

Abstract
Human ribosomal protein S3 (RPS3) has previously been shown to have alternative roles beyond its participation in protein synthesis. For example, our in vitro studies have shown that RPS3 has an extraordinarily high binding affinity for 7, 8-dihydro-8-oxoguanine (8-oxoG). Notably, in cells exposed to oxidative stress RPS3 translocates to the nucleus where it co-localizes with foci of 8-oxoG. We have engineered transgenic mice over expressing RPS3 in an attempt to determine the outcome of RPS3 translocation in a whole animal. Mouse embryonic fibroblasts (MEF's) isolated from these transgenic mice showed an increased accumulation of DNA damage in cells exposed to oxidative damage when compared to MEF's from wild-type mice. In MEF's exposed to oxidative stress we observed the translocation of RPS3 from the cytoplasm to the nucleus and co-localizing to 8-oxoG foci, an observation that could involve the blocking of the repair of this mutagenic base and thereby explain why transgenic MEF's exposed to oxidative stress have higher levels of DNA damage.
Keywords: Ribosomal protein S3, Oxidative DNA damage, Transgenic mice
1. Introduction
Human S3 (RPS3) (Accession No. NM_001005) is a component of the 40S ribosomal subunit and forms a part of the domain on the ribosome where the initiation of translation occurs [1]. It has roles in protein translation and ribosomal maturation [2]. In addition, RPS3 has been shown to possess several extra-ribosomal activities. For example we recently have shown that RPS3 interacts with the p65 subunit of NF-κB and mediates selective gene regulation by NF-κB [3]. Our in vitro studies using Surface Plasmon Resonance (SPR) analysis has shown that RPS3 also has a very high binding affinity for the mutagenic lesion 8-oxoG [4] formed by oxidative stress, but lacks the ability to remove 8-oxoG. We hypothesized that this high binding affinity could create an obstacle to the efficient repair of an 8-oxoG DNA lesion. Indeed, we found that when a 37-mer oligonucleotide containing 8-oxoG was preincubated with RPS3, it blocked repair of 8-oxoG by the N-glycosylase hOGG1 [5]. Additionally, a single amino acid change of a lysine residue at position 132 to an alanine (K132A) not only abrogated the high binding affinity of RPS3 but also stimulated repair by hOGG1 almost 2 fold [5]. Despite the absence of DNA binding activity for K132A the stimulation of 8-oxoG removal suggests positive interactions of RPS3 with other BER proteins. This is in agreement with our earlier findings [6] that RPS3 interacts with BER proteins, hOGG1 and APE/ref-1. On the other hand we recently showed that a 40% knockdown of RPS3 by iRNA protected cells from a variety of DNA damaging agents, which suggests that the expression of RPS3 can have a detrimental effect on cell survival [7]. Finally our more recent data shows that RPS3 translocates to the nucleus from the cytosol after phosphorylation by ERK1/2 in cells exposed to oxidative stress and co-localizes with 8-oxoG foci in the nucleus, suggesting the availability of RPS3 to participate in nuclear events, such as DNA repair [8].
In the present study, we generated transgenic (Tg) mice over expressing RPS3 driven by a CMV promoter showing transgene expression in all the major tissues studied. Similar to our in vitro observations, we observed in transgenic MEF's that cytoplasmic S3 translocates to the nucleus and co-localizes with 8-oxoG in the presence of oxidative damage. We further show that over expression of RPS3 causes an increase in DNA damage in Tg MEF's when exposed to oxidative damage. The Tg-RPS3 mouse therefore is a potentially valuable animal model for studying the role of this ribosomal protein in DNA repair.
2. Materials and methods
2.1 Generation of transgenic RPS3 mice
The RPS3 over expressing mice in a C57BL/6 background were generated by the transgenic core facility at the University of Texas Health Sciences Center, San Antonio, Texas. Briefly, RPS3 coding sequence cloned into pcDNA3.1 vector was purified by cesium chloride gradient and isolated along with the CMV promoter and polyA sequence by restriction digestion with MluI and DraIII. This fragment was injected into fertilized eggs from a super ovulated female and 227 embryos that survived the process were then transferred into 9 pseudo pregnant recipient females, of which 5 females carried the litters to term. Out of the 33 pups born, 5 founder males were identified by digesting tail DNA with BamHI and analyzed by Southern blotting with a 32P labeled DNA probe, which contained the RPS3 coding sequence along with the CMV promoter and polyA sequence. All procedures for handling the mice in this study were reviewed and approved by the IACUC (Institutional Animal Care and Use Committee) of Pennington Biomedical Research Center.
2.2 Detection of RPS3 and 8-oxoG for expression and co-localization analysis by Immunofluorescence Microscopy
For expression analysis, fixed tissues were used; for co-localization microscopy, wild-type and RPS3 over expressing transgenic MEF cells were treated with 1 mM H2O2 (CAS# 7722-84-1) for 48 hours, fixed in 10% neutral buffer formalin for 30 min and permeabilized in 0.1% Triton X-100 for 10 min. After washing in PBS, samples were blocked in 1% BSA for 1 h and incubated with anti-RPS3 and anti-8-oxoG (clone 2E2, Trevigen) antibodies for an additional hour. Samples were then washed in PBS, and further incubated with rhodamine red-anti-rabbit (Molecular Probes) and Alexa 488-anti-mouse (Invitrogen) secondary antibodies for 1 h. After washing again in PBS, samples were mounted with 4′-6-diamidino-2-phenylindole (DAPI) containing vectashield mounting media. Localization of RPS3, 8-oxoG, and nuclei were performed by fluorescence microscopy using Zeiss 510 Meta multiphoton confocal microscope equipped with argon (458, 477, 488, 514 nm), Helium (543 and 633 nm) and Chameleon (720-950 nm) laser lines. Captured images were processed using ImageJ software.
2.3 Detection of RPS3 expression in tissues by western blot analysis
Total protein from tissues was extracted by adding 250 μl of lysis buffer (50 mM KCl, 1% NP-40, 25 mM HEPES (pH 7.8), 10 μg/ml leupeptin, 20 μg/ml, aprotinin, 125 μM DTT, 1mM PMSF and 1mM Na3VO4) per 25 mg of tissue. The tissue bits were homogenized in lysis buffer and then incubated on ice for 20 min. The lysed tissue sample was then centrifuged at 13,000 rpm for 10 min at 4 °C and supernatant collected as extracted protein. Protein concentration was determined by Bradford assay [9]. Western blot analysis was performed as described previously [7]. Briefly the total protein from tissues was resolved by SDS-PAGE and transferred onto nitrocellulose membrane by electro transfer technique. Membranes were then blocked in 5% non-fat dairy milk and immunoblotted with anti-RPS3 (custom-made by Proteintech Group, Chicago, IL) and anti-GAPDH (Chemicon International, Temecula, CA). After treating the membranes with appropriate secondary antibodies, antigen bands were visualized by enhanced chemiluminescence technique using western lightning chemiluminescence reagent plus (Perkin-Elmer). Densitometric tracing of the protein bands was performed by using AlphaEaseFc software (Alpha Innotech Corporation).
2.4 Mouse embryonic fibroblast isolation for in vitro experiments
For isolation of MEFs one male and two females (8 - 10 weeks old) of appropriate genotype were set up for breeding. Females were visually observed daily for a vaginal plug to verify breeding. The date a plug was observed the female was removed from the male's cage and this day noted as day 0. On day 13 of pregnancy the female was euthanized by CO2 asphyxiation, and the ventral abdomen thoroughly moistened with 70% isopropyl alcohol and opened with sterile scissors to expose the uterus. The gravid uterus was removed and placed in a petri dish containing sterile PBS. The embryos were removed from the uterus, decapitated and minced in 5 ml of 1× trypsin. The petri dish containing the minced embryos was placed in a 37 °C incubator for 15 min. The cell and tissue bits were then transferred to a 15 ml conical tube and pipetted several times to further dissociate the tissue bits. DMEM supplemented with 10% FBS media (5 ml) was added and centrifuged at 200 g for 5 min. The supernatant was aspirated and the embryonic cell pellet resuspended in 10 ml of fresh media and transferred to a clean sterile P100 dish. The cells and tissue bits were evenly distributed by horizontal agitation and then incubated at 37 °C in 5% CO2. Further cultures were maintained in DMEM supplemented with 15% FBS, 1 mM glutamine and antibiotics.
2.5 Evaluation of MEF cells for DNA Damage by the Comet Assay
The alkaline single cell gel electrophoresis assay (comet assay) was performed using a commercially available kit (Trevigen, Gaithersburg, MD). Wild-type and RPS3 transgenic MEF cells were treated with 1 mM H2O2 for 48 h and used for comet assay. Aliquots of 1 × 105 cells were collected and washed twice with 1 ml of PBS. Cell pellets were then re-suspended in 150 μl of 0.5% low melting agarose (Fisher) and an aliquot of this suspension (75 μl) was immediately layered onto comet slides (Trevigen). Two slides were prepared from each sample, one of which was used for the normal comet assay and the other one processed for the Fpg (E.coli Formamidopyrimidine-DNA Glycosylase) FLARE (Fragment Length Analysis using Repair Enzymes) assay. After the agarose gels were allowed to solidify at 4 °C for 10 min, slides were placed in pre-chilled lysis solution (Trevigen) and incubated in the dark for 55 min at 4 °C. FLARE comet assay slides were then placed in wash buffer (10 mM HEPES, pH 7.4, 100 mM KCL, 10 mM EDTA, pH 8, 0.1 mg/ml BSA) for 10 min at room temperature. After drip-drying, 5-7 units of Fpg enzyme (Trevigen) was added to each sample and incubated at 37 °C for 55 min. The slides for the normal comet assay were stored in the dark at room temperature during this time. Both normal comet and FLARE comet slides were then immersed in alkaline buffer (1 mM EDTA, 300 mM NaOH) and incubated at room temperature in the dark for 55 min. After rinsing the slides twice with TBE, gels were subjected to electrophoresis in TBE for 10 min at 17 volts. Thereafter, gels were fixed by placing them in ice-cold methanol for 5 min, followed by ethanol for additional 5 min. Gels were then stained with 50 μl SYBR green dye (Trevigen, 1:10,000) and viewed under a fluorescence microscope (Zeiss Axiophot research microscope) using a FITC filter. Images of 25 randomly selected fields from each gel were acquired using a SPOT-RT slider digital camera and SPOT software (Diagnostic Instruments, Inc).
The extent of DNA damage was determined by calculating the comet moment, which is the integrated density of the comet tail multiplied by the distance from the center of the nucleus to the center of mass of the tail. Computations scored 25 cells for each sample using a macro available from Herbert M. Geller at http://www2.umdnj.edu/∼geller/lab/comet-Scoring-Macro.txt. As the rapidly changing intensities of the individual cells are difficult to control, a large variance within each experiment is an unavoidable consequence of using this method. Therefore, a normalizing and variance-stabilizing logarithmic transformation was then applied to the calculated tail moments [10-13]. After the data were transformed, analysis of variance (ANOVA) with multiple comparisons was applied with respect to the different treatment groups as previously described [10-13]. Cell frequency was determined by a visual scoring method [11] classifying DNA damage into the following categories: type1- intact nucleus, smooth outer edges; type2- intact nucleus, small amount of tailing; type3- intact nucleus, large amount of tailing; type4- shrinking nucleus, large amount of tailing.
2.6 Histopathology of tissue sections
For tissue sections, animals were sacrificed by asphyxiation with carbon dioxide and necropsied within 5 min of death. All organs were fixed in 10% buffered formalin, trimmed, embedded in paraffin, sectioned to a thickness of 5 to 6 μm, stained with H & E and examined microscopically. Preparation of slides for histopathology evaluation was done at the Dept. of Pathobiological Sciences, School of Veterinary Medicine, Louisiana State University.
2.7 In situ Apoptosis detection in tissue sections
DNA labeling of fixed tissue sections was performed using the TACS (Trevigen Apoptotic Cell System) 2 TdT in situ apoptosis detection kit as per the manufacturers instructions (Trevigen; Gaithersburg, MD). This kit utilizes a TdT enzyme that incorporates biotinylated nucleotides at the 3′ OH ends of the DNA fragments that are formed during apoptosis. For fluorescent detection, tissue sections were treated with a fluorescein conjugate of streptavidin and visualized by utilizing the DAPI and FITC filter sets of a Zeiss Axioplan2 microscope. Imaging was performed with a photometric coolsnap HQ CCD camera and slide book software.
3. Results
3.1 Expression analysis of RPS3 in tissues from transgenic mice
To detect expression of RPS3, wild-type and F1 transgenic litter male and female animals obtained from the founder males were euthanized to analyze RPS3 expression in various tissues using confocal microscopy. The custom antibody towards human RPS3 also cross reacts to the endogenous mouse RPS3 hence over expression with fixed tissues was determined by first setting the microscope parameters to view S3 expression in each of the control (wild-type) tissues. The transgenic tissues were then viewed with the same settings and increase in the rodamine red intensity was concluded as S3 over expression due to the transgene. RPS3 was found to be over expressed in the stomach, liver, pancreas, kidney, lungs, heart, brain, adrenal, spleen and intestines of both male and female Tg mice as compared with wild-type controls. A representative result is shown for over expression of transgenic RPS3 in liver and kidney (Fig. 1A). RPS3 over expression was also determined by western blot analysis of protein extracts from liver and kidney tissues. Whole protein extracts (20 μg) from liver and kidney tissues of wild-type and transgenic (male and female) animals were immunoblotted using anti-RPS3 and anti-GAPDH antibodies. Each RPS3 protein band was normalized to GAPDH from the same extract and over expression determined by comparing normalized levels of wild-type and transgenic protein extracts. RPS3 was found to be over expressed in both liver and kidney tissues from male and female transgenic mice as compared with wild-type mice (Fig. 1B).
Figure 1. Expression analysis of RPS3.
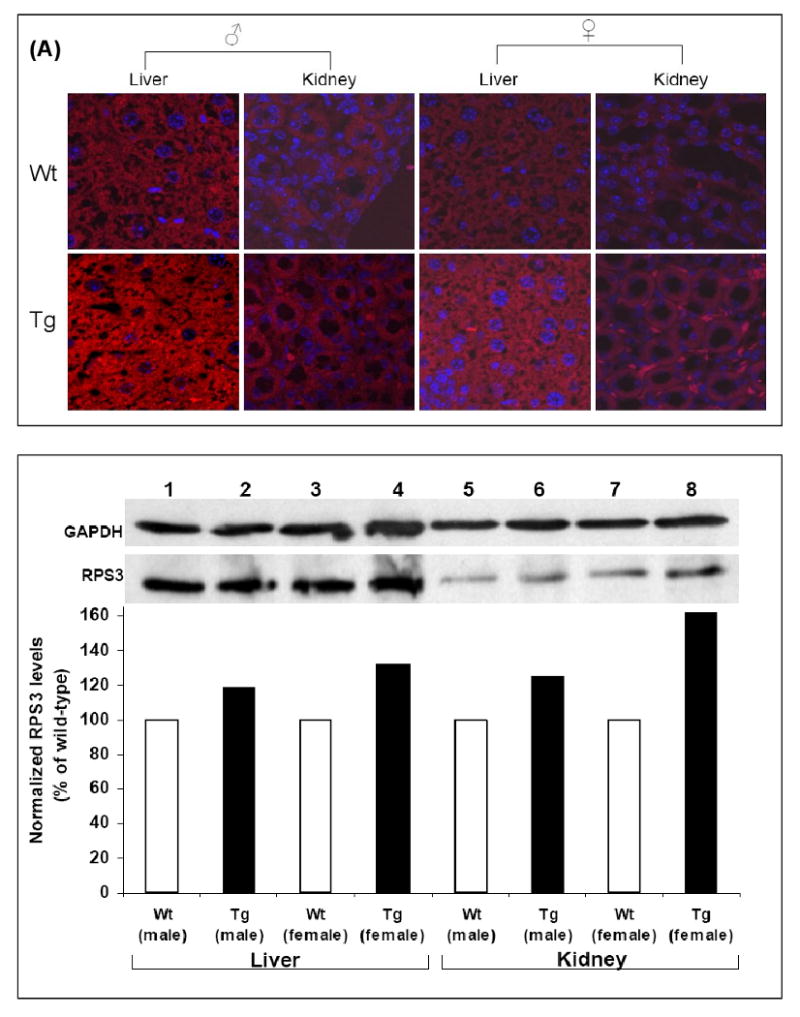
(A) Human RPS3 levels were analyzed in tissues from wild-type and transgenic mice by laser scanning confocal microscopy after staining with custom made RPS3 primary antibody and rodamine red tagged anti-rabbit secondary antibody. Nuclei were stained by DAPI. A representative image of immunofluorescence detection of RPS3 over expression in liver and kidney of male and female transgenic as well as wild-type mice is shown. (B) Over expression of RPS3 in liver and kidney of transgenic male and female mice was also determined by immobilizing an aliquot (20 μg) of protein extract from each tissue by SDS-PAGE. After immunoblotting using anti-RPS3 and anti-GAPDH antibodies the levels of RPS3 were evaluated from integrated density values (IDV), obtained by densitometry tracing of each band. Each RPS3 band was normalized by dividing the IDV of a protein band by the IDV of the GAPDH within the same sample. The normalized IDV values for wild-type was set as 100% and over expression of RPS3 was determined as a percent of wild-type for each tissue.
3.2 Over expression of RPS3 results in increased DNA damage in MEF cells exposed to oxidative stress
Mouse embryonic fibroblasts (MEF's) from wild-type and RPS3 transgenic animals were analyzed by Comet assay to determine whether over expression of RPS3 increases DNA damage due to oxidative stress. As seen in Fig. 2(A) (bar graph, untreated Normal comet), in unexposed cells the presence or absence of the RPS3 transgene had little effect on the calculated comet tail moment, even under conditions where cells were exposed to the Fpg DNA N-glycosylase to reveal oxidized purines and pyrimidines (Fig. 2(A); bar graph, untreated FLARE comet). However, in RPS3 transgenic MEF cells exposed to 1 mM hydrogen peroxide for 48 h, Fpg treatment revealed a greater amount of DNA damage (FLARE comet) in the form of oxidized DNA bases in addition to single-strand breaks and abasic sites when compared to wild-type cells (Fig. 2(A); bar graph, Treated FLARE comet). Moreover, in MEF cells expressing the RPS3 transgene, the amount of single-strand breaks or, abasic sites (Treated Normal comet) significantly increased from that observed in exposed, wild-type cells (Fig. 2(A); bar graph, Treated Normal comet). However, the tail moment increased only slightly in H2O2 exposed, transgenic MEF cells treated with Fpg (Fig. 2(A); bar graph, Treated FLARE comet). One explanation for this could be that the comet assay described here uses alkaline lysis followed by electrophoresis at neutral conditions which has been shown to detect double and single strand breaks in addition to alkali-labile lesions [14]. This would suggest that transgenic RPS3 expression resulted in greater amounts of DNA damage accumulating in cells exposed to oxidative stress in vivo as seen with Treated Normal comet. Subsequent Fpg treatment of transgenic cells (Treated FLARE comet) revealed only a modest increase in DNA damage from that observed in the Treated Normal comet for transgenic MEF cells. DNA damage to individual cells was subsequently conducted using a visual scoring method [11] classifying in this case the extent of DNA damage into the 4 different categories as described in the materials and methods section. Cell frequency graphs show that in untreated MEF cells (Fig. 2B) there are more undamaged cells (type 1 and 2) as compared to damaged cells (type 3 and 4) in both normal and FLARE comet for wild-type and transgenic MEF cells. However in exposed MEF cells (Fig. 2C) the transgenic MEF cells have more DNA damage (type 3 and 4) as compared to wild-type MEF cells in both normal and FLARE comet (Fig. 2C). The observations made by comet assay analysis of transgenic and wild-type MEF cells are consistent with the increased accumulation of DNA damage in cells harboring over expressed RPS3, and is suggestive, but not conclusive that RPS3 interferes with BER [5].
Figure 2. Increased incidence of oxidative DNA damage in transgenic cells after exposure to H2O2.
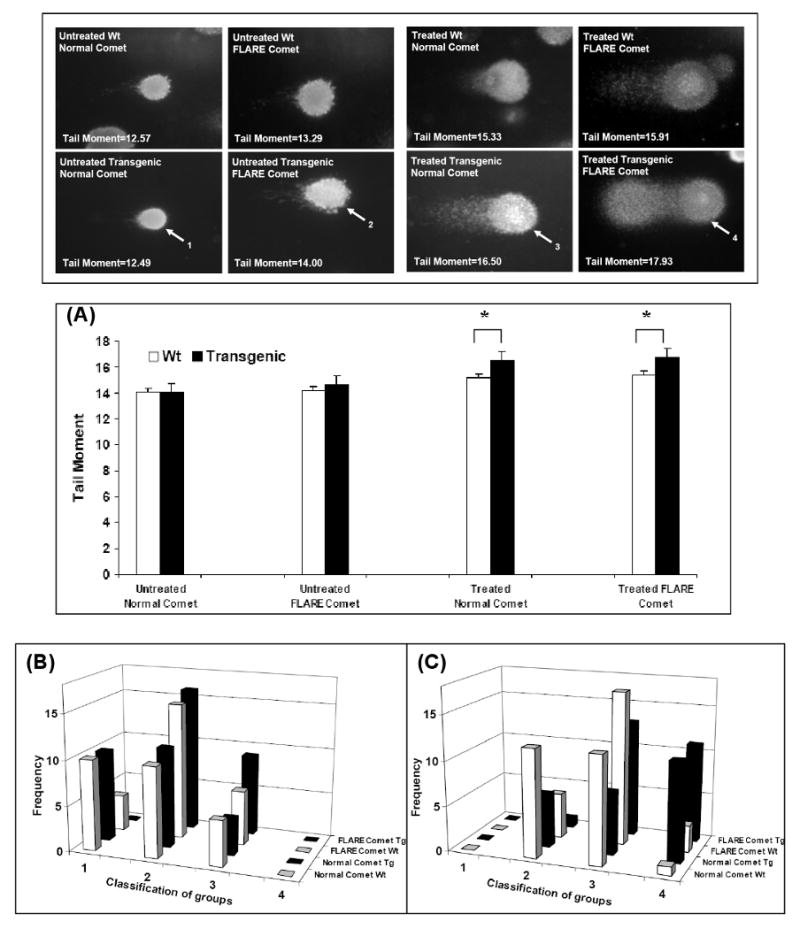
MEF wild-type cells or RPS3 transgenic cells were treated with 1 mM H2O2 for 48 h, and evaluated for the extent of DNA damage by normal comet assay and FLARE comet assay. (A) For each sample, tail moments expressed as adjusted tail moment (normalizing and variance-stabilizing logarithmic transformation applied to the calculated tail moments) of 25 cells from randomly selected fields were calculated and statistically significant differences determined by ANOVA analysis are represented with P-values. *, P=<0.01. DNA damage to individual cells was also determined by a visual scoring method [11] and plotted as cell frequency graphs where DNA damage to cells was divided into the following categories: type1- intact nucleus, smooth outer edges; type2- intact nucleus, small amount of tailing; type3- intact nucleus, large amount of tailing; type4- shrinking nucleus, large amount of tailing. (B) Unexposed wild-type and transgenic MEF cells show more number of cells with minimal damage (type1 and 2) for both normal comet and Fpg treated FLARE comet analysis. However in H2O2 exposed MEF cells we see a significant number of damaged cells (type 3 and 4) in transgenic MEF cells for both normal and FLARE comet compared to wild-type MEF cells (C).
3.3 Oxidative stress results in nuclear translocation of RPS3 and co-localization to sites of 8-oxoG in MEF's
Our earlier studies with RPS3 had shown that under conditions of oxidative stress cytoplasmic RPS3 translocates to the nucleus and co-localizes with foci of 8-oxoG [8]. In order to determine this in MEF cells, we performed immunofluorescence microscopy with wild-type and transgenic RPS3 cells that had been exposed to H2O2 (1 mM), or left untreated, for 48 h. Human RPS3 was subsequently detected by staining the cells with a rabbit monoclonal anti-RPS3 primary antibody and rhodamine red labeled secondary antibody (red fluorescence). For 8-oxoG localization, anti-8-oxoG primary antibody and Alexa 488 labeled secondary antibody (green fluorescence) were used. Samples were also stained with the nuclear fluorochrome, DAPI. Representative sets of images obtained with rhodamine (RPS3), FITC (8-oxoG), and UV filters (nuclei) are shown in Fig. 3. Additionally, a merge of rhodamine, FITC, and UV images is also shown (Fig. 3). Exposure of cells to hydrogen peroxide resulted in translocation of the predominantly cytoplasmic RPS3 into the nucleus where it co-localizes with 8-oxoG (green) foci in the nuclei (blue), which appear as white spots highlighted by arrows in the merged image (Fig. 3). RPS3 expression has been suggested to cause apoptosis in cells [15] and the MEF cells exposed to 1 mM H2O2 for 48 h show a distinct condensed nucleus (DAPI panel; Fig. 3) typical of cells undergoing apoptosis [16,17]. The translocation and co-localization of RPS3 with foci of 8-oxoG observed is specific to the condensed nucleus (merge panel; Fig. 3). As the human RPS3 antibody cross reacts with endogenous mouse RPS3, we also see translocation and co-localization of S3 with foci of 8-oxoG even in wild-type MEF cells following oxidative stress (Fig. 3). These observations confirm our previous results [8] that oxidative stress causes translocation of RPS3 to the nucleus and results in the co-localization to foci of 8-oxoG.
Figure 3. Human RPS3 co-localizes with 8-oxoG residues in MEF cells exposed to H2O2.
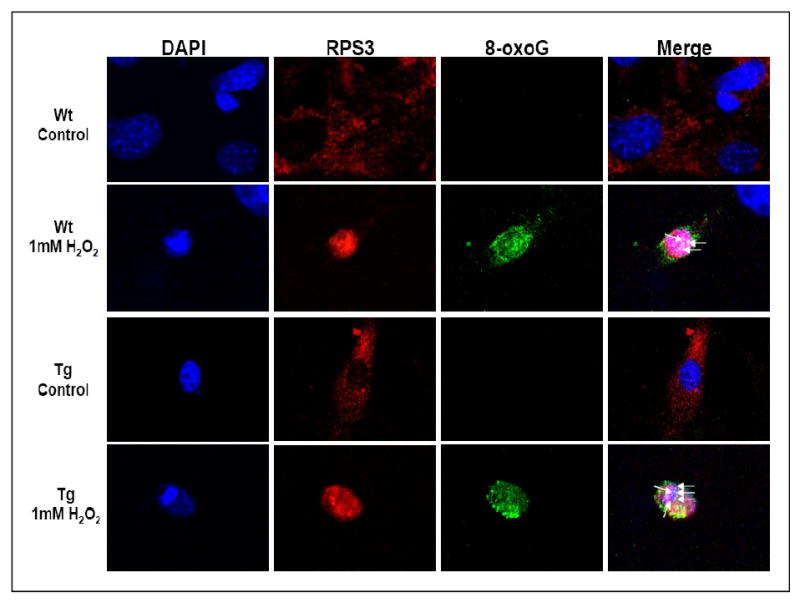
Wild-type and transgenic MEF cells were exposed to 1 mM H2O2 for 48 h, and 8-oxoG/RPS3 co-localization was evaluated by immunofluorescence microscopy. A representative of images obtained with rhodamine (RPS3), FITC (8-oxoG), and UV (nuclei) filters are shown. Additionally, a merge of rhodamine, FITC, and UV images in which the co-localization of RPS3 and foci of 8-oxoG (white spots) within the nuclei is indicated by arrows in both wild-type and transgenic MEF cells.
3.4 Does over expression of S3 predispose transgenic animals to tumorigenesis?
Tissues from 13-month-old age-matched male wild-type C57BL/6 (N=3) and RPS3 over expressing transgenic (N=6) animals submitted for microscopic examination included liver, spleen, kidneys, adrenal glands, thymus, intestine, stomach, heart, lung, reproductive tract, urinary bladder, mesenteric and perirenal white and brown adipose tissue. Morphologic changes in lung, kidney and liver were noted in transgenic animals compared with the wild type. Lungs of transgenic animals were observed to have increased numbers of lymphocytes and plasma cells around vessels and airways as compared to wild-type controls (data not shown). Transgenic animals exhibited a more severe and widespread degree of hepatic fatty change, although all animals including wild-type exhibited some degree of fatty change through out the kidneys. Transgenic animals were found to have larger aggregates of lymphocytes and plasma cells than wild-type controls, located perivascularly throughout the cortex and perirenal adipose tissue, as opposed to the small scattered interstitial and pelvic aggregates found in the wild-type. The morphological changes observed in lung, kidney and liver due to over expression of RPS3 in transgenic mice would suggest a possible propensity in these animals of developing tumors. Immunohistochemical staining of fixed lung, kidney and liver tissues from transgenic mice for B-cell and T-cell markers, CD20 and CD3 respectively, revealed that the lymphoid aggregates were of mixed cell origin (data not shown), with both B-cells and T-cells present throughout, suggesting an inflammatory rather than neoplastic origin.
The high binding affinity of RPS3 could have either of two consequences. First, the repair of closely associated 8-oxoG residues could lead to the formation of double strand breaks in which the binding of RPS3 could moderate the ambitious repair of these sites by OGG1 and its associated AP lyase activity. Alternatively, the block to repair of 8-oxoG could result in an age dependant accumulation of cells with damaged DNA which could be channeled to undergo apoptosis. To investigate this possibility we stained fixed liver and kidney tissue sections from the age-matched 13-month-old wild-type and transgenic male animals with a commercially available in situ apoptosis analysis kit for apoptotic activity detection in these tissues. The kidney tissue sections for transgenic animals showed distinct apoptosis positive cells spread throughout the tissue as determined by the incorporated labeled biotinylated nucleotides (green) (Fig. 4 E-F). The wild-type sections also showed some apoptosis positive cells (Fig. 4 C-D). However both transgenic and wild-type kidney tissues showed significant amount of apoptosis positive cells in the urethra area which might be due to the fact that these cells have a faster turnover rate. Liver cells in both transgenic and wild-type animals did not show any significant apoptosis positive cells (not shown).
Figure 4. In situ apoptosis detection in fixed tissue.
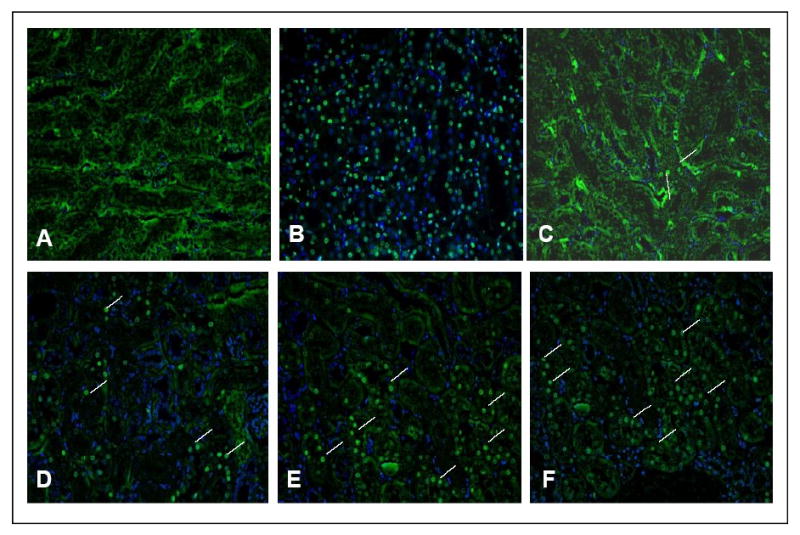
Kidney and liver tissue sections from age-matched 13-month-old wild-type and Tg (RPS3) male mice were labeled in situ to detect apoptotic cells in these tissues. DAPI was used to stain the nuclei. Merged images of FITC and DAPI channels that represent wild-type (panel C-D; different fields) and Tg (panel E-F; different fields) kidney respectively are shown. Apoptotic cells are indicated by white lines. Panel (A) is negative control where the TdT labeling enzyme was omitted in the staining process and panel (B) shows positive control treated with the TACS-nuclease to generate DNA strand breaks.
4. Discussion
Our previous in vitro observations that RPS3 possesses an apparent high binding affinity for 8-oxoG residues that could block the liberation of this modified base has led us to question what effect RPS3 expression has in vivo on the whole animal and cells derived from transgenic mice. Here we show that significant transgene expression was observed, and that the transgene was expressed in most of the major tissues. Using MEF's from Tg (RPS3) mice, we found that over expression of RPS3 increased DNA damage after oxidative stress compared with MEF's from wild-type mice. Our results using the comet assay suggest that the over expression of RPS3 in the Tg MEF cells exposed to H2O2 leads to increased level of oxidized bases, concomitant with a significant increase in DNA repair intermediates such as single-strand breaks and AP sites. These results therefore support a model in which RPS3 is indeed blocking the repair of oxidative DNA base lesions. Our unpublished in vitro observations have shown an interaction between RPS3 and MYH (E.coli MutY homolog). MYH is involved in the removal of adenines mismatched with guanines or 8-oxoG that arise through replication errors [18], which suggests a possibility of RPS3 binding to sites of 8-oxoG:A mispairs to prevent removal of the 8-oxoG base by OGG1 glycosylase before MYH can correct the misincorporated adenine. Therefore the block to DNA repair by RPS3 binding to sites of DNA damage could be to moderate the formation of double strand breaks (DSB's) as a consequence of promiscuous repair. As we had reported previously [8], the S3 imposed block to repair of oxidized DNA bases remains a possibility since the predominantly cytoplasmic RPS3 translocates to the nucleus and co-localizes to 8-oxoG foci upon oxidative stress.
Block to DNA repair would result in a DNA repair deficiency which is associated with nearly all human diseases of segmental premature aging, supporting the argument that aging results from a gradual accumulation of DNA damage [19]. Microscopic examination of tissues from age-matched 13-month-old wild-type and transgenic mice showed increased amounts of lymphocytic to lymphoplasmacytic infiltration throughout the lungs, kidney, and liver of transgenic animals as compared to controls. In cattle that eventually develop multicentric lymphoma due to endogenous retrovirus, individuals frequently progress through an initial inflammatory phase incorporating lymphoid hyperplasia and persistent lymphocytosis as they age and as the neoplastic process progresses [20]. It is possible that the lesions described in these transgenic mice represent a similar process, although additional studies with larger cohorts of mice and longer study times would be necessary to definitively confirm this theory.
The involvement of ribosomal protein genes in cancer promotion was previously shown by a large-scale insertional mutagenesis screen in zebra fish that revealed the ability of ribosomal proteins to contribute to tumorigenesis [21], raising the possibility of many ribosomal protein genes being cancer genes in humans. Previous observations have shown that in human transformed-cells from adenomatous polyps and colorectal carcinoma, elevated levels of human RPS3 mRNA was detected [22]. Also in a recent study, 2700 essential genes in Caenorhabditis elegans were screened for increased adult lifespan by initiating the gene knockdown once the animal had reached adulthood [23]. A total of 64 genes were identified that can extend lifespan when inactivated and genes involved in protein synthesis caused the most potent lifespan change. Interestingly this study found that RPS3 was one of the three clones that target components of the 40S subunit of the ribosome and increased mean lifespan. In summary, studies presented here clearly indicate over expression of RPS3 having an effect by increasing DNA damage intermediates when cells are exposed to oxidative damage. Aging transgenic mice (13-month-old) also show lymphocytic infiltrations in lung, kidney and liver over that seen in wild-type mice. The mechanisms by which RPS3 affects tumor development remains to be determined, as does the mechanism of its interaction with other proteins. Future studies to elucidate these relationships are thus warranted.
Acknowledgments
This work utilized the facilities of the Cell Biology and Bioimaging Core that are supported in part by COBRE (NIH P20-RR021945) and CNRU (NIH 1P30-DK072476) center grants from the National Institutes of Health. This work was supported in part by a supplement to US Public Health Service grant CA 109798 to WAD and a pilot and feasibility grant from the division of Nutrition and Chronic Diseases, Pennington Biomedical Research center to VH. This research was conducted while Vijay Hegde was an AFAR Research grant recipient. We would like to acknowledge and thank one of the reviewers of our manuscript for suggesting the possibility of RPS3 binding to 8-oxoG:A mispairs and blocking OGG1 until MYH can remove the misincorporated adenine.
Footnotes
Conflicts of Interest Statement: The authors declare that there are no conflicts of interest.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Westermann P, Heumann W, Bommer UA, Bielka H, Nygard O, Haltin T. Crosslinking of initiation factor eIF-2 to proteins of the small subunit of rat liver ribosomes. FEBS Lett. 1979;97:101–104. doi: 10.1016/0014-5793(79)80061-7. [DOI] [PubMed] [Google Scholar]
- 2.Schäfer T, Maco B, Petfalski E, Tollervey D, Böttcher B, Aebi U, Hurt E. Hrr25-dependent phosphorylation state regulates organization of the pre-40S subunit. Nature. 2006;441:651–655. doi: 10.1038/nature04840. [DOI] [PubMed] [Google Scholar]
- 3.Wan F, Anderson DE, Barnitz RA, Snow A, Bidere N, Zheng L, Hegde V, Lam LT, Staudt LM, Levens D, Deutsch WA, Lenardo MJ. Ribosomal protein S3: a KH Domain protein found in NF-κB complexes that mediates selective gene regulation. Cell. 2007;131:927–939. doi: 10.1016/j.cell.2007.10.009. [DOI] [PubMed] [Google Scholar]
- 4.Hegde V, Wang M, Deutsch WA. Characterization of human ribosomal protein S3 binding to 7,8-dihydro-8-oxoguanine and abasic sites by surface plasmon resonance. DNA Repair (Amsterdam) 2004;3:121–126. doi: 10.1016/j.dnarep.2003.10.004. [DOI] [PubMed] [Google Scholar]
- 5.Hegde V, Wang M, Mian IS, Spyres L, Deutsch WA. The high binding affinity of human ribosomal protein S3 to 7, 8-dihydro-8-oxoguanine is abrogated by a single amino acid change. DNA Repair (Amsterdam) 2006;5:810–815. doi: 10.1016/j.dnarep.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 6.Hegde V, Wang M, Deutsch WA. Human Ribosomal Protein S3 Interacts with DNA Base Excision Repair Proteins hAPE/Ref-1 and hOGG1. Biochemistry. 2004;43:14211–14217. doi: 10.1021/bi049234b. [DOI] [PubMed] [Google Scholar]
- 7.Hegde V, Yadavilli S, Deutsch WA. Knockdown of ribosomal protein S3 protects human cells from genotoxic stress. DNA Repair (Amsterdam) 2007;6:94–99. doi: 10.1016/j.dnarep.2006.09.004. [DOI] [PubMed] [Google Scholar]
- 8.Yadavilli S, Hegde V, Deutsch WA. Translocation of human ribosomal S3 to sites of DNA damage is dependent on ERK-mediated phosphorylation following genotoxic stress. DNA Repair (Amsterdam) 2007;6:1453–1462. doi: 10.1016/j.dnarep.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bradford MM. A Rapid and Sensitive Method for the Quantitation of Microgram Quantities of Protein Utilizing the Principle of Protein-Dye Binding. Anal Biochem. 1976;72:248–254. doi: 10.1016/0003-2697(76)90527-3. [DOI] [PubMed] [Google Scholar]
- 10.Deutsch WA, Kukreja A, Shane B, Hegde V. Phenobarbital, Oxazepam, and Wyeth 14,643 cause DNA damage as measured by the Comet assay. Mutagenesis. 2001;16:439–442. doi: 10.1093/mutage/16.5.439. [DOI] [PubMed] [Google Scholar]
- 11.Dellinger B, Pryor WA, Cueto R, Squadrito GL, Hegde V, Deutsch WA. The role of free radicals in the toxicity of airborne fine particulate matter. Chem Res Toxicol. 2001;14:1371–1377. doi: 10.1021/tx010050x. [DOI] [PubMed] [Google Scholar]
- 12.Vicente MG, Nurco DJ, Shetty SJ, Osterloh J, Ventre E, Hegde V, Deutsch WA. Synthesis, dark toxicity and induction of in vitro DNA photodamage by a tetra(4-nido-carboranylphenyl) porphyrin. J of Photochem Photobiol B: Biol. 2002;68:123–132. doi: 10.1016/s1011-1344(02)00383-4. [DOI] [PubMed] [Google Scholar]
- 13.Heilbronn LK, de Jonge L, Frisard MI, Delany JP, Larson-Meyer DE, Rood J, Nguyen T, Martin CK, Volaufova J, Most M, Greenway FL, Smith SR, Deutsch WA, Williamson DA, Ravussin E. Effect of 6-month calorie restriction on biomarkers of longevity, metabolic adaptation, and oxidative stress in overweight individuals: a randomized controlled trial. JAMA. 2006;295:1539–1548. doi: 10.1001/jama.295.13.1539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Olive PL, Banath JP, Durand RE. Heterogeneity in radiation induced DNA damage and repair in tumor and normal cells using comet assay. Radiat Res. 1990;122:86–94. [PubMed] [Google Scholar]
- 15.Jang CY, Lee JY, Kim J. RpS3, a DNA repair endonuclease and ribosomal protein, is involved in apoptosis. FEBS Lett. 2004;560:81–85. doi: 10.1016/S0014-5793(04)00074-2. [DOI] [PubMed] [Google Scholar]
- 16.Arends MJ, Wyllie AH. Apoptosis: mechanisms and roles in pathology. Intern Rev Exp Pathol. 1991;32:223–254. doi: 10.1016/b978-0-12-364932-4.50010-1. [DOI] [PubMed] [Google Scholar]
- 17.Raff MC. Social controls on cell survival and cell death. Nature. 1992;356:397–400. doi: 10.1038/356397a0. [DOI] [PubMed] [Google Scholar]
- 18.Takao M, Zhang QM, Yonei S, Yasui A. Differential subcellular localization of human MutY homolog (hMYH) and the functional activity of adenine:8-oxoguanine DNA glycosylase. J Biol Chem. 1991;266:6480–6484. doi: 10.1093/nar/27.18.3638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kirkwood TB. Evolution of Ageing. Nature. 1977;270:301–304. doi: 10.1038/270301a0. [DOI] [PubMed] [Google Scholar]
- 20.Ferret JF, Marshak RR, Abt DA, Kenyon SJ. Relationship between lymphosarcoma and persistent lymphocytosis in cattle: a review. J Am Vet Med Assoc. 1979;175:705–708. [PubMed] [Google Scholar]
- 21.Amsterdam A, Sadler KC, Lai K, Farrington S, Bronson RT, Lees JA, Hopkins N. Many Ribosomal Protein Genes Are Cancer Genes in Zebrafish. PLoS Biol. 2004;2:0690–0698. doi: 10.1371/journal.pbio.0020139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Pogue-Geile K, Geiser JR, Shu M, Miller C, Wood IG, Meisler AI, Pipas JM. Ribosomal protein genes are over expressed in colorectal cancer: isolation of a cDNA clone encoding the human S3 ribosomal protein. Mol Cell Biol. 1991;11:3842–3849. doi: 10.1128/mcb.11.8.3842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Curran SP, Ruvkun G. Lifespan regulation by evolutionarily conserved genes essential for viability. PloS Genetics. 2007;3:0479–0487. doi: 10.1371/journal.pgen.0030056. [DOI] [PMC free article] [PubMed] [Google Scholar]