [(A), (C), and (E)] and Celsr3| Emx1 [(B), (D), and (F)] mice. In sagittal sections at P6 stained with an antibody against the L1 molecule (A) and (B), corticospinal axons are labeled in control [arrow in (A)] but not in the mutant ventral hindbrain. Crosses were carried out with Thy1-YFP mice, a transgene that labels neurons in cortical layer 5 and corticospinal axons (C) to (F). At P20, layer 5 is well populated in control mice (C), and the corticospinal tract is clearly defined [arrows in (E)], whereas cortical layer 5 is very diminutive (D) and no corticospinal axons are detected in the hindbrain (F) of Celsr3|Emx1 mice.
In contrast to subcerebral projections, CTA and TCA developed normally in Celsr3|Emx1 mice (Fig. 2). At E14.5, fibers from dorsal thalamus turned at the diencephalon-telencephalon border and progressed along the ganglionic eminences before passing the pallial subpallial boundary and growing toward the cortex, similar to fibers of control mice. Injections of DiI in the cortex and thalamus resulted in labeling of thalamic and cortical neurons, respectively (fig. S2). To assess the cortical distribution of TCA, we studied cortical barrels by using cytochrome oxidase and Nissl staining of parietal cortex. As shown in fig. S8, barrels failed to form in mice that had no TCA, such as Celsr3|Foxg1 and Celsr3|Dlx5/6 mice, and developed normally in Celsr3|Emx1 mice, indicating normal TCA mapping. Thus, inactivation of Celsr3 in CTA did not prevent them from navigating to the thalamus, nor did it perturb the growth and cortical mapping of TCA.
Why would Celsr3 be required in AC and subcerebral axons but not in CTA? First, a few subplate cells could escape Celsr3 inactivation in Celsr3|Emx1 mice and provide pioneering axons to thalamus (15, 16). However, Celsr3 is inactivated early in the cortex in those mice (fig. S1), making this rather unlikely. Second, other Celsr proteins may act redundantly with Celsr3 in CTA neurons and mediate their interactions with Celsr3-positive guidepost cells. Alternatively, normal Celsr3 expression in dorsal thalamus and basal forebrain in Celsr3|Emx1 mice allows progression of TCA, which could encounter Celsr3-deficient CTA and help them travel to the thalamus, as predicted by the “handshake hypothesis” (17). Celsr3|Rora mice were produced to inactivate Celsr3 in dorsal thalamic nuclei and thereby assess their role in TCA growth. The IC developed normally in those mice, indicating a situation reciprocal to that in Celsr3|Emx1 mice. However, studies of ROSA26R|Rora mice showed that Cre expression was restricted to a subset of dorsal thalamic cells. Thus being unable to test the function of Celsr3 in thalamic neurons in vivo, we addressed the question using explant cultures. We co-cultured explants from normal or Celsr3-mutant thalamus that expressed the GFP transgene ubiquitously (18) with explants of normal ventral diencephalons at E13.5. As shown in fig. S9, normal dorsal thalamic axons were repelled by explants from ventral diencephalon (32/57 cases) (19). However, almost no repulsive activity was detected for Celsr3-defective thalamic axons (4/34 cases; P < 0.01, χ2), suggesting that Celsr3 expression in TCA was required for their response to ventral diencephalic cues. Thus, Celsr3 expression is probably necessary both in TCA and in cells along their pathway (Celsr3|Dlx5/6 mice).
Our results have implications for the mechanisms of brain wiring and the function of Celsr3. Demonstrated in invertebrates (20), a role of guidepost cells in axonal navigation in mammals has been repeatedly proposed (21–24). Our results demonstrate that they indeed play a crucial function that requires Celsr3 expression (the role of other molecules implicated in thalamocortical and CST fiber nagivation is discussed briefly in SOM). Altogether, our data indicate that Celsr3 is required both in axons and guidepost cells, consistent with its mediating homophilic interactions. Normal CTA development in Celsr3|Emx1 mice indicates that Celsr3-independent cues are also involved in their growth. Candidate mechanisms are CTA-TCA fiber interactions like the handshake (17, 21), fiber-fiber interactions between CTA and pioneer subplate axons (15, 16), and adhesion of Celsr3 in guidepost cells with other Celsr molecules present in growth cones. Furthermore, Celsr3|Emx1 mice provide a unique model to study how subcerebral projections segregate from CTA when they reach the medial aspect of the IC en route to the cerebral peduncles, an important developmental event that hitherto received little attention.
In Drosophila wing cells, symmetrically expressed Fmi/stan proteins are thought to undergo homophilic interactions, bringing distal and proximal cell membranes in contact and thereby fostering signaling by asymmetrically located Frizzled on the distal and Van Gogh on the proximal side (25). Axonal anomalies in Celsr3 and Fzd3 mutant mice are similar (13), suggesting that corresponding proteins also act together in mice (25). Moreover, Fzd3 and Vangl2 are co-expressed with Celsr3 in postmigratory neurons (26). Like in the fly, Celsr3 expressed on the membranes of growth cones and guidepost cells may promote adhesion and allow Fzd3 and Vangl2 to interact and signal. This model predicts that the expression and action of Fzd3 and Vangl2 should be asymmetric, one in axons and the other in guidepost cells. Conditional Fzd3 and Vangl2 alleles should allow testing that model further.
Footnotes
Supporting Online Material
www.sciencemag.org/cgi/content/full/320/5878/946/DC1
Materials and Methods
SOM Text
Figs. S1 to S9
Table S1
References
References and Notes
- 1.Tessier-Lavigne M. Harvey Lect. 2002;98:103. [PubMed] [Google Scholar]
- 2.Tissir F, Bar I, Jossin Y, Goffinet AM. Nat Neurosci. 2005;8:451. doi: 10.1038/nn1428. [DOI] [PubMed] [Google Scholar]
- 3.Usui T, et al. Cell. 1999;98:585. doi: 10.1016/s0092-8674(00)80046-x. [DOI] [PubMed] [Google Scholar]
- 4.Chae J, et al. Development. 1999;126:5421. doi: 10.1242/dev.126.23.5421. [DOI] [PubMed] [Google Scholar]
- 5.Tissir F, De-Backer O, Goffinet AM, Lambert de Rouvroit C. Mech Dev. 2002;112:157. doi: 10.1016/s0925-4773(01)00623-2. [DOI] [PubMed] [Google Scholar]
- 6.Materials and methods are available on Science Online.
- 7.Soriano P. Nat Genet. 1999;21:70. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- 8.Hebert JM, McConnell SK. Dev Biol. 2000;222:296. doi: 10.1006/dbio.2000.9732. [DOI] [PubMed] [Google Scholar]
- 9.Gorski JA, et al. J Neurosci. 2002;22:6309. doi: 10.1523/JNEUROSCI.22-15-06309.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kessaris N, et al. Nat Neurosci. 2006;9:173. doi: 10.1038/nn1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Stenman J, Toresson H, Campbell K. J Neurosci. 2003;23:167. doi: 10.1523/JNEUROSCI.23-01-00167.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Molyneaux BJ, Arlotta P, Menezes JR, Macklis JD. Nat Rev Neurosci. 2007;8:427. doi: 10.1038/nrn2151. [DOI] [PubMed] [Google Scholar]
- 13.Wang Y, Thekdi N, Smallwood PM, Macke JP, Nathans J. J Neurosci. 2002;22:8563. doi: 10.1523/JNEUROSCI.22-19-08563.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Feng G, et al. Neuron. 2000;28:41. doi: 10.1016/s0896-6273(00)00084-2. [DOI] [PubMed] [Google Scholar]
- 15.Ghosh A, Antonini A, McConnell SK, Shatz CJ. Nature. 1990;347:179. doi: 10.1038/347179a0. [DOI] [PubMed] [Google Scholar]
- 16.McConnell SK, Ghosh A, Shatz CJ. Science. 1989;245:978. doi: 10.1126/science.2475909. [DOI] [PubMed] [Google Scholar]
- 17.Molnar Z, Blakemore C. Nature. 1991;351:475. doi: 10.1038/351475a0. [DOI] [PubMed] [Google Scholar]
- 18.Okabe M, Ikawa M, Kominami K, Nakanishi T, Nishimune Y. FEBS Lett. 1997;407:313. doi: 10.1016/s0014-5793(97)00313-x. [DOI] [PubMed] [Google Scholar]
- 19.Braisted JE, Tuttle R, O’Leary D. Dev Biol. 1999;208:430. doi: 10.1006/dbio.1999.9216. [DOI] [PubMed] [Google Scholar]
- 20.Bentley D, Caudy M. Nature. 1983;304:62. doi: 10.1038/304062a0. [DOI] [PubMed] [Google Scholar]
- 21.Hevner RF, Miyashita-Lin E, Rubenstein JL. J Comp Neurol. 2002;447:8. doi: 10.1002/cne.10219. [DOI] [PubMed] [Google Scholar]
- 22.Mitrofanis J, Guillery RW. Trends Neurosci. 1993;16:240. doi: 10.1016/0166-2236(93)90163-g. [DOI] [PubMed] [Google Scholar]
- 23.Lopez-Bendito G, et al. Cell. 2006;125:127. [Google Scholar]
- 24.Tuttle R, Nakagawa Y, Johnson JE, O’Leary DD. Development. 1999;126:1903. doi: 10.1242/dev.126.9.1903. [DOI] [PubMed] [Google Scholar]
- 25.Strutt D, Strutt H. Dev Biol. 2007;302:181. doi: 10.1016/j.ydbio.2006.09.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tissir F, Goffinet AM. Eur J Neurosci. 2006;23:597. doi: 10.1111/j.1460-9568.2006.04596.x. [DOI] [PubMed] [Google Scholar]
- 27.We thank V. Bonte, I. Lambermont, and E. Paitre for technical assistance; G. Hamard for help with embryonic stem cell injections; M. Okabe for GFP mice; F. G. Rathjen for antibodies against L1; P. Soriano for ROSA26R mice; and L. Nguyen, A. Stoykova, and P. Vanderhaeghen for help or discussion. This work was supported by grants Actions de recherches concertées (ARC-186), FRFC 2.4504.01, FRSM 3.4501.07, and FRSM 3.4529.03; by Interuniversity Poles of Attraction (SSTC, PAI p6/20); and by the Fondation Médicale Reine Elisabeth, all from Belgium. F.T. is a Research Associate of the Fonds National de la Recherche Scientifique.