

Abstract
Background
Recent studies have identified critical roles for microRNAs (miRNAs) in a variety of cellular processes, including regulation of cardiomyocyte death. However, the signature of miRNA expression and possible roles of miRNA in the ischemic heart have been less well-studied.
Methods and Results
Here we performed miRNA arrays to detect the expression pattern of miRNAs in murine hearts subjected to ischemia/reperfusion (I/R) in vivo and ex vivo. Surprisingly, we found that only miR-320 expression was significantly decreased in the hearts upon I/R in vivo and ex vivo. This was further confirmed by Taqman RT-PCR. Gain-of-function and loss-of-function approaches were employed in cultured adult rat cardiomyocytes to investigate the functional roles of miR-320. Overexpression of miR-320 enhanced cardiomyocyte death and apoptosis, while knock-down was cytoprotective, upon simulated I/R. Furthermore, transgenic mice with cardiac-specific overexpression of miR-320 revealed an increased extent of apoptosis and infarction size in the hearts upon I/R in vivo and ex vivo, relative to the WT controls. Conversely, in vivo treatment with antagomir-320 reduced the infarction size, relative to the administration of mutant antagomir-320 and saline controls. Using Target-Scan software and proteomic analysis, we identified Hsp20, a known cardioprotective protein, as an important candidate target for miR-320. This was validated experimentally by utilizing a luciferase/GFP reporter activity assay and examining the expression of Hsp20 upon miR-320 overexpression and knockdown in cardiomyocytes.
Conclusions
Our data demonstrate that miR-320 is involved in the regulation of I/R-induced cardiac injury and dysfunction via antithetical regulation of Hsp20. Thus, miR-320 may constitute a new therapeutic target for ischemic heart diseases.
Keywords: MicroRNA-320, Ischemia/Reperfusion, Hsp20, Apoptosis, Infarction
There are more than 1 million Americans who suffer from myocardial infarction (MI) every year.1 Both human autopsy data and evidence from rodent models of MI indicate that most cell death happens by apoptosis during the initial 2–4 h following coronary occlusion.2,3 Clinical treatment of MI by thrombolytic therapy and revascularization by percutaneous coronary intervention or coronary artery bypass graft surgery are effective.1, 3 However, given the health, economic, and personal burden caused by ischemic heart disease, research into additional treatment modalities is imperative. Furthermore, the molecular mechanisms that regulate gene expression during myocardial ischemia/reperfusion (I/R) are still not completely understood.
MicroRNAs (miRNAs) are a class of endogenous non-protein-coding RNAs, comprising about 22 nucleotides. 4-6 They regulate gene expression via RNA-induced silencing complexes (RISC), targeting them to mRNAs where they inhibit translation or direct destructive cleavage.4-6 Increasing evidence indicates the importance of miRNAs in the regulation of cardiac developmental and pathological processes.7-10 For example, inhibition of miR-133 was sufficient to induce cardiomegaly in vivo;9 similary, targeted deletion of miR-1-2 revealed numerous functions in the heart, including regulation of cardiac morphogenesis, electrical conduction, and cell-cycle control. 10 More recently, miR-1 and miR-133 were shown to produce opposing effects on oxidative stress-induced apoptosis in H9c2 cells, with miR-1 being pro-apoptotic and miR-133 being anti-apoptotic. 11 Van Rooij et al. reported a signature pattern of stress-responsive miRNAs that could evoke cardiac hypertrophy and heart failure.12 Accordingly, miR-208 deficiency resulted in blunted hypertrophic and fibrotic responses to transverse aortic constriction (TAC).13 These results suggest that miRNAs have a fundamental role in the development of heart disease. However, the signature of miRNA expression and possible roles of miRNAs in myocardial infarction are less well-studied.
In this study, we performed miRNA arrays to detect the expression pattern of miRNAs in murine hearts subjected to I/R in vivo and ex vivo. Surprisingly, we observed that only miR-320 expression was consistently decreased in murine hearts upon I/R in vivo and ex vivo. Overexpression of miR-320 in cardiomyocytes resulted in increased sensitivity to I/R injury, whereas knock-down of endogenous miR-320 using antisense methodology ex vivo and antagomir administration in vivo was cytoprotective. Using Target-Scan software, proteomic analysis and a luciferase/GFP reporter assay in vitro, we identified Hsp20 as a real target for miR-320. Taken together, our findings implicate miR-320 as a potential therapeutic target for ischemic heart disease.
Materials and Methods
An expanded Materials and Methods section is available in the online data supplement at http://circ.ahajournals.org.
microRNA Extraction, miRNA-Microarray and Quantitative RT-PCR
miRNAs were isolated from mouse hearts (B6129SF2/JF2, 10-12-weeks old) after 24-h reperfusion preceded by 30-min ischemia, via left anterior descending (LAD) coronary artery occlusion, or from mouse hearts (FVB/N) subjected to 45-min no-flow global ischemia and 2-h reperfusion ex vivo, using the mirVana miRNA isolation kit (Ambion, Inc., Austin, TX), according to the manufacturer's protocol. The concentration of RNA was determined by a NanoDrop ND-1000 Spectrophotometer (NanoDrop Tech., Rockland, DE). miRNA expression profiling was determined by miRNA microarray analysis using the mouse miRNA array probes (mirVana™ miRNA Bioarrays Version 2, Ambion, Inc., Austin, TX) that include 266 mature mouse miRNAs. Dysregulated miRNAs were validated by using the mirVana™ qRT-PCR miRNA Detection Kit TaqMan miRNA assays. Primer sets for these miRNAs including control snoRNA412 were purchased from Ambion, Inc. (Austin, TX). Microarrays were performed in the Genomics and Microarray Laboratory, University of Cincinnati Medical Center. The data generated by GenePix® Pro version 5.0 software were analyzed to identify differentially expressed miRNAs. Data normalization was performed in two separate steps for each microarray, as previously described (details are available in the online data supplement). 14 All RT reactions, including no-template controls and RT minus controls, were run in triplicate in a GeneAmp PCR 9700 Thermocycler (Applied Biosystems). Relative expression was calculated using the comparative threshold cycle (Ct) method, as previous described.15
Cell Culture and Construction of Adenoviral Vectors
Adult ventricular cardiomyocytes were isolated from 2-month-old male Sprague–Dawley rats (Harlan Laboratory), as previously described. 16 Primary miR-320 DNA was PCR-amplified, using high fidelity AccuPrime Taq DNA polymerase (Invitrogen) from mouse genomic DNA. After sequencing, the amplified fragment (470 bp) was inserted under the CMV promoter into the AdEasy-1/Shuttle backbone, similar to our previous construction of adenoviral vectors.17 Antisense miR-320 adenovirus (named as AdasmiR-320) was generated by cloning the primary miR-320 DNA in the reverse orientation relative to the CMV promoter.
Generation of a miR-320 Transgenic Mouse Model
Transgenic (TG) mice were constructed by using a 470 bp DNA fragment containing murine primary miR-320 under the control of the α-myosin heavy chain promoter (α-MHCp). The expression levels of miR-320 were detected by Northern-blot as described in the Methods.
Global Ischemia Ex Vivo and Cardiac Injury Analysis
The cellular and functional responses to I/R were assessed in mice by using an isolated perfused heart model, as previously described. 18
In Vivo Administration of Antagomir-320
Chemically modified antisense oligonucleotides (antagomir) have been used to inhibit microRNA expression in vivo 9, 19-21. Antagomirs were synthesized by Dharmacon (www.dharmacon.com). Sequences are 5′-uscsgcccucucaacccagcusususus- Chol-3′ (antagomir-320), 5′-uscsgcccucucaaccgcagascscsus- Chol-3′ (antagomir-320-mutant as control). Lower case letters represent 2′-O-Methyl-modified oligonucleotides, subscript ‘s’ represents a phosphorothioate linkage, ‘Chol’ represents linked cholesterol, and underlined letters are a mutated seed sequence. Antagomir oligonucleotides were deprotected, desalted and purified by high-performance liquid chromatography. FVB/N male mice (6-weeks old) received either antagomir-320 or mutant antagomir-320 at a dose of 80 mg/kg body weight or a comparable volume of saline (200 μl) through tail vein injection. Regional ischemia in vivo was performed at 3 days after treatment.
Additional Methods
An expanded Materials and Methods section containing details regarding simulated ischemia/reperfusion treatment and cell survival assay, Northern blot detection of miR-320 expression, Western-blot analysis, GFP repression experiments, and luciferase reporter assay for targeting Hsp20 3′-UTRs is available in the online data supplement.
Statistical Analysis
All values are expressed as mean ± SEM. Student's t-test was used for two-group comparisons. Comparisons of parameters among 3 or more groups were analyzed by 1-way ANOVA for single factor, or 2-way ANOVA for two-factor variables with repeated measures, followed by Student's t-test with Bonferroni's correction for multiple-comparison. Differences were considered statistically significant at a value of P<0.05.
The authors had full access to and take full responsibility for the integrity of the data. All authors have read and agree to the manuscript as written.
Results
Aberrant Expression of miRNAs in Ischemic/Reperfused (I/R) Hearts
Successful ischemia after 30 min of LAD occlusion was confirmed by visual observation (cyanosis) and continous ECG monitoring. After 24-h reperfusion, the hearts were perfused with 1% TTC, followed by perfusion with 5% pthalo blue. As expected, the I/R group displayed significant cardiac infarction (infarction size, 21.7 ± 2.3%, n=6), whereas the sham group showed no infarction (Fig.1A). To further evaluate cardiac injury, we measured cardiomyocyte apoptosis, using two quantitative assays: TUNEL-staining and an ELISA-based nucleosome assay. As shown in Figure 1B, the proportion of TUNEL-positive nuclei in the myocardium of mice subjected to I/R was significantly increased, compared to the shams (7.6 ± 1.5% vs 0.6 ± 0.2%, P<0.01, n=6). Furthermore, DNA fragmentation, measured by the levels of mono-and oligo-nucleosomes, was significantly higher in the lysates of I/R hearts, relative to the shams (Fig.1C). These data indicate that I/R in vivo induces cardiac injury.
Figure 1.
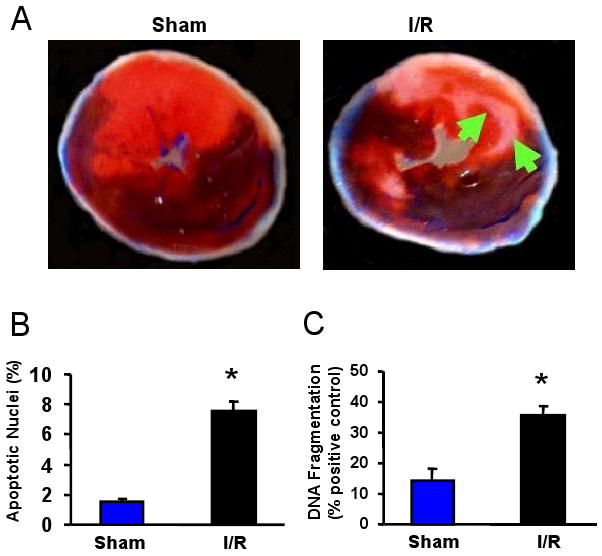


miRNA expression in the I/R hearts. (A-C) Occlusion of the left anterior descending coronary artery (LAD) for 30 min, followed by 24-h reperfusion (I/R), induced cardiac injury (infarction and apoptosis) in mice. (A) Infarct zone was observed in I/R hearts (white-gray zone, indicated by arrows), but not in sham samples. (B) The number of apoptotic nuclei and (C) the degree of DNA fragmentation were greatly increased in I/R hearts, compared with the shams (n=6, * P<0.01). (D) A partial heat-map of the upregulated and downregulated miRNAs (labeled by arrows). All of the microRNA array raw data is available in the Online supplemental data. (E) Dysregulated expression of miRNAs was confirmed by Taqman RT-PCR (normalized to control snoRNA412). RT-PCR primer sets for these miRNAs and control snoRNA412 were purchased from Ambion, Inc. We did not validate ambi-miR-9651 due to no available primer sets for this miRNA. (n=6, * P<0.05).
To determine the potential involvement of miRNAs in cardiac I/R injury, we used microarray analysis to determine miRNA levels in murine hearts after I/R in vivo (30-min LAD occlusion followed by 24-h reperfusion). We were excited to find that, compared with the sham group, only 6 of 640 probed-miRNAs were differentially expressed in I/R hearts (P < 0.01); 5 miRNAs were up-regulated, and only miR-320 was down-regulated (Fig. 1D and Table 1). These results were further validated by TaqMan RT-PCR assay (Fig.1E). Notably miR-7 expression was not detectable (Fig.1E), in agreement with its very low microarray intensity (Table 1). Furthermore, we extended our studies to an ex vivo model of no-flow global ischemia (45min) followed by 2-h reperfusion, and found that miRNAs upregulated in vivo were not dysregulated in ex vivo I/R hearts, compared with shams (online data supplement). This may be due to in vivo confounding effects, such as systemic circulation and a host of peripheral complications, activating different signaling pathways from ex vivo models, or may be related to the time points examined. However, miR-320 was consistently down-regulated in ex vivo I/R hearts, suggesting that miR-320 is an I/R-related microRNA. Therefore, we chose miR-320 for further determination of its potential roles in I/R-induced cardiac injury.
Table 1. MicroRNAs Significantly changed in the mouse hearts upon ischemia/reperfusion in vivo.
| MicroRNA | Intensities Averaged Over Probes | Fold Change | Std error (SE) (log2 scale) | P values | |
|---|---|---|---|---|---|
| IR | Sham | ||||
| Upregulated | |||||
| Ambi-miR-9651 | 101 | 33 | 2.72 | 0.31 | 0.006 |
| mmu-miR-146b | 353 | 135 | 2.64 | 0.24 | 0.0003 |
| mmu-miR-21 | 6436 | 2541 | 2.54 | 0.27 | 0.001 |
| mmu-miR-491 | 246 | 71 | 3.76 | 0.36 | 0.001 |
| mmu-miR-7 | 59 | 21 | 3.35 | 0.33 | 0.001 |
| Downregulated | |||||
| mmu-miR-320 | 1039 | 1814 | -1.84 | 0.26 | 0.005 |
MiR-320 Sensitizes Adult Rat Cardiomyocytes to Simulated I/R-Induced Apoptosis
To investigate the functional significance of miR-320 in ischemic heart, gain-of-function and loss-of function approaches were employed in cultured adult rat cardiomyocytes. We generated two adenoviral vectors encoding primary miR-320 in the sense or antisense direction, designated as AdmiR-320 and AdasmiR-320, respectively (Fig. 2A). Following infection of the cardiomyocytes with these recombinant adenoviral vectors for 48-h, we observed nearly 100% infection efficiency (Fig. 2B). Importantly, there were no apparent morphological alterations or differences in the number of adherent cells and rod-shaped cells among the AdmiR-320-, AdasmiR-320- and AdGFP-infected groups (Fig. 2B). After 60-h of adenoviral infection, Taqman real-time PCR (RT-PCR) clearly showed overexpression of the exogenous miR-320 in AdmiR-320-infected cardiomyocytes, while endogenous miR-320 was successfully knocked-down in AdasmiR-320-cells by ∼40% (Fig. 2C). We next examined the effects of miR-320 on cell survival upon 1-h simulated ischemia, followed by 3-h reperfusion. Cell viability analysis showed that ectopic expression of miR-320 reduced cell survival by ∼15%; while knock-down of endogenous miR-320 revealed opposite effects (i.e. increased survival by ∼20%), relative to GFP-cells (Fig. 2D and E). Furthermore, upon I/R, AdmiR-320-infected myocytes exhibited a significant increase in nuclear fragmentation, compared to Ad.GFP-infected myocytes (48 ± 4% vs 35± 3%, Fig. 2F). In contrast, infection with AdasmiR-320 significantly reduced the number of I/R-induced condensed nuclei (22± 5%, Fig. 2F). Consistently, the levels of DNA fragmentation in cell lysates upon I/R were significantly higher in miR-320-cells. Conversely, asmiR-320-cells presented with reduced DNA fragmentation, compared to the Ad.GFP group (Fig. 2G). Taken together, these data suggest that miR-320 may play an important role in I/R-mediated cardiac injury, probably through the regulation of cell death/apoptosis process.
Figure 2.
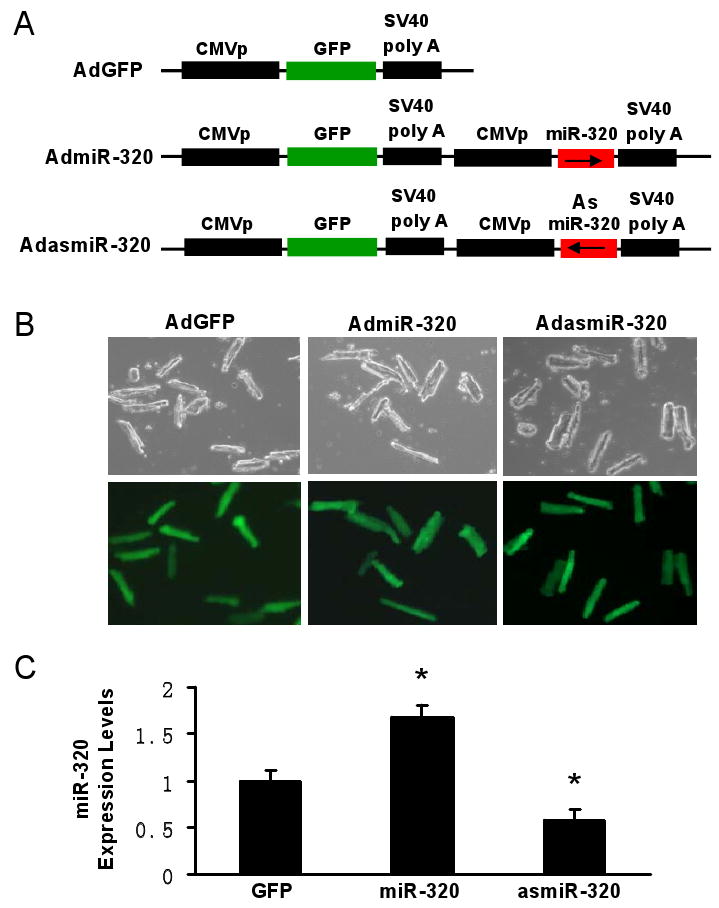
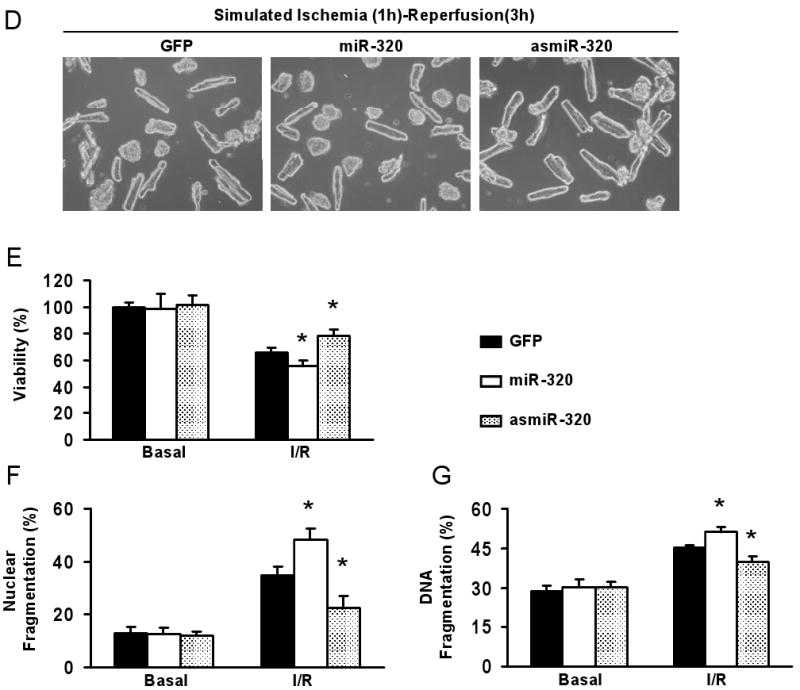
Overexpression of miR-320 enhanced cell death and apoptosis in cultured adult rat cardiomyocytes upon simulated ischemia/reperfusion, while knock-down was cytoprotective. (A) Diagram of recombinant adenoviral vectors. The primary miR-320 (470 bp) was PCR-amplified and inserted in the sense or antisense direction to generate AdmiR-320 or AdasmiR-320. (B) Nearly 100% cardiomyocytes were infected after 48-h adenoviral infection, as indicated by GFP fluorescence, and there were no morphological changes among GFP-, miR-320-, and asmiR-320-infected cells. (C) The levels of miR-320 were determined after 60-h infection by Taqman RT-PCR, as described in Figure 1, which confirmed the expression of mature miR-320 in AdmiR-320-infected cells, and knockdown of endogenous miR-320 in AdasmiR-320-cells. (D-G) miR-320-overexpressing cardiomyocytes were sensitive, while knockdown cells resistant to simulated ischemia/reperfusion (I/R)-induced cell death (D and E), and apoptosis (F and G), as determined by MTS incorporation (E), nuclear fragmentation by Hoechst staining (F), and cell-death-detection ELISA kit (G) (*P < 0.05 versus control AdGFP). Similar results were observed in three additional, independent experiments.
The Effects of miR-320 overexpression on I/R Cardiac Injury In Vivo
To further elucidate the in vivo effects of miR-320 upon I/R, we generated 6 transgenic (TG) mouse lines that carry the mouse primary miR-320 DNA under the control of the α–MHC mouse promoter (Fig.3A). All miR-320 TG mice were healthy and showed no apparent cardiac morphological or pathological abnormalities. Northern blot analysis (Fig.3B) revealed that miR-320 was successfully overexpressed in the TG hearts from Lines #1, #5, and #6 (2-3 fold increases). We selected Lines #5 and #6 for further I/R studies. These hearts were subjected to in vivo 30 min myocardial ischemia, via coronary artery occlusion, followed by 24-h reperfusion. We observed that the ratio of infarct-to-risk region was 39.4 ± 3.5% in Line #5 hearts and 44.5 ± 2.4% in Line #6 hearts (n=6 for each Line), compared to 22.7 ± 4.1% in WT hearts (n=8, P<0.001) under in vivo I/R conditions (Fig. 3C and D); whereas the region at risk was not significantly different among groups (Fig. 3E).
Figure 3.
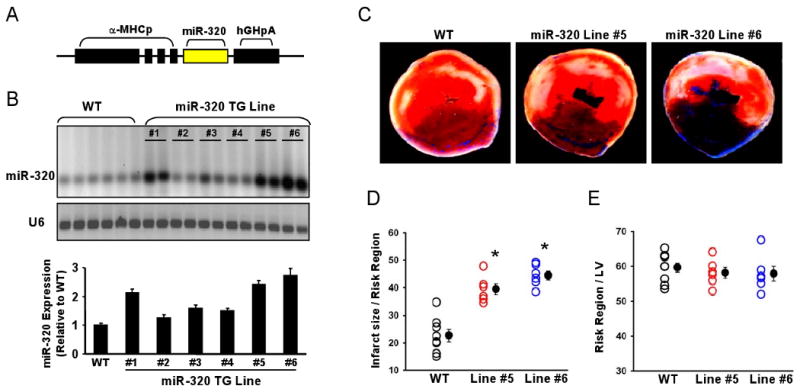
Overexpression of miR-320 increased I/R-induced cardiac injury in vivo. (A) Diagram of miR-320 TG construct. Transgenic mice were generated using a mouse primary miR-320 DNA under the control of the α-myosin heavy chain promoter (α-MHCp). (B) Northern blots showed that mature miR-320 was overexpressed in the transgenic hearts. Loading control was performed with an RNA probe against the small housekeeping RNA, U6 (106 nt). (C-D) miR-320 overexpression greatly increased myocardial infarct size after 30 min myocardial ischemia, via left anterior descending (LAD) coronary artery occlusion, followed by 24-h reperfusion, (E) while the region at risk was not significantly different among groups. (n=8, WT; n=6, miR-320 TG. *P<0.05 vs WT).
Increased Cardiac Injury and Dysfunction in the miR-320 Hearts upon I/R Ex Vivo
To further examine the effects of increased expression of miR-320 on cardiac functional recovery during I/R, we used an isolated perfused-heart preparation. Hearts from TG Line #6 were stabilized for 30 minutes, and subjected to 30 minutes of global ischemia and 1-h reperfusion. During reperfusion, the transgenic hearts exhibited significantly depressed functional recovery compared to the WTs (Fig. 4A-D), evidenced by the decreased rates of contraction (+dP/dt) and relaxation (-dP/dt) in miR-320 hearts during reperfusion, relative to WT hearts (Fig. 4A and B). Similarly, the left ventricular developed pressure (LVDP) recovered to 55 ±5% of pre-ischemic values after 1-h reperfusion in miR-320 hearts, while being 87 ±3 % in WT hearts (Fig. 4C). The left ventricular end-diastolic pressure (LVEDP) was also markedly increased in the miR-320 hearts during reperfusion (Fig. 4D), relative to WT hearts.
Figure 4.

miR-320 overexpression depressed post-ischemic cardiac performance in isolated perfused hearts. After 30 min stabilization with KH buffer, hearts were subjected to 30 minutes of global ischemia and 1-h reperfusion. (A-C) Recovery of post-ischemic contractile function (±dP/dt) and left ventricular developed pressure (LVDP) during reperfusion was significantly depressed in TG mice; (D) Left ventricular end-diastolic pressure (LVEDP) was significantly higher in TG than that of WT hearts, (n=10, WT; n=11, miR-320 TG. *P<0.05 vs WT). (E and F) miR-320 overexpression enhanced ischemia/reperfusion-induced necrosis, measured by LDH release in the outflow of first 10-min of reperfusion; and apoptosis, measured by DNA fragmentation in the I/R hearts. The cumulative release of LDH in the outflow was normalized to the total tissue LDH content. (n=10-11 for LDH activity measurement; n=5 for DNA fragmentation measurement, *P<0.05 vs WT).
To determine the degree of necrosis in these I/R hearts, we assessed the level of lactate dehydrogenase (LDH) released during the first 10 min of reperfusion after global ischemia. We observed that the total LDH was 3-fold higher in miR-320 hearts compared to WTs (Fig. 4E), demonstrating increased necrosis in miR-320-overexpressing hearts. Furthermore, heart lysates from a subset of experimental WT and TG hearts were assayed for DNA fragmentation, using a quantitative nucleosome assay. Transgenic hearts exhibited a 1.7-fold increase over WT hearts (Fig. 4F), indicating overexpression of miR-320 enhanced I/R-induced cardiac apoptosis.
MiR-320 Acts Directly at the Hsp20 3′UTR
To elucidate the potential mechanism of miR-320 in the regulation of cardiac I/R injury, we first identified the putative targets of miR-320, using computational predictions, as detailed at TargetScan (http://genes.mit.edu/targetscan/). Even though this analysis yielded 482 potential candidates, prediction does not consider the secondary structure of the target mRNA, which controls the accessibility of miRNA binding, suggesting some predicted targets are not real. Given the major function of miRNAs as protein-expression regulators, we searched the protein expression profile in I/R hearts. Excitingly, one study reported that the expression levels of only 12 proteins were altered in the murine heart proteome after I/R, relative to sham hearts.22 After matching the proteomic data with the TargetScan result, we found that only Hsp20 (also named as HspB6) was listed among the assumed targets for miR-320 in the murine heart (Fig. 5A), and the near perfect complimentary base-paring is located at 193-211bp of mouse Hsp20 3-UTR (Fig. 5B). More interestingly, the seed sequence of Hsp20 3′UTR targeting to miR320 is highly conserved among the species of mouse, human, rat and dog (Fig. 5C), suggesting a critical role in their physiology.
Figure 5.

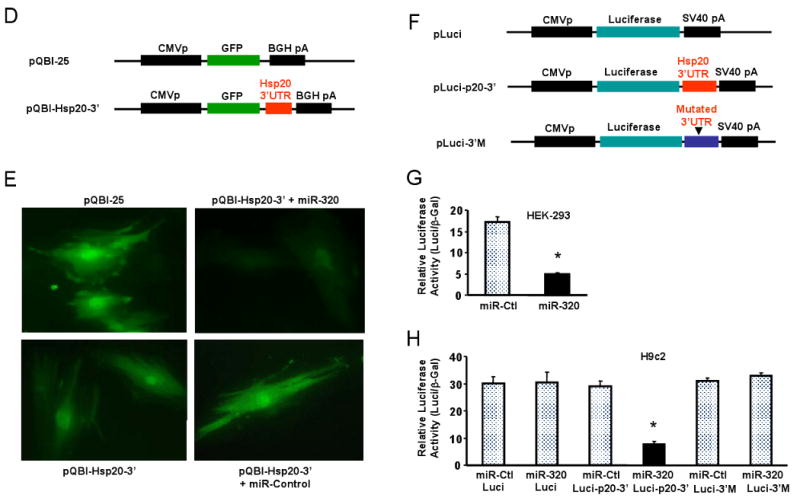
Hsp20 is a target of miR-320 in the murine heart. (A) TargetScan predicts that miR-320 has 482 targets, however, proteomic data showed that only 12 protein spots were altered in the proteome of I/R hearts (* Ref.# 22), and Hsp20 was listed in both the TargetScan results and the proteomics analysis. (B) Sequence alignment of miR-320 and 3′UTR of Hsp20. Note the complementarity at the 5′ and 3′ end of miR-320, where the crucial seed regions are located. (C) The putative miR-320-binding sites within the Hsp20 3′UTR are conserved among mammalian species (mouse, human, rat and dog). (D) Diagram of plasmid construction. A segment of Hsp20 3′ UTR was inserted downstream of the GFP-encoding sequence. (E) H9c2 cells were co-transfected with the plasmid containing the segment of Hsp20 3′ UTR and either miR-320 or a control oligoribonucleotide. Pictures were taken 48 h after transfection. (F) Diagram of plasmid construction. A segment of Hsp20 3′ UTR or a mutated segment was cloned downstream of the luciferase-encoding region. (G) Dual luciferase activity assay of HEK-293 cells co-transfected with the plasmid containing the segment of Hsp20 3′ UTR and either miR-320 or a control oligoribonucleotide showed that miR-320 inhibited luciferase activity, compared with controls. (H) Luciferase activity in H9c2 cells co-transfected with the various vectors indicated. (* P<0.05 relative to respective controls). Similar results were observed in three additional, independent experiments.
To validate whether miR-320 directly recognizes the 3′-UTR of Hsp20, we cotransfected H9c2 cells with a construct (Fig. 5D) containing the 3′-UTR of Hsp20 fused downstream to the GFP coding sequence along with miR-320 or a negative control miRNA. Overexpression of miR-320 resulted in a marked reduction of the GFP fluorescence intensity (Fig. 5E). This result was subsequently confirmed in both HEK-293 and H9c2 cells using a luciferase assay (Fig. 5F-H). Co-transfection of miR-320 in H9c2 cells strongly inhibited the luciferase activity from the reporter construct containing the 3′UTR segment of Hsp20, whereas no effect was observed with a construct containing a mutated segment of Hsp20 3′UTR (seed sequence CAGCUUU was mutated to GACACAA, Fig. 5H). This effect was specific because there was no change in luciferase reporter activity when a negative control miRNA was cotransfected with either reporter construct (Fig. 5H). Collectively, these data indicate that Hsp20 transcripts may represent a genuine target of miR-320.
MiR-320 Downregulates Hsp20 Protein In Vitro and In Vivo
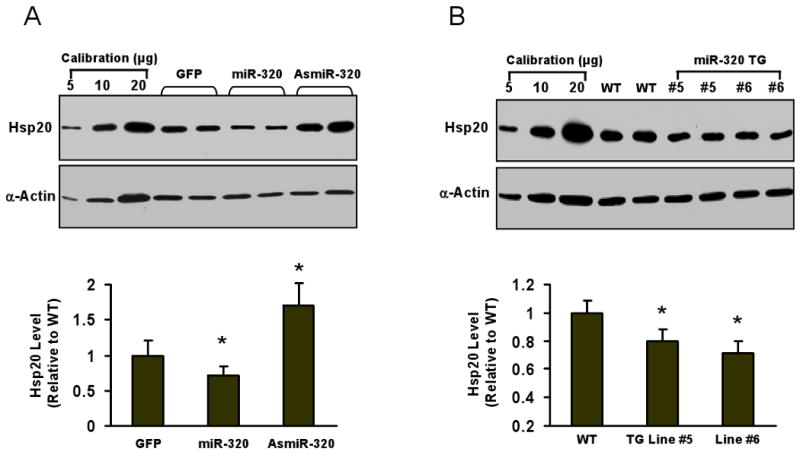
To ascertain if miR-320 regulates Hsp20 expression, we harvested adult rat cardiomyocytes ∼60-h after infection with AdmiR-320 or AdasmiR-320. Although real-time PCR demonstrated that Hsp20 messenger RNA copy numbers were similar among these groups (data not shown), cardiomyocytes infected with AdmiR-320 had reduced Hsp20 protein by ∼30%; whereas knockdown of endogenous miR-320 by AdasmiR-320 infection increased the levels of Hsp20 protein by ∼60% (Fig. 6A), indicating miR-320 acts as a negative regulator of Hsp20 translation.
Figure 6.
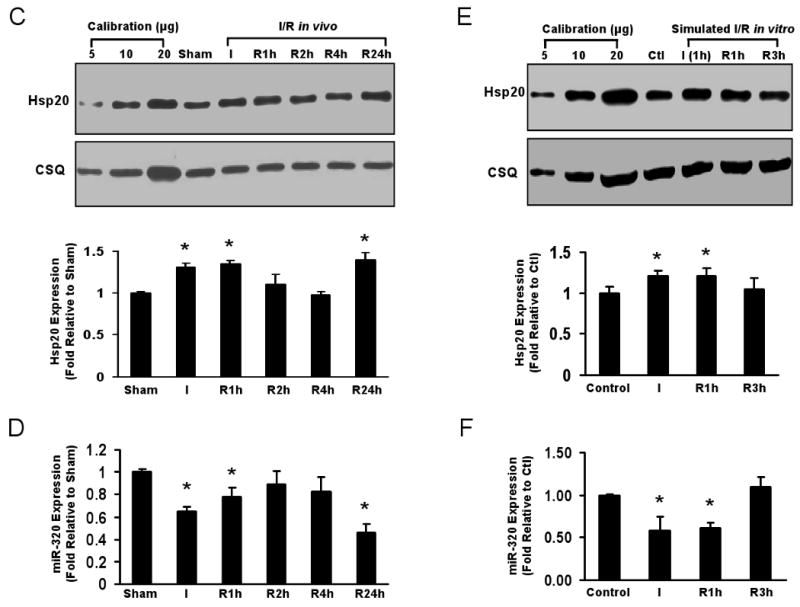
Reciprocal expression of miR-320 and Hsp20. (A) Total cellular protein from various adenoviral-infected cardiomyocytes after 60-h infection was probed using an Hsp20 antibody. Actin was used as a loading control. (B) Immunoblots for determination of Hsp20 expression in miR-320 TG hearts. * P<0.05 relative to controls, n=6-8 hearts. (C) Time course expression of Hsp20 in hearts during in vivo I/R determined by Western blots. Calsequestrin (CSQ) was used as a loading control. (D) Time course expression of miR-320 in murine hearts during I/R was determined by Taqman RT-PCR, as described in Figure 1 legend. * P<0.05 relative to shams, n=3 hearts. (E) Time course expression of Hsp20 and (F) miR-320 in ex vivo cultured adult rat cardiomyocytes subjected to simulated ischemia (1h)/reperfusion (3h). * P<0.05 relative to controls, n=3 hearts.
In miR-320 transgenic hearts, Hsp20 levels were reduced by 21% in Line #5 and 30% in Line #6, respectively (Fig. 6B). Furthermore, we examined the time course for both Hsp20 and miR-320 expression during I/R in vivo and ex vivo (Fig.6C-F). The expression levels of Hsp20 were significantly increased at the end of ischemia, after 1-h reperfusion and 24-h reperfusion (in vivo), which was negatively correlative with the reduced expression of miR-320 in the hearts (in vivo, Fig. 6D) and cardiomyocytes (ex vivo, Fig. 6F). Taken together, these data indicate that miR-320 expression is correlated to Hsp20 levels in the heart, and miR-320 efficiently represses Hsp20 expression in vivo and ex vivo.
Knockdown of miR-320 Decreases the In Vivo Cardiac Infarction Size
To further evaluate the biological role of downregulation of miR-320 on myocardial infarction, we knocked down miR-320 via a single tail vein injection of cholesterol-modified antagomir-320 (80 mg/kg). Mutant antagomir-320 and saline were used as controls. Three days after administration of antagomir-320, Taqman RT-PCR analysis showed a dramatic reduction of miR-320 expression in the heart tissue (Fig. 7A). In contrast, mutant antagomir-320 had no effect on the expression level of miR-320 compared with the saline control (Fig. 7A). These results indicate that antagomir-320 efficiently downregulate miR-320 expression in the heart, consistent with previous reports, using antagomirs. 9, 19-21 In parallel, administration of antagomir-320, but not antagomir-320 mutant, was associated with greatly increased levels of Hsp20 in the hearts (Fig.7B). Furthermore, these hearts treated with antagomirs for 3 days were also subjected to a 30-minute coronary occulusion, followed by 24-h reperfusion. We observed that the infarct size was greatly reduced in the antagomir-320-treated hearts (6.2 ± 1.5% vs. 16.9 ± 3.8%, 20.8 ±4.3% area at risk in the antagomir-320 mutant-treated and saline-treated controls, respectively; P < 0.05, Fig. 7C). In addition, region at risk was not significantly different among groups (Fig. 7C). Taken together, these results suggest that systemically or locally applied inhibitory miR-320 molecules (i.e. antagomir-320) may protect the heart against I/R injury in vivo.
Figure 7.
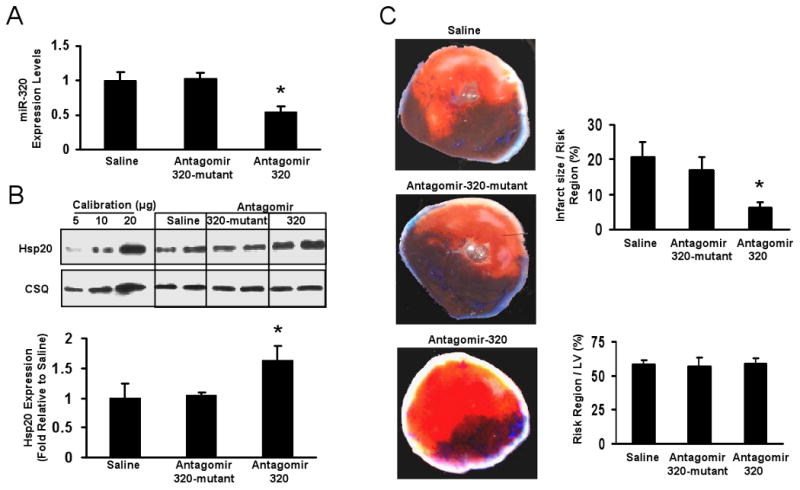
In vivo effects of antagomir-320 administration on myocardial ischemia/reperfusion. (A) The levels of miR-320 expression were determined in murine hearts 3 days after administration of antagomir-320, antagomir-320 mutant and saline control by Taqman RT-PCR, as described in Figure 1 legend. (B) Western blot and densitometric analysis of Hsp20 expression in murine hearts 3 days after treated with these anatgomirs. Calsequestrin (CSQ) was used as a loading control. (C) Antagomir-320 treatment greatly reduced myocardial infarct size after 30 min myocardial ischemia followed by24-h reperfusion, while the region at risk was not significantly different among groups. (n=7, saline; n=5, mutant antagomir-320; n=7, antagomir-320. *P<0.05 vs saline control).
Discussion
Cardiomyocyte death/apoptosis is a key cellular event in ischemic hearts.2 It is well established that multiple genes are aberrantly expressed in infarct hearts, which are responsible for cardiac remodeling after ischemia/reperfusion (I/R).22 Because miRNAs are endogenous regulators of gene expression, it is reasonable to hypothesize that they may be involved in I/R-induced cardiac injury. We therefore, for the first time, applied a well-established mouse cardiac I/R model in vivo and ex vivo to determine the miRNA expression signature in ischemic hearts. We were excited to find that only miR-320 expression was consistently dysregulated in ischemic hearts in vivo and ex vivo, suggesting miR-320 is an I/R-related microRNA in the murine heart. Furthermore, knockdown of endogenous miR-320 expression reduced cardiomyocyte death and apoptosis induced by simulated I/R, while overexpression of miR-320 increased sensitivity to I/R-triggered cell death. Thus, at the cellular level, both loss-of function and gain-of function experiments indicate that miR-320 is a negative regulator of cardioprotection against I/R injury. These cellular effects were further confirmed in vivo with cardiac-specific overexpression of miR-320 mouse models and antagomir-320 treatment, in which miR-320 hearts were sensitive, while antagomir-320-treated hearts being resistant, to I/R-induced cardiac injury.
It should be noted that several studies have demonstrated aberrant expression of miR-320 in both animal heart hypertrophy and human heart disease.23-25 Expression of miR-320 was downregulated in murine hearts after aortic banding at day 7, but not at day 14 and 28. 23 In end-stage human failing hearts, expression of miR-320 was increased by 3.4 fold.24 Ikeda et al. examined the expression of 428 microRNAs in 67 human left ventricular samples including control, ischemic cardiomyopathy (ICM), dilated cardiomyopathy (DCM), and aortic stenosis (AS) diagnostic groups.25 They observed that miR-320 was upregulated in ICM and AS samples, but no significant alterations were noted in DCM samples.25 Collectively, these data suggest that miR-320 could be involved in the regulation of multiple independent pathophysiological processes in the heart, especially in the ischemic heart.
While the results from computational miRNA target prediction algorithms revealed miR-320 had 482 potential targets, previous proteomic data showed that 12 proteins were altered in the heart upon I/R.22 Surprisingly, among 12 altered proteins, only Hsp20 was listed in miR-320 target-scan results. Considering that proteomic approaches have restrictions due to technical limitations and different turnover times for proteins, there could be more than 12 proteins altered in I/R hearts, compared to sham hearts. Therefore, it is impossible to exclude other potential targets of miR-320, which may also contribute to modulation of cardiac I/R injury. Indeed, a new proteomic approach using SILAC (stable isotope labeling with amino acids in cell culture), which directly measures genome-wide alterations in protein synthesis induced by miRNAs, indicated that overexpression of a miRNA in HeLa cells mildly downregulated thousands of proteins.26, 27 In the present study, we validated that Hsp20 was one of miR-320-targeted proteins in the heart. Furthermore, mouse Hsp20 3′UTR has 11 potential miRNA binding regions including a miR-320 binding site (Online supplemental data, Figure S2); however, the microarray intensity of miR-320 in the heart was far higher than those of 10 other targeted miRNAs (Online supplemental data). These results suggest that Hsp20 may be regulated to a great extend by miR-320 in the heart, which is an important addition to other possible mechanisms regulating Hsp20 protein synthesis such as transcription and protein degradation.
It is noteworthy that Hsp20, compared to other small heat-shock proteins, is mostly up-regulated in animal hearts upon ischemic conditions, 22 exercise training 28 and rapid right ventricular pacing.29 This suggests that Hsp20 may play a major role in cellular stress-resistance and development of tolerance as an adaptive response after exposure to various stimuli. Our data presented in this study indicate that downregulation of miR-320 might represent an important adaptive mechanism to upregulate the expression levels of Hsp20 during the I/R, because previous reports from our laboratory and others have shown that Hsp20: 1) protects hearts against I/R-, doxorubicin-, and chronic isoproterenol-induced apoptosis and remodeling; 18, 30, 31 2) inhibits platelet aggregation; 32 3) regulates activities of vasorelaxation; 33 and 4) enhances contractile function. 18, 30
In conclusion, our data suggest that dysregulation of miR-320 expression contributes to ischemic heart disease. Knockdown of endogenous miR-320 provides protection against I/R-induced cardiomyocyte death and apoptosis by targeting Hsp20, a well-studied cardioprotector. Future studies using an inducible system will be helpful to identify the exact therapeutic role for miR-320 in ischemic heart disease. As miRNAs often have numerous targets, it is very important to further explore the target-network of miR-320, which may be involved in maintaining cardiac output after infarction. This may lead to rational target selection for therapeutic intervention in patients suffering from heart disease.
Clinical Perspective
MicroRNAs (miRNA) are a class of small non-coding RNAs with important post-transcriptional regulatory functions. Recent data suggest that miRNAs are aberrantly expressed in many human diseases including cardiovascular disease, which leads to an increasing interest in miRNA regulation as a therapeutic and diagnostic approach. Of note, multiple genes/proteins have been shown to be aberrantly expressed in infarcted hearts, which are responsible for cardiac remodeling after ischemia/reperfusion (I/R). In the present study, we used microarrays to profile the expression of 640 probed-miRNAs in murine hearts upon I/R in vivo and ex vivo. Several miRNAs were differentially expressed between shams and I/R-hearts, with miR-320 showing down-regulation consistently in I/R-hearts ex vivo and in vivo relative to the shams. Gain-of-function and loss-of-function approaches were employed in cultured adult rat cardiomyocytes and in mouse hearts to investigate the functional roles of miR320. Our data indicate the increased levels of miR-320 may be responsible for cardiac I/R injury, whereas downregulation of miR-320 may be protective. Administration of antagomir-320 that specifically knocked-down endogenous miR-320 significantly decreased cardiac infarction size. Thus, systemically or locally applied mimic or inhibitory miRNA molecules (i.e. antagomir) that influence specific cardiac miRNAs may finally open novel miRNA-based therapies for heart disease.
Supplementary Material
Acknowledgments
Funding Sources: This study was supported by NIH grant HL-087861 (Dr. G. C. Fan).
We are grateful to Dr. Evangelia G. Kranias for helpful discussion of this manuscript.
Footnotes
Conflict of Interest Disclosures: None
References
- 1.Cannon RO., 3rd Mechanisms, management and future directions for reperfusion injury after acute myocardial infarction. Nat Clin Pract Cardiovasc Med. 2005;2:88–94. doi: 10.1038/ncpcardio0096. [DOI] [PubMed] [Google Scholar]
- 2.Abbate A, Bussani R, Amin MS, Vetrovec GW, Baldi A. Acute myocardial infarction and heart failure: role of apoptosis. Int J Biochem Cell Biol. 2006;38:1834–1840. doi: 10.1016/j.biocel.2006.04.010. [DOI] [PubMed] [Google Scholar]
- 3.Cokkinos DV, Pantos C. Myocardial protection in man--from research concept to clinical practice. Heart Fail Rev. 2007;12:345–362. doi: 10.1007/s10741-007-9030-5. [DOI] [PubMed] [Google Scholar]
- 4.van Rooij E, Olson EN. MicroRNAs: powerful new regulators of heart disease and provocative therapeutic targets. J Clin Invest. 2007;117:2369–2376. doi: 10.1172/JCI33099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Latronico MV, Catalucci D, Condorelli G. Emerging role of microRNAs in cardiovascular biology. Circ Res. 2007;101:1225–1236. doi: 10.1161/CIRCRESAHA.107.163147. [DOI] [PubMed] [Google Scholar]
- 6.Callis TE, Wang DZ. Taking microRNAs to heart. Trends Mol Med. 2008;14:254–260. doi: 10.1016/j.molmed.2008.03.006. [DOI] [PubMed] [Google Scholar]
- 7.Zhao Y, Samal E, Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature. 2005;436:214–220. doi: 10.1038/nature03817. [DOI] [PubMed] [Google Scholar]
- 8.Sayed D, Hong C, Chen IY, Lypowy J, Abdellatif M. MicroRNAs play an essential role in the development of cardiac hypertrophy. Circ Res. 2007;100:416–424. doi: 10.1161/01.RES.0000257913.42552.23. [DOI] [PubMed] [Google Scholar]
- 9.Carè A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MV, Høydal M, Autore C, Russo MA, Dorn GW, 2nd, Ellingsen O, Ruiz-Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G. MicroRNA-133 controls cardiac hypertrophy. Nat Med. 2007;13:613–618. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- 10.Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, Tsuchihashi T, McManus MT, Schwartz RJ, Srivastava D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell. 2007;129:303–317. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 11.Xu C, Lu Y, Pan Z, Chu W, Luo X, Lin H, Xiao J, Shan H, Wang Z, Yang B. The muscle-specific microRNAs miR-1 and miR-133 produce opposing effects on apoptosis by targeting HSP60, HSP70 and caspase-9 in cardiomyocytes. J Cell Sci. 2007;120:3045–3052. doi: 10.1242/jcs.010728. [DOI] [PubMed] [Google Scholar]
- 12.van Rooij E, Sutherland LB, Liu N, Williams AH, McAnally J, Gerard RD, Richardson JA, Olson EN. A signature pattern of stress-responsive microRNAs that can evoke cardiac hypertrophy and heart failure. Proc Natl Acad Sci U S A. 2006;103:18255–18260. doi: 10.1073/pnas.0608791103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–579. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- 14.Sartor M, Schwanekamp J, Halbleib D, Mohamed I, Karyala S, Medvedovic M, Tomlinson CR. BioTechniques. 2004;36:790–796. doi: 10.2144/04365ST02. [DOI] [PubMed] [Google Scholar]
- 15.Zhang X, Leung YK, Ho SM. AP-2 regulates the transcription of estrogen receptor (ER)-beta by acting through a methylation hotspot of the 0N promoter in prostate cancer cells. Oncogene. 2007;26:7346–7354. doi: 10.1038/sj.onc.1210537. [DOI] [PubMed] [Google Scholar]
- 16.Fan GC, Yuan Q, Zhao W, Chu G, Kranias EG. Junctin is a prominent regulator of contractility in cardiomyocytes. Biochem Biophys Res Commun. 2007;352(3):617–22. doi: 10.1016/j.bbrc.2006.11.093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fan GC, Chu G, Mitton B, Song Q, Yuan Q, Kranias EG. Small heat-shock protein Hsp20 phosphorylation inhibits beta-agonist-induced cardiac apoptosis. Circ Res. 2004;94:1474–1482. doi: 10.1161/01.RES.0000129179.66631.00. [DOI] [PubMed] [Google Scholar]
- 18.Fan GC, Ren X, Qian J, Yuan Q, Nicolaou P, Wang Y, Jones WK, Chu G, Kranias EG. Novel cardioprotective role of a small heat-shock protein, Hsp20, against ischemia/reperfusion injury. Circulation. 2005;111:1792–1799. doi: 10.1161/01.CIR.0000160851.41872.C6. [DOI] [PubMed] [Google Scholar]
- 19.Krützfeldt J, Rajewsky N, Braich R, Rajeev KG, Tuschl T, Manoharan M, Stoffel M. Silencing of microRNAs in vivo with ‘antagomirs’. Nature. 2005;438:685–689. doi: 10.1038/nature04303. [DOI] [PubMed] [Google Scholar]
- 20.van Rooij E, Sutherland LB, Thatcher JE, DiMaio JM, Naseem RH, Marshall WS, Hill JA, Olson EN. Dysregulation of microRNAs after myocardial infarction reveals a role of miR-29 in cardiac fibrosis. Proc Natl Acad Sci U S A. 2008;105:13027–13032. doi: 10.1073/pnas.0805038105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Krützfeldt J, Kuwajima S, Braich R, Rajeev KG, Pena J, Tuschl T, Manoharan M, Stoffel M. Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res. 2007;35:2885–2892. doi: 10.1093/nar/gkm024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.De Celle T, Vanrobaeys F, Lijnen P, Blankesteijn WM, Heeneman S, Van Beeumen J, Devreese B, Smits JF, Janssen BJ. Alterations in mouse cardiac proteome after in vivo myocardial infarction: permanent ischaemia versus ischaemia-reperfusion. Exp Physiol. 2005;90:593–606. doi: 10.1113/expphysiol.2005.030296. [DOI] [PubMed] [Google Scholar]
- 23.Cheng Y, Ji R, Yue J, Yang J, Liu X, Chen H, Dean DB, Zhang C. MicroRNAs are aberrantly expressed in hypertrophic heart: do they play a role in cardiac hypertrophy? Am J Pathol. 2007;170:1831–1840. doi: 10.2353/ajpath.2007.061170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thum T, Galuppo P, Wolf C, Fiedler J, Kneitz S, van Laake LW, Doevendans PA, Mummery CL, Borlak J, Haverich A, Gross C, Engelhardt S, Ertl G, Bauersachs J. MicroRNAs in the human heart: a clue to fetal gene reprogramming in heart failure. Circulation. 2007;116:258–267. doi: 10.1161/CIRCULATIONAHA.107.687947. [DOI] [PubMed] [Google Scholar]
- 25.Ikeda S, Kong SW, Lu J, Bisping E, Zhang H, Allen PD, Golub TR, Pieske B, Pu WT. Altered microRNA expression in human heart disease. Physiol Genomics. 2007;31:367–373. doi: 10.1152/physiolgenomics.00144.2007. [DOI] [PubMed] [Google Scholar]
- 26.Baek D, Villén J, Shin C, Camargo FD, Gygi SP, Bartel DP. The impact of microRNAs on protein output. Nature. 2008;455:64–71. doi: 10.1038/nature07242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Selbach M, Schwanhäusser B, Thierfelder N, Fang Z, Khanin R, Rajewsky N. Widespread changes in protein synthesis induced by microRNAs. Nature. 2008;455:58–63. doi: 10.1038/nature07228. [DOI] [PubMed] [Google Scholar]
- 28.Boluyt MO, Brevick JL, Rogers DS, Randall MJ, Scalia AF, Li ZB. Changes in the rat heart proteome induced by exercise training: Increased abundance of heat shock protein Hsp20. Proteomics. 2006;6:3154–3169. doi: 10.1002/pmic.200401356. [DOI] [PubMed] [Google Scholar]
- 29.Dohke T, Wada A, Isono T, Fujii M, Yamamoto T, Tsutamoto T, Horie M. Proteomic analysis reveals significant alternations of cardiac small heat shock protein expression in congestive heart failure. J Card Fail. 2006;12:77–84. doi: 10.1016/j.cardfail.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 30.Fan GC, Yuan Q, Song G, Wang Y, Chen G, Qian J, Zhou X, Lee YJ, Ashraf M, Kranias EG. Small heat-shock protein Hsp20 attenuates beta-agonist-mediated cardiac remodeling through apoptosis signal-regulating kinase 1. Circ Res. 2006;99:1233–1242. doi: 10.1161/01.RES.0000251074.19348.af. [DOI] [PubMed] [Google Scholar]
- 31.Fan GC, Zhou X, Wang X, Song G, Qian J, Nicolaou P, Chen G, Ren X, Kranias EG. Heat shock protein 20 interacting with phosphorylated Akt reduces doxorubicin-triggered oxidative stress and cardiotoxicity. Circ Res. 2008;103:1270–9. doi: 10.1161/CIRCRESAHA.108.182832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Niwa M, Kozawa O, Matsuno H, Kato K, Uematsu T. Small molecular weight heat shock-related protein, HSP20, exhibits an anti-platelet activity by inhibiting receptor-mediated calcium influx. Life Sci. 2000;66:7–12. doi: 10.1016/s0024-3205(99)00566-4. [DOI] [PubMed] [Google Scholar]
- 33.McLemore EC, Tessier DJ, Thresher J, Komalavilas P, Brophy CM. Role of the small heat shock proteins in regulating vascular smooth muscle tone. J Am Coll Surg. 2005;201:30–36. doi: 10.1016/j.jamcollsurg.2005.03.017. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.