

Abstract
The signal transduction pathways involved in neuronal death are not well understood. Neuroglobin (Ngb), a recently discovered vertebrate globin expressed predominantly in the brain, shows increased expression in neurons in response to oxygen deprivation and protects neurons from ischemic and hypoxic death. The mechanism of this neuroprotection is unclear. We examined the surface distribution of raft membrane microdomains in cortical neuron cultures during hypoxia using the raft marker cholera toxin B (CTx-B) subunit Mechanistically, we demonstrate that hypoxia induces rapid polarization of somal membranes and aggregation of microdomains with the subjacent mitochondrial network. This signaling complex is formed well before neurons commit to die, consistent with an early role in death signal transduction. Ngb-expressing neurons and neurons from Ngb-overexpressing transgenic (Ngb-Tg) mice do not undergo microdomain polarization or mitochondrial aggregation in response to, and are resistant to death from hypoxia. We link the protective actions of Ngb to inhibition of Pak1 kinase activity and Rac1-GDI disassociation, and inhibition of actin assembly and death-signaling module polarization.
Keywords: soma, polarity, disc-death inducing signaling complex
Introduction
The death of neurons is poorly understood, especially the extent to which different mechanisms are linked to different death-inducing stimuli, such as ischemia, excitotoxicity, and degenerative disorders. Neuroglobin (Ngb), an evolutionarily ancient member of the vertebrate globin family (1), is induced by neuronal hypoxia and cerebral ischemia and protects neurons from hypoxic and ischemic cell death (2). How Ngb protects neurons is unclear. Although Ngb belongs to the globin family, it shares little amino-acid sequence similarity with vertebrate myoglobins and hemoglobins, suggesting a distinct evolution and physiological functions separate from O2 storage and transport.
In ischemic brain, two forms of neuronal death are observed (3). Necrotic neuronal death occurs in regions of severe ischemic injury, while programmed cell death is observed in less injured areas. Current understanding of cell-death signaling in non-neural cells may provide clues to the mechanisms that regulate the balance between these two forms of cell death in neurons.
In immune cells, death-signal transduction involves the spatial organization of signaling molecules into higher order, death-inducing signaling complexes (DISC). The death-signaling specificity of DISC is regulated by the lateral movement and compartmentalization of membrane proteins, with Fas/CD95 aggregation and dynamic polarization of lipid microdomains an essential event in DISC formation (4). Polarization of membrane microdomains, in turn, is shaped by activation of Rho GTPases and reorganization of the actin cytoskeleton; accordingly, inhibitors of actin assembly, such as cytochalasin D, block microdomain aggregation (5). Disruption of lipid rafts also blocks plasma membrane compartmentalization and inhibits death-signal transduction in the immune system.
The role of raft microdomains and spatial regulation of death-signaling pathways in neurons is unknown. Cultured cortical neurons respond rapidly to oxygen deprivation, demonstrating pronounced changes in morphology within minutes. These changes include bleb formation, process retraction and a change in shape of the soma from pyramidal to round (6). Disruption of cytoskeletal architecture during hypoxia in epithelial cells is mediated in large part by Rho proteins (7). The Rho GTPase family includes several members, but RhoA, Rac1 and Cdc42 are the best characterized, and unique effects on actin are associated with each (8). Rho GTPases are GDP-bound and maintained inactive in the cytosol, complexed with a GDP-dissociation inhibitor (GDI). Activation of Rho GTPases requires GDI dissociation, replacement of GDP with GTP, and intracellular translocation of GTPases from the cytoplasm to the plasma membrane (9). Recent evidence indicates that that human Ngb may function as a GDI and regulate receptor-mediated G protein signaling (10).
We examined the role of Ngb in modulating Rho GTPase-RhoGDI complex dynamics and cytoskeletal re-organization during neuroprotection against hypoxia. We demonstrate that cytoskeletal changes in hypoxia are not random or chaotic, but part of a polarity-based signaling response. In this context, Ngb appears to protect against hypoxia by interfering with cytoskeletal polarization and lipid raft-dependent death signaling through the Rho GTPase pathway.
Materials and methods
Western blotting
To evaluate interactions between endogenous RhoGDI or Ngb, we carried out coimmunoprecipitation experiments. Cell lysates were extracted in PBS containing 1% Nonidet P-40, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 1 μg/ml of aprotinin and 100 μg/ml of phenylmethylsulfonyl fluoride, and protein concentration was determined using a Bio-Rad protein assay. RhoGDI or Ngb was immunoprecipitated from cultured control and Ngb-Tg cortical neurons before and after 6 h hypoxia using anti-RhoGDI or anti-Ngb antibody. Precipitated protein was washed four times with lysis buffer, separated by SDS-PAGE, transferred to polyvinyldifluoridine membrane, and probed with anti-Rac1, anti-RhoA, anti-RhoGDI, anti-Pak1 and anti-Flotillin-1 antibodies (Santa Cruz Biotechnology) for coimmunoprecipitated proteins. The level of RhoGDI or Ngb expression in whole-cell lysates was analyzed by Western blotting with anti-RhoGDI or anti-Ngb antibodies. Membranes were washed with PBS containing 0.1% Tween-20, incubated with horseradish peroxidase conjugated anti-mouse, anti-rabbit or anti-goat secondary antibody (Santa Cruz Biotechnology) at 4° for 60 min, and washed three times for 15 min with PBS/Tween-20. Peroxidase activity was visualized with a chemiluminescence substrate system (NEN Life Science Products Inc.).
Pak1 kinase assay
Kinase reactions contained recombinant GST-Pak1 (1 μg per reaction) in kinase buffer (50 mM Hepes, pH 7.5, 10 mM MgCl2, 2 mM MnCl2, 0.2 mM DTT) in a 70-μl reaction mixture, with 5 μg of purified recombinant Ngb, 20 μM ATP and 0.5 μCi/reaction radiolabeled [γ-32P]ATP (specific activity 4500 mCi/mmol, from ICN, Costa Mesa, CA). As a positive control, the kinase reaction was performed in the presence of 1 μg of myelin basic protein (Sigma M-1891). The reactions were incubated for 30 min at 30 °C and stopped by addition of 15 μl of 4× concentrated Laemmli’s SDS-PAGE sample buffer. Phosphorylation reactions were resolved by 12% SDS-polyacrylamide gel electrophoresis (SDS-PAGE), then gels were stained with Coomassie Brilliant Blue R-250, destained, dried, and subjected to autoradiography for 1–24 h.
Measures for cortical neuronal culture viability (LDH release, chromatin fragmentation)
Cortical cultures enriched in cells of neuronal lineage were prepared from 16-day mouse embryos with Neurobasal medium containing B27 supplement, 2 mM glutamate, and 1% penicillin and streptomycin. After 4 days, one-half of the medium was replaced with Neurobasal medium containing 2% B27, and experiments were conducted at 8 days in vitro.
The LDH release value obtained from cortical neurons exposed to 1% Triton X-100-containing lysis buffer served as 100% LDH release, and data obtained in other groups were calculated as percent of this value accordingly. Data are expressed as the mean ± S.E.M (n = 3).
We determined the percent nuclei with condensed or fragmented chromatin for control and Ngb-Tg cortical neurons by counting 500 nuclei in several random fields. We also determined the percentage of neurons with aggregated and polarized raft microdomains (CTx-B raft staining) induced by hypoxia for control and Ngb-Tg neurons by counting 500 neurons in several random fields. Following 6 h hypoxia the percentage of neurons with polarized raft microdomains is significantly lower in Ngb-Tg neurons. Following 24 h hypoxia treatment the percentage of neurons with condensed and fragmented chromatin is significantly lower in Ngb-Tg neurons. The staining and scoring of all samples was performed in a single-blinded fashion.
In lymphocytes, reorganization of membrane microdomains (lipid rafts) mediates formation of death-inducing signaling complexes (DISC) involved in transducing Fas/CD95 death signals. Here, uniformly distributed small rafts form larger aggregates, which then coalesce to form a polarized signal transduction domain in one section of the plasma membrane and mediate Fas death signaling. As such, Fas/C95 microdomain polarization and chromatin condensation and fragmentation are considered objective measurements of Fas mediated apoptotic death.
To establish criterion for neuronal death signal transduction, we describe somal microdomain polarization and chromatin fragmentation as integral parts of the neuronal death cascade. As in the case of Fas DISC signaling in lymphocytes, inhibition of raft polarization is sufficient to block neuronal death and downstream chromatin fragmentation. Thus, we relate somal raft polarization directly to LDH release and chromatin fragmentation as an early stage of neuronal death signaling.
Transgenic Mice
To generate Ngb transgenic mice, we introduced full-length mouse Ngb cDNA into the Spe1 and EcoRV restriction sites of the pTR-UF12d vector, upstream of the chicken β-actin promoter and the distal enhancer region, and downstream of green fluorescent protein (GFP), to generate a Ngb-GFP vector. All final plasmids were verified by sequencing and overexpression of Ngb and GFP proteins in the 293 cell line and confirmed by Western analysis.
Transgenic mice were produced by pronuclear injection of BDF1 × BDF1 embryos. Founders were identified by PCR analysis of lysates from tail biopsies with two different primer pairs. For genotyping of Ngb-GFP transgenic mice, mouse tail DNA was screened by PCR using specific primers (5′-GGGTTACTCCCACAGGTGAG-3′ and 5′-CAAGCTGGTCAGGTACTCCTCC-3′ for Ngb 506-bp product; 5′-GCGGTCACAAACTCCAGCAGGACCA-3′ and 5′-GGCGTGGTCCCAATCTCGTGGAA-3′ for GFP 664-bp product). Founder animals were intercrossed with CD1 mice to establish lines. Of 5 independent transgenic lines, 3 had comparable expression levels as determined by immunoblot analysis. Ngb-Tg mice used in the present study were offspring of intersibling matings over at least 6 generations. Ngb++ mice displayed no overt phenotypic abnormalities based on visual inspection, dissection of the major organs, brain histology, or simple tests of behavior. Ngb was constitutively overexpressed in multiple cells types and in multiple organs, including both neurons and astrocytes in brain (26).
Ngb siRNA target DNA sequence (ATGGCGCTGCATGTGCGTTGA)
Ngb-Tg cortical cultures were pre-treated with 4 μM control siRNA or 4 μM Ngb siRNA 24 h before hypoxia. Positively transfected cortical neurons showed decreases in Ngb expression as measured by decreased Ngb immunfluorescence staining intensity.
Target DNA sequence: ATGGCGCTGCATGTGCGTTGA. Positively transfected cortical neurons showed decreases in Ngb expression as measured by decreased Ngb immunfluorescence staining intensity. Transfection of Ngb-Tg cortical cultures with siNgb RNA resulted in 71 ± 14% knockdown of Ngb protein. A similar reduction in Ngb protein levels and results was obtained with Ngb siRNA sequence ATGGAGCGCCCGGAGTCAGAG. Images were processed by the Imaris imaging interphase (Bitplane AG). The IsoSurface module was used in Imaris to quantify intensity signals between samples.
Results
Polarized Hypoxia Signaling
First, we examined actin polymerization and the surface distribution of raft membrane microdomains in cultured cortical neurons using FITC-phalloidin to label polymeric actin, and the Alexa-labeled raft marker cholera toxin B (CTx-B) subunit to label the glycosphingolipid GM1 (11) which is enriched in lipid rafts. GM1 gangliosides are abundant in the exoplasmic leaflet of the plasma membrane and CTx-B is frequently used to visualize GM1 enriched microdomains on the plasma membrane surface. In normoxic cortical neurons, GM1 is evenly distributed along the somal surface (Fig 1) and axonal and dendritic processes. By contrast, in cortical neurons exposed to hypoxia, GM1 is redistributed into clusters and dense polarized regions (Fig 1), and axodendritic CTx-B staining is reduced. In addition, neuronal mitochondria aggregate underneath the polarized lipid microdomain (Fig 2). This occurs within minutes following hypoxia and precedes cytochrome c release during neuronal death, suggesting that raft polarization and mitochondrial aggregation are early signaling responses to neuronal hypoxia (Fig 3). Raft polarization and mitochondrial aggregation are accompanied by an active reorganization of actin and the establishment of discrete foci of actin polymerization underneath the lipid microdomain (Fig 1).
Fig 1. Polarized Hypoxic Signal Transduction: Hypoxia induces membrane microdomain polarization in cultured cortical neurons.

Cortical neurons at 9 DIV stained with Alexa-CTx-B (red/left top panel) and FITC-phalloidin (green/middle top panel) were analyzed by confocal microscopy. (Panel A) Lipid rafts are symmetrically dispersed on resting neurons. CTx-B raft (red) staining is uniformly distributed around the cell body in resting neurons. Phalloidin actin (green) is faint with the exception of several spontaneous actin clusters indicating primarily dispersed unpolymerized actin in resting neurons. 4,6-diamidino-2-phenylindole (DAPI)-labeled nuclei are in blue. Hypoxia induces lipid raft polarization and actin polymerization. CTx-B raft surface staining (red/left, panels B and C) and actin (green/middle, panels B and C) 30 min after hypoxia treatment is initially localized to patched membrane regions (middle panel), which aggregate into a polarized membrane complex (panel C). Arrow indicates aggregated raft membrane microdomains and underlying co-aggregated polymerized actin patches. DAPI-labeled nuclei are in blue. Yellow indicates co-localization of CTx-B raft (red) and phalloidin actin (green) staining (Merge/right panels).
Fig 2. Hypoxia induces mitochondria aggregation within the cytoplasm of cortical neurons, which is localized beneath polarized lipid rafts.

Mitochondria are symmetrically distributed within the cytoplasm of resting cortical neurons. (Panel D) CTx-B raft staining (red/left top panel), mitochondria (MitoTracker® green/middle top panel), and Dapi-labeled nuclei (blue) in control neurons. Merged image of CTx-B raft staining (red), mitochondria (MitoTracker® green), and Dapi-labeled nuclei (blue) in control neurons (Merge/right panel D). CTx-B raft staining (red/left panel E), mitochondria (MitoTracker® green/middle panel E), and Dapi-labeled nuclei (blue) following 3 h of hypoxia. Merged image of CTx-B raft staining (red), mitochondria (MitoTracker® green), and Dapi-labeled nuclei (blue) following 3 h of hypoxia (Merge/right panel E).
Fig 3. Hypoxia induced lipid raft and mitochondria aggregation precede cytochrome c release during hypoxia-induced neuronal death.

Mitochondria are symmetrically distributed within the cytoplasm of resting cortical neurons (a). Areas of co-localization of mitochondria and cytochrome c are indicated in yellow by the blending of mitochondria (green) and cytochrome c (red) (left panels) and in white by the blending of rafts (blue), mitochondria (green) and cytochrome c (red) (right panels). Polarized cytoplasmic distribution of mitochondria within hypoxia treated cortical neurons (b). Merged image of aggregated lipid rafts (blue), mitochondria (green) and cytochrome c (red) following 3 h of hypoxia. Areas of co-localization are indicated in yellow by the blending of mitochondria (green) and cytochrome c (red) (left panels) and in white by the blending of rafts (blue), mitochondria (green) and cytochrome c (red) (right panels). Mitochondria (green) and cytochrome c (red) co-localization (yellow) indicates that cytochrome c is still inside mitochondria. Uncondensed DAPI-labeled nuclei are in purple. Merged image of aggregated mitochondria (green) and cytochrome c (red) following 6 h of hypoxia (c). Arrow indicates neurons with aggregated mitochondria (green) and lack of co-localization (yellow) of cytochrome c (red). The separation of mitochondria (green) and cytochrome c (red) staining suggests that cytochrome c is no longer within the mitochondria and has been released into the cytoplasm. DAPI-labeled nuclei are in purple. Arrow points to areas of condensed chromatin. Bar represents 10 m. The experiments in Fig 1–3 were repeated three times with similar results.
Mitochondria play a central role in apoptotic death signaling pathways. Following exposure to death-inducing stimuli, mitochondria release cytochrome c into the cytoplasm, and activate caspase cascades leading to cell death (12). In non-neural cells undergoing apoptosis, mitochondria also aggregate in response to toxic insults (13). We examined the distribution of mitochondria in cortical neurons before and after hypoxia. In normoxic cortical neuron cultures, mitochondria are distributed broadly and uniformly in the cytoplasm, but are concentrated at the nuclear periphery (Fig 2). In response to hypoxia, neuronal mitochondria dynamically aggregate at a locus underneath the polarized lipid microdomain, before nuclear changes associated with hypoxic death are detected (Fig 2).
Hypoxia can produce apoptotic neuronal death mediated partly by caspase-3, and the competitive caspase-3 inhibitor, carbobenzoxy-Glu-Val-Asp-CH2-2, 6-dichlorobenzolate (ZEVD), suppresses hypoxia-induced nuclear changes (14). However, ZEVD does not inhibit hypoxia-induced raft polarization or mitochondrial aggregation, suggesting that these processes are caspase-3-independent, and precede chromatin condensation and fragmentation.
Mitochondria-mediated caspase-3 activation is initiated by the release of cytochrome c from mitochondria to the cytoplasm (15). We examined the distribution of cytochrome c before and after mitochondrial aggregation. In resting neurons, cytochrome c and mitochondria are uniformly distributed and co-localized, consistent with an intramitochondrial location of cytochrome c (Fig 3). In hypoxic cells, mitochondria aggregate, but cytochrome c and mitochondria remain co-localized, indicating that aggregation of lipid rafts and mitochondria precedes cytochrome c release (Fig 3). At later times, cytochrome c and mitochondria are clearly separated (Fig 3), indicating that most cytochrome c has been released. These results suggest that lipid raft polarization, mitochondrial aggregation, and actin reorganization are upstream signaling events preceding cytochrome c release and caspase-3 activation in hypoxic neuronal death. Our data are consistent with observations that mitochondria in mammalian cells can aggregate and form a physically interconnected network (16). This network is responsive to local high microdomains (“hotspots”) of calcium within the soma and may represent an attempt to buffer and channel calcium during death signaling.
We reasoned that if mitochondria aggregate to a Ca2+ entry source within the soma, proteins involved in ion homeostasis and ion flux during hypoxia may occur within the clustered raft complex. To explore the protein composition of the hypoxia-induced polarized raft- signaling complex, we probed cultured neurons with antibodies against key signaling molecules. Kv2.1 potassium channels, which mediate neuronal responses to ischemia and apoptosis (17,18,19) and target to lipid rafts in normoxic neurons (20), were distributed in clusters along the somal surface and co-localized with CTx-B (Fig 1D). Hypoxia induces polarization of Kv2.1 immunoreactivity within the cell membrane and its co-localization with Rac1 and CTx-B (Fig 4). Also co-localized within this complex are the Na+, K+ ATPase β2 subunit, Na+, Ca2+ exchanger (NCX1) and TRPC5 transient receptor potential cation channel (Fig 4). Our data imply location-dependent roles for mitochondria, and are consistent with observations that a subset of mitochondria near a Ca2+ source preferentially takes up Ca2+ and is a target for Ca2+- induced toxicity (21).
Fig 4. Panel 4 Hypoxia induces mobilization and co-aggregation of membrane microdomains and signaling molecules.

Cortical neuron cultures were stained with combinations of CTx-B, and mAbs against Kv2.1, and Na+, K+ ATPase β2, before and 3 hr after hypoxia and were analyzed by confocal microscopy. (panel a) Lipid rafts, Kv2.1, and Na+, K+ ATPase β2 are symmetrically dispersed on resting neurons. CTx-B staining (red), Kv2.1 (green) and Na+, K+ ATPase β2 (blue) immunoreactivity (green) are dispersed around the cell body and processes in resting neurons. DAPI-labeled nuclei are in blue. (panel b) Hypoxia induces lipid raft, Kv2.1, and Na+, K+ ATPase β2 aggregation. CTx-B raft, Kv2.1 and Na+, K+ ATPase β2 surface staining 3 hr after hypoxia treatment is co-localized to compartmentalized membrane regions. Co-localization is indicated in white by the blending of aggregated raft microdomain staining (CTx-B red), Kv2.1 (green), and Na+, K+ ATPase β2 (blue) immunoreactivity. Similar CTx-B co-localization results following hypoxia were obtained with Abs against the Na+, Ca2+ exchanger and TRPC5 cation channel. Bar represents 10 μm. Arrow indicates hypoxia-induced polarized raft microdomain co-localized with Kv2.1 and Na+, K+ ATPase β2.
Membrane microdomain polarization is mediated by actin cytoskeleton reorganization in a variety of cell types (22). To examine the role of cytoskeletal changes during hypoxia we pretreated cortical cultures with latrunculin-A (Lat-A), a toxin that inhibits actin assembly by sequestering monomeric actin (G-actin), resulting in net depolymerization of actin polymer (F-actin) (23). In response to hypoxia, cortical neurons pre-treated with Lat-A do not undergo cytoskeleton reorganization, microdomain polarization or mitochondrial aggregation and are protected against cell death (Fig 5, Fig 8a). Similar protection was seen using cytochalasin D, which also depolymerizes actin filaments (24). Our results are in accord with data, obtained from the immune system, which demonstrate that polarization of lipid microdomains into signaling complexes depends on Rho GTPase-induced actin polymerization (22).
Fig 5. Latrunculin-A, an actin assembly inhibitor, prevents microdomain polarization and protects cortical neurons from hypoxia-induced death.
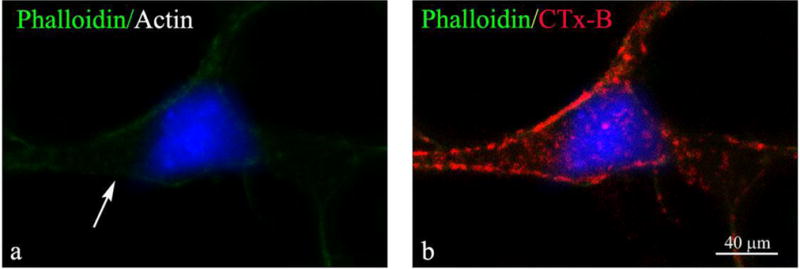
Lat-A 1 μM treated cortical neurons exposed to 6 h hypoxia. Arrow in Lat-A-treated neurons indicates diffuse actin (green) staining (left panel) and uniform distribution of CTx-B (red) (right panel merge). See Fig 1A, panel b, for actin polymerization comparison with Lat-A untreated culture. DAPI-labeled nuclei are in blue. Bar represents 40 μm. The experiments in Fig 4, 5 were repeated three times with similar results.
Fig 8. Ngb-Tg mice cortical neurons are protected from hypoxia-induced death as measured by LDH release and chromatin fragmentation.

(panel a) Resting control cultured cortical neurons (control group, CNTL, white bars) and Ngb-Tg cortical neurons (Ngb over-expression group, Ngb-Tg, white bars) at 9 DIV were untreated or exposed to hypoxia for 24 h (Hypoxia group, blue bars). Ngb over-expression induced resistance to hypoxic neuronal death was prevented by co-incubation of Ngb-Tg cortical neurons with 4 μM Ngb siRNA to reduce neuroglobin protein levels (neuroglobin depleted group, siNgb, control siRNA group, siCNTL).
Latrunculin-A, an actin assembly inhibitor, prevents microdomain polarization and protects cortical neurons from hypoxia-induced death as measured by LDH release and chromatin fragmentation (Hypoxia group, teal bars). Cultured cortical neurons at 9 DIV were untreated (control group, CNTL, white bars), exposed to hypoxia for 24 h (hypoxia group, HYP, black bars) or pre-treated with 1 μM latrunculin-A for 1 h (Lat-A, teal bars) before 24 h of hypoxia. At the end of 24 h of hypoxia, culture medium was collected for LDH release assay. The LDH release value obtained from cortical neurons exposed to 1% Triton X-100-containing lysis buffer served as 100% LDH release, and data obtained in other groups were calculated as percent of this value accordingly. Data are expressed as the mean ± S.E.M (n = 3). Control and 1 μM Lat-A treated cortical neurons were assessed for raft distribution (Ctx-B staining) and chromatin integrity (DAPI staining) following 6 h and 24 h of hypoxia, respectively. We determined the percentage of neurons with aggregated and polarized Ctx-B staining and the percent nuclei with condensed and fragmented chromatin for each experimental group of neurons by counting 500 neurons in several random fields. Following 6 h hypoxia treatment, the incidence of neurons with polarized raft microdomains is significantly lower and following 24 h hypoxia treatment the incidence of neurons with condensed and fragmented chromatin is significantly lower in Ngb-Tg and Lat-A treated neurons.
Ngb over-expression inhibits hypoxia-induced membrane microdomain polarization (CTx-B raft staining) in Ngb-Tg cortical neurons. (panel b) Control cortical neuronal cultures stained with CTx-B (red) and anti-Ngb ab (green). CTx-B (red) is uniformly distributed around the cell body in resting control neurons. (panel c) Control neurons exposed to 6 h hypoxia. Arrow in control hypoxia-treated neurons indicates aggregated CTx-B (red) with Ngb staining (green) beneath polarized CTx-B. (panel d) Resting Ngb-Tg cortical neuronal cultures stained with CTx-B (red) and anti-Ngb ab (green) have uniform distributions of CTx-B (red) and increased levels of Ngb immunoflourescence (green). Yellow indicates co-localization of CTx-B raft (red) and Ngb (green) staining. (panel e) Ngb-Tg cortical neurons exposed to 6 h hypoxia maintain uniform distributions of CTx-B (red). Yellow indicates co-localization of CTx-B raft (red) and Ngb (green) staining. (panel f) Ngb over-expression induced resistance to microdomain polarization was prevented by pre-treatment of Ngb-Tg cultures with 4 μM Ngb siRNA 24 h before hypoxia. Positively transfected cells show siNgb RNA-induced decreased levels of Ngb protein staining (green). Arrow in Ngb siRNA treated Ngb-Tg neurons exposed to 6 h hypoxia indicates polarized distribution of CTx-B (red). The experiments in Fig 8 were repeated three times with similar results.
Ngb Inhibits RhoGTPase-RhoGDI Dissociation
To examine the role of Rho GTPases in hypoxic reorganization of the actin cytoskeleton, we monitored the subcellular distribution of Rac1 and RhoA GTPases. Rho GTPases are normally GDP-bound and maintained inactive in the cytosol complexed with a GDI. Activation of Rho GTPases requires GDI dissociation, replacement of GDP with GTP and translocation of the GTPases from cytoplasm to plasma membrane (9). In normoxic neurons, Rac1 and RhoA are uniformly distributed throughout the cytoplasm, but RhoA is enriched in processes and co-localizes with Ctx-B staining (Fig 6A). With hypoxia, somal Rac1 staining becomes asymmetric and co-localizes with the polarized membrane microdomain signaling complex, whereas RhoA is distributed uniformly through the cell membrane (Fig 6A). Our results demonstrate that Rho family GTPases are differentially regulated by hypoxia and partitioned into distinct spatial pools. Hypoxic targeting of Rac1 to raft microdomains indicates a role for Rac1 in microdomain polarization in accord with the microdomain targeting of Rac1 by integrins in shaping adhesion-mediated signaling complexes (25).
Fig 6. Fig 6A Rho GTPases exist in distinct spatial pools in cortical neurons following hypoxia treatment. Rac1 co-localizes with polarized raft microdomains following hypoxia.

Confocal images showing topographical distribution of CTx-B (red), Rac1 (green) and Rho A (blue) in control cultured cortical neurons before (panel A) and after 3 h hypoxia (panel B). Purple indicates areas of overlap between Rho A (blue) and CTx-B (red). Yellow indicates areas of overlap between Rac1 (green) and CTx-B (red). Cyan indicates areas of overlap between Rac1 (green) and Rho A (blue). Co-localization of CTx-B (red), Rac1 (green) and Rho A (blue) is indicated in white. Dapi-labeled nuclei are pseudo-colored grey and are condensed following hypoxia. Arrows indicate hypoxia-induced Rac1 co-localization with polarized raft microdomains.
Fig 6B,C Rac1 and RhoA exist in Rac1-GDI and RhoA-GDI complexes in resting neurons which are dissociated by hypoxia. Increases in Neuroglobin protein levels induce Ngb-RhoGDI complexes and increase Rac1 association with RhoGDI. (panel B) RhoGDI was immunoprecipitated from cultured control and Ngb-Tg cortical neurons before (C, CNgb-Tg) and after 6 h hypoxia (Hyp, HypNgb-Tg) using anti-RhoGDI antibody. Precipitated RhoGDI was separated by SDS-PAGE, transferred to nitrocellulose membrane, and probed with anti-Rac1 and anti-RhoA antibody for coimmunoprecipitated Rac1 and RhoA. The level of RhoGDI protein expression in whole-cell lysates (bottom panel) was analyzed by Western blotting with anti-RhoGDI antibodies. Data shown are representative of three independent experiments. (panel C) Ngb was immunoprecipitated from cultured control and Ngb-Tg cortical neurons before (C, CNgb-Tg) and after 6 h hypoxia (Hyp, HypNgb-Tg) using anti-Ngb antibody. Precipitated Ngb protein was separated by SDS-PAGE, transferred to nitrocellulose membrane, and probed with anti-RhoGDI, anti-Rac1, anti-RhoA, anti-Pak1 and anti-Flotillin-1 antibodies for coimmunoprecipitated proteins. The level of Ngb expression in whole-cell lysates (bottom panel) was analyzed by Western blotting with anti-Ngb antibodies. Data shown are representative of three independent experiments. Control immunoprecipitations using goat and mouse IgG did not result in specific bands following Western analysis.
We next examined the role of Ngb in Rho GTPase-RhoGDI dynamics and cytoskeletal re-organization during hypoxia, using transgenic mice (Ngb-Tg) that over-express Ngb protein (26). Ngb-Tg mice are resistant to ischemia in vivo (26) and neurons from these mice are resistant to hypoxia in vitro (Fig. 8). We compared the binding of Rac1 and RhoA to RhoGDI in wild type and Ngb-Tg cortical neuron cultures before and during hypoxia. In normoxic wild type neurons, Rac1 and RhoA coprecipitate with RhoGDI, whereas hypoxia decreases RhoGDI-associated Rac1 complexes reflecting the release of Rac1 from RhoGDI, a permissive step for GTPase activation and actin assembly (Fig. 6B).
Total RhoGDI, Rac1 and RhoA levels are similar in normoxic wild-type and Ngb-Tg neurons. However, increased amounts of Rac1 and RhoA coprecipitate with RhoGDI in Ngb-Tg neurons. Further, in hypoxic Ngb-Tg cortical neurons, less Rac1 is released from Rac1-RhoGDI complexes, indicating that Rac1 is maintained in an inert Rac1-GDI complex (Fig. 6C). Wild type neurons express low levels of Ngb, which are up-regulated by hypoxia (2), while Ngb-Tg neurons express substantially greater levels of Ngb, which do not increase during hypoxia (Fig. 6C). We used immunoprecipitation to further examine in vivo interactions between Ngb and Rho family proteins before and during hypoxia. In wild type neurons, coprecipitation of Ngb with RhoGDI, Rac1 and RhoA is low in the normoxic state, but increases during 6 h of hypoxia. In contrast, Ngb-Tg neurons show substantial coprecipitation, which decreases during hypoxia. Ngb in hypoxic control or Ngb-Tg neurons also coprecipitates with the raft microdomain marker flotillin-1, indicating that Rho family GTPases interact with Ngb associated with plasma membrane microdomains. Flotillins are localized at the cytoplasmic face of membrane microdomains and are thought to act as scaffolds for the formation of multiprotein complexes. Further, Ngb coprecipitates with Pak1, a kinase that is a key regulator of actin assembly during polarity establishment, and as part of the RhoGDI-GTPase signaling complex modulates GDI-Rac1GTPase release (27) (Fig. 6C). Pak1 signaling cascades involve the phosphorylation of LIM kinase, which then phosphorylates its substrate, cofilin, and regulate the generation of cytoskeletal structures. In vitro kinase assays using purified recombinant Pak1 and Ngb demonstrate Ngb binds directly to Pak1 and inhibits Pak1’s kinase activity as part of its neuroprotective action, and that Ngb is also phosphorylated by Pak1 (Fig. 7).
Fig 7. Purified Recombinant Ngb inhibits Pak1 kinase activity.

Purified recombinant free Ngb (5 μg) was added to a recombinant GST-Pak1 (1 μg) in vitro kinase assay using purified myelin basic protein (MBP 1 μg) as a positive control, as in Experimental Procedures. Autoradiography: Lanes C- control Pak1 kinase assay in the absence of Ngb and with MBP 1 μg as a positive target. Lane N-Pak1 kinase assay in the presence of 5 μg of Ngb. Ngb inhibits Pak1 phosphorylation of MBP and is also phosphorylated by Pak1. Arrows indicate GST-Pak1, MBP and Ngb, respectively.
Pak1 autophosphorylates (GST-Pak1)—top arrow indicates levels of GST-Pak1 in control lanes C, C and Ngb lane N. MBP phosphorylation levels (control lanes C, C) are reduced by Ngb (lane N); molecular mass standards in kDa are indicated. Results shown are representative of three independent experiments.
We next examined the subcellular distribution of Ngb within wild-type and Ngb-Tg neurons before and after hypoxia. In normoxic wild-type neurons, low levels of Ngb are diffusely distributed throughout the cytoplasm unassociated with the raft membrane marker CTx-B. Following 6 h of hypoxia, Ngb staining is localized beneath the polarized raft signaling module with minimal CTx-B co-localization (Fig. 8). In contrast, Ngb staining in normoxic Ngb-Tg neurons is increased compared to wild type, and distributed throughout the cytoplasm in discrete puncta, and a portion of Ngb staining co-localizes with raft membrane CTx-B staining. Following 6 h of hypoxia, Ngb staining in Ngb-Tg neurons co-localizes predominantly with CTx-B, and the cell membrane and cytoskeleton remain unpolarized. Addition of Ngb siRNA restores hypoxia-induced microdomain polarization, reverses Ngb-induced neuroprotection, and suggests that Ngb-mediated inhibition of microdomain polarization may be essential to its neuroprotective mechanism (Fig. 8).
Discussion
Somal Polarity Mediates Hypoxic Neuronal Death Signal Transduction
Our data show that hypoxia cause the formation of a polarized neuronal signaling module composed of a lipid raft-mitochondrial-actin cytoskeletal lattice that by its coupling architecture vectorially propagates the death signal. The formation of this signaling complex is regulated by Ngb through Pak1 kinase inhibition, enhancement of Rac1-RhoGDI association and inhibition of actin cytoskeleton assembly. Increased Ngb levels, produced by hypoxia or constitutive over-expression, facilitate the interaction of Ngb with RhoGDI and RhoGTPase family members, modulating their activity and their roles in signaling module assembly. We propose that, in response to hypoxia and other neuronal insults, raft polarization, mitochondrial aggregation, and actin cytoskeleton reorganization act in concert to establish somal polarity, in order to sense and mount a response to the insult (Fig. 9). Similar RhoGTPase regulation of microdomain polarization occurs during Fas/CD95 DISC formation in lymphocytes (4) and may represent a mechanism that is conserved across the immune and nervous systems for death signal transduction. As in Fas DISC death signaling, disruption of aggregation and polarization of this module in neurons, mediated through increases in Ngb expression dampens death signaling and protects against injury. Other receptors involved in cell death induction, such as TNFR1, DR5, and DCC are also located in lipid microdomains (28,29,30,31,32) and this localization seems to be a prerequisite for their ability to trigger cell death. Disruptors of lipid rafts and inhibitors of raft aggregation abrogate death signaling, presumably by interfering with the topological relationship of death receptors and caspases in death signaling complexes.
Fig 9.

A model of the formation of polarized somal signaling in neurons following exposure to neurotoxic stimuli (hypoxia, Aβ, NMDA). The model shows the effects of Ngb on the Rho GTPase–GDI cycle and more specifically on the release and activation of Rac1.
Rho GTPases such as Rac1 act as molecular switches to regulate downstream biological responses. To perform this function, they must cycle between GDP-bound inactive states and GTP-bound active states. GDP dissociation inhibitors (GDIs: RhoGDI) sequester the inactive GTPase, preventing the dissociation of GDP and interactions with regulatory and effector molecules. This inhibitory action of GDIs requires that they be dissociated from their partner GTPases for the GTPases to become activated and elicit their biological effects.
In response to cell stimulation, Rac1 is induced to dissociate from GDI through Pak1 kinase-mediated phosphorylation of GDI. The GDI-free Rac1 is converted to the active GTP-bound form, which is then able to bind to effectors, promote actin polymerization and associate with membranes. The interaction of Rac1 with the membrane is terminated by the conversion of Rac1 to the GDP form and re-association with GDI.
Our data suggests that Ngb over-expression promotes Rac1 association with GDI maintaining Rac1 in an inactive state and retarding actin polymerization and subsequent cytoskeletal and microdomain aggregation. Our data also demonstrates that Ngb associates with Pak1, a kinase that phosphorylates GDI and promotes Rac1 release. Our results demonstrate that Ngb also inhibits Pak1 kinase activity further inhibiting Rac1 release and that Ngb is also a target for Pak1 phosphorylation. Maintaining Rac1 in an inactive state inhibits action polymerization, microdomain aggregation and polarized signal transduction.
Figure 9. Proposed role of Ngb in Rac1-mediated formation of hypoxia-induced signaling complex in neurons. (a) Ngb inhibits and Pak1 stimulates GDP/GTP exchange and release of Rac1 from Rho-GDI-Rac1 complexes. Rac1 release and activation and subsequent signaling complex formation is triggered by hypoxia, Aβ or NMDA. (b) This generates active (GTP-bound) Rac1, which promotes actin polymerization, leading to aggregation of lipid rafts.
Brain ischemia is associated with at least two forms of neuronal death. Necrotic death occurs in regions of severely reduced blood flow (ischemic core), while in the surrounding ischemic penumbra, where oxygen levels are higher and blood flow is less compromised, programmed cell death is observed (3). Ngb immunoreactivity is increased in murine cortical neurons after focal cerebral ischemia (2), especially in the penumbra, suggesting that Ngb expression might be particularly increased in neurons that are ischemic, but capable of survival. Our results suggest that somal microdomain polarization is part of a programmed signaling response to hypoxia that parallels cytoskeletal microdomain polarization observed during DISC formation in immune cells. Our data demonstrate that increases in Ngb expression in Ngb-Tg cortical neurons retard microdomain polarization through inhibition of Rac1-GDI dissociation and Rac1-mediated actin assembly, and suggest a novel mechanism for Ngb-mediated neuroprotection. Pak1 kinase activity, a regulator of actin cytoskeleton polarization (33), is also inhibited by Ngb as part of its neuroprotective action (Fig 7), suggesting further parallels between polarization mechanisms and death signal transduction in neural and immune systems.
Our in vitro Pak1 kinase results demonstrate Ngb itself is phosphorylated by Pak1. Globins display a high degree of functional plasticity by creating or deleting docking sites and cavities within the protein moiety. 3D crystallographic data of murine CO-ligated Ngb shows substantial structural changes upon binding of CO to the ferrous heme iron, involving a sliding motion of the heme and a topological reorganization of Ngb’s large internal cavity (34). It is possible that these conformational changes may be dynamically regulated by Pak1 phosphorylation and modulate Ngb’s protein-protein interactions as part of the signal encoding hypoxic information.
It is unclear whether death mechanisms may be linked between different forms of neuronal death (ischemia, trauma, excitotoxicity, or degenerative disease) and different death inducing stimuli. We show here that hypoxia causes the formation of a conserved neuronal-death signaling module composed of a lipid raft-mitochondrial-actin cytoskeletal lattice. We extend our signaling module observations and the range of neuroglobin’s protective actions to encompass Aβ- and NMDA-induced neuronal death (35), and suggest a common signaling pathway for neurons to die and a common mechanism for neuroprotection.
The role of raft microdomains in the formation of death-signaling modules may help explain the sensitivity of a variety of cytotoxic processes to cholesterol-depleting drugs. We demonstrate that lowering cholesterol levels impairs formation of polarized microdomain death signaling as part of its neuroprotective mechanism (35). Thus, signaling module formation and neuroglobin itself are relevant drug targets that lend themselves to novel screens for the identification of new drugs and therapeutics that would retard death module formation and protect against neuronal injury and death. Our research provides insight into possible mechanisms of neuroprotection by Ngb, and may have therapeutic implications for stroke and neurodegenerative disease.
Acknowledgments
We thank M. Khan for discussions. We are grateful to W. Schilling and W. G. Sinkins for TRPC3 and TRPC5 antibodies, and A. Ruknudin for NCX1 antibody. This work was supported by NIH grant NS35965 (to D.A.G.).
Abbreviations
- Ngb
neuroglobin
- GDI
guanine dissociation inhibitor
- DISC
death inducing signaling complex
- Ngb-Tg
neuroglobin transgenic mouse
- Pak1
p21 activated kinase 1
References
- 1.Burmester T, Weich B, Reinhardt S, Hankeln T. A vertebrate globin expressed in the brain. Nature. 2000;407:520–523. doi: 10.1038/35035093. [DOI] [PubMed] [Google Scholar]
- 2.Sun Y, Jin K, Mao XO, Zhu Y, Greenberg DA. Neuroglobin is up-regulated by and protects neurons from hypoxic-ischemic injury. Proc Natl Acad Sci U S A. 2001;98:15306–15311. doi: 10.1073/pnas.251466698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dirnagl U, Iadecola C, Moskowitz MA. Pathobiology of ischemic stroke: an integrated view. TINS. 1999;22:391–397. doi: 10.1016/s0166-2236(99)01401-0. [DOI] [PubMed] [Google Scholar]
- 4.Kischkel FC, Hellbardt S, Behrmann I, Germer M, Pawlita M, Krammer PH, Peter ME. Cytotoxicity-dependent APO-1 (Fas/CD95)-associated proteins form a death-inducing signaling complex (DISC) with the receptor. EMBO J. 1995;14:5579–5588. doi: 10.1002/j.1460-2075.1995.tb00245.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Subauste MC, Herrath MV, Benard V, Chamberlain CE, Chuang TH, Chui K, Bokoch GM, Hahn KM. Rho family proteins modulate rapid apoptosis induced by cytotoxic T lymphocytes and Fas. J Biol Chem. 2000;275:9725–97233. doi: 10.1074/jbc.275.13.9725. [DOI] [PubMed] [Google Scholar]
- 6.Friedman JE, Chow EJ, Haddad GG. State of actin filaments is changed by anoxia in cultured rat neocortical neurons. Neurosci. 1998;82:421–427. doi: 10.1016/s0306-4522(97)00217-0. [DOI] [PubMed] [Google Scholar]
- 7.Raman N, Atkinson SJ. Rho controls actin cytoskeletal assembly in renal epithelial cells during ATP depletion and recovery. Am J Physiol. 1999;276:C1312–C1324. doi: 10.1152/ajpcell.1999.276.6.C1312. [DOI] [PubMed] [Google Scholar]
- 8.Bishop AL, Hall A. Rho GTPases and their effector proteins. Biochem J. 2000;348:241–245. [PMC free article] [PubMed] [Google Scholar]
- 9.Van Aelst L, D’Souza-Schorey C. Rho GTPases and signaling networks. Genes Dev. 1997;11:2295–2322. doi: 10.1101/gad.11.18.2295. [DOI] [PubMed] [Google Scholar]
- 10.Wakasugi K, Nakano T, Morishima I. Oxidized human neuroglobin acts as a heterotrimeric Galpha protein guanine nucleotide dissociation inhibitor. J Biol Chem. 2003;278:36505–36512. doi: 10.1074/jbc.M305519200. [DOI] [PubMed] [Google Scholar]
- 11.Schon A, Freire E. Thermodynamics of intersubunit interactions in cholera toxin upon binding to the oligosaccharide portion of its cell surface receptor, ganglioside GM1. Biochemistry. 1989;28:5019–5024. doi: 10.1021/bi00438a017. [DOI] [PubMed] [Google Scholar]
- 12.Green DR, Reed JC. Mitochondria and apoptosis. Science. 1998;281:1309–1312. doi: 10.1126/science.281.5381.1309. [DOI] [PubMed] [Google Scholar]
- 13.Haga N, Fujita N, Tsuruo T. Mitochondrial aggregation precedes cytochrome c release from mitochondria during apoptosis. Oncogene. 2002;22:5579–5585. doi: 10.1038/sj.onc.1206576. [DOI] [PubMed] [Google Scholar]
- 14.Namura S, Zhu J, Fink K, Endres M, Srinivasan A, Tomaselli KJ, Yuan J, Moskowitz MA. Activation and cleavage of caspase-3 in apoptosis induced by experimental cerebral ischemia. J Neurosci. 1998;18:3659–3668. doi: 10.1523/JNEUROSCI.18-10-03659.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liu X, Kim CN, Yang J, Jemmerson R, Wang X. Induction of apoptotic program in cell-free extracts: requirement for dATP and cytochrome c. Cell. 1996;86:147–157. doi: 10.1016/s0092-8674(00)80085-9. [DOI] [PubMed] [Google Scholar]
- 16.Rizzuto R, Pinton P, Carrington W, Fay FS, Fogarty KE, Lifshitz LM, Tuft RA, Pozzan T. Close Contacts with the Endoplasmic Reticulum as Determinants of Mitochondrial Ca2+ Responses. Science. 1998;280:1763–1766. doi: 10.1126/science.280.5370.1763. [DOI] [PubMed] [Google Scholar]
- 17.Yu SP, Yeh CH, Sensi SL, Gwag BJ, Canzoniero LM, Farhangrazi ZS, Ying HS, Tian M, Dugan LL, Choi DW. Mediation of Neuronal Apoptosis by Enhancement of Outward Potassium Current. Science. 1997;278:114–117. doi: 10.1126/science.278.5335.114. [DOI] [PubMed] [Google Scholar]
- 18.Pal S, Hartnett KA, Nerbonne JM, Levitan ES, Aizenman E. Mediation of neuronal apoptosis by Kv2.1-encoded potassium channels. J Neurosci. 2003;23:4798–4802. doi: 10.1523/JNEUROSCI.23-12-04798.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Conforti L, Bodi I, Nisbet JW, Millhorn DE. O2-sensitive K+ channels: role of the Kv1.2 -subunit in mediating the hypoxic response. J Physiol. 2000;524:783–793. doi: 10.1111/j.1469-7793.2000.00783.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Martens JR, Navarro-Polanco R, Coppock EA, Nishiyama A, Parshley L, Grobaski TD, Tamkun MM. Differential targeting of Shaker-like potassium channels to lipid rafts. J Biol Chem. 2000;275:7443–7446. doi: 10.1074/jbc.275.11.7443. [DOI] [PubMed] [Google Scholar]
- 21.Zhu LP, Yu XD, Ling S, Brown RA, Kuo TH. Mitochondrial Ca(2+) homeostasis in the regulation of apoptotic and necrotic cell deaths. Cell Calcium. 2000;28:107–117. doi: 10.1054/ceca.2000.0138. [DOI] [PubMed] [Google Scholar]
- 22.Villalba M, Bi K, Rodriguez F, Tanaka Y, Schoenberger S, Altman A. Vav1/Rac-dependent actin cytoskeleton reorganization is required for lipid raft clustering in T cells. J Cell Biol. 2001;155:331–338. doi: 10.1083/jcb.200107080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coue M, Brenner SL, Spector I, Korn ED. Inhibition of actin polymerization by latrunculin A. FEBS Lett. 1987;213:316–318. doi: 10.1016/0014-5793(87)81513-2. [DOI] [PubMed] [Google Scholar]
- 24.Acuto O, Cantrell D. T cell activation and the cytoskeleton. Ann Rev of Immunol. 2000;18:165–184. doi: 10.1146/annurev.immunol.18.1.165. [DOI] [PubMed] [Google Scholar]
- 25.del Pozo MA, Alderson NB, Kiosses WB, Chiang HH, Anderson RG, Schwartz MA. Integrins regulate Rac targeting by internalization of membrane domains. Science. 2004;303:839–842. doi: 10.1126/science.1092571. [DOI] [PubMed] [Google Scholar]
- 26.Khan AA, Wang Y, Sun Y, Mao XO, Xie L, Miles E, Graboski J, Chen S, Ellerby LM, Jin K, Greenberg DA. Neuroglobin-overexpressing transgenic mice are resistant to cerebral and myocardial ischemia. Proc Natl Acad Sci U S A. 2006;47:17944–17948. doi: 10.1073/pnas.0607497103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.DerMardirossian C, Schnelzer A, Bokoch GM. Phosphorylation of RhoGDI by Pak1 mediates dissociation of Rac GTPase. Mol Cell. 2004;15:7–127. doi: 10.1016/j.molcel.2004.05.019. [DOI] [PubMed] [Google Scholar]
- 28.Hueber AO, Bernard AM, Herincs Z, Couzinet A, He HT. An essential role for membrane rafts in the initiation of Fas/CD95-triggered cell death in mouse thymocytes. EMBO Rep. 2002;3:190–196. doi: 10.1093/embo-reports/kvf022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hueber AO. Role of membrane microdomain rafts in TNFR-mediated signal transduction. Cell Death Differ. 2003;10:7–9. doi: 10.1038/sj.cdd.4401155. [DOI] [PubMed] [Google Scholar]
- 30.Delmas D, Rebe C, Micheau O, Athias A, Gambert P, Grazide S, Laurent G, Latruffe N, Solary E. Redistribution of CD95, DR4 and DR5 in rafts accounts for the synergistic toxicity of resveratrol and death receptor ligands in colon carcinoma cells. Oncogene. 2004;23:8979–8986. doi: 10.1038/sj.onc.1208086. [DOI] [PubMed] [Google Scholar]
- 31.Muppidi JR, Tschopp J, Siegel RM. Life and death decisions: secondary complexes and lipid rafts in TNF receptor family signal transduction. Immunity. 2004;21:461–465. doi: 10.1016/j.immuni.2004.10.001. [DOI] [PubMed] [Google Scholar]
- 32.Furne C, Corset V, Hérincs Z, Cahuzac N, Hueber AO, Mehlen P. The dependence receptor DCC requires lipid raft localization for cell death signaling. Proc Natl Acad Sci U S A. 2006;103:4128–4133. doi: 10.1073/pnas.0507864103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li Z, Hannigan M, Mo Z, Liu B, Lu W, Wu Y, Smrcka AV, Wu G, Li L, Liu M, Huang CK, Wu D. Directional sensing requires G beta gamma-mediated PAK1 and PIX alpha-dependent activation of Cdc42. Cell. 2005;114:215–227. doi: 10.1016/s0092-8674(03)00559-2. [DOI] [PubMed] [Google Scholar]
- 34.Vallone B, Nienhaus K, Matthes A, Brunori M, Nienhaus GU. The structure of carbonmonoxy neuroglobin reveals a heme-sliding mechanism for control of ligand affinity. PNAS. 101:17351–17356. doi: 10.1073/pnas.0407633101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Khan AA, Mao XO, Banwait S, Jin K, Greenberg DA. Neuroglobin Attenuates β-Amyloid Neurotoxicity in Vitro and Transgenic Alzheimer Phenotype in Vivo. PNAS. doi: 10.1073/pnas.0706167104. In Press. [DOI] [PMC free article] [PubMed] [Google Scholar]