

Abstract
The telencephalic premotor nucleus HVC is situated at a critical point in the pattern-generating premotor circuitry of oscine songbirds. A striking feature of HVC's premotor activity is that its projection neurons burst extremely sparsely. Here we present a computational model of HVC embodying several central hypotheses: 1) sparse bursting is generated in bistable groups of recurrently connected robust nucleus of the arcopallium (RA)–projecting (HVCRA) neurons; 2) inhibitory interneurons terminate bursts in the HVCRA groups; and 3) sparse sequences of bursts are generated by the propagation of waves of bursting activity along networks of HVCRA neurons. Our model of sparse bursting places HVC in the context of central pattern generators and cortical networks using inhibition, recurrent excitation, and bistability. Importantly, the unintuitive result that inhibitory interneurons can precisely terminate the bursts of HVCRA groups while showing relatively sustained activity throughout the song is made possible by a specific constraint on their connectivity. We use the model to make novel predictions that can be tested experimentally.
INTRODUCTION
In oscine songbirds, the telencephalic nucleus HVC (used as the proper name; Reiner et al. 2004) is a key structure of the premotor pathway, projecting both to the premotor nucleus RA (the robust nucleus of the arcopallium) and to a basal ganglia nucleus, area X, which forms the first step of a basal ganglia–thalamocortical pathway essential for song learning (the anterior forebrain pathway; Abarbanel et al. 2004a,c; Bottjer et al. 1984; Brainard and Doupe 2000; Perkel 2004; Scharff and Nottebohm 1991; Sohrabji et al. 1990).
HVC contains three broad classes of neuron: RA-projecting (HVCRA), X-projecting (HVCX), and interneurons (HVCI) (Dutar et al. 1998; Fortune and Margoliash 1995; Kubota and Taniguchi 1998; Mooney 2000; Nixdorf et al. 1989; Rauske et al. 2003; Shea 2004). Using paired intracellular recordings and antidromic stimulation in slices, Mooney and Prather (2005) found connections between members of all three of these classes and among HVCRA neurons.
HVCRA neurons, which are the HVC projection neurons that participate directly in the adult song control pathway, have been shown to exhibit temporally sparse bursting during singing in zebra finches (Hahnloser et al. 2002; Kozhevnikov and Fee 2007): in this study, each neuron bursts at most once per song motif. Each burst consists of 4.3 ± 1.3 spikes and has a duration of 5.1 ± 1.8 ms (Kozhevnikov and Fee 2007). Similar sparse bursting also occurs spontaneously during sleep (Hahnloser and Fee 2007; Hahnloser et al. 2002, 2006). By contrast, HVCI neurons spike and burst densely throughout the song (Hahnloser et al. 2002; Kozhevnikov and Fee 2007). We and others have previously used this sparse bursting in models of birdsong (Abarbanel et al. 2004b; Fiete et al. 2004, 2007). The basis of sparse bursting in the neuronal circuitry of HVC remains unknown and is the focus of the model that we describe herein. In the following companion paper (Gibb et al. 2009), we present a model of the potential role of neural feedback to HVC in syllable sequencing.
The present study has the goal of developing a numerical model of HVC sparse bursting in which inhibitory interneurons play a central role. In our HVC model, the sparse bursting associated with any syllable is generated by the propagation of a wave of activity along a network of locally excitatory HVCRA neurons interacting with globally inhibitory HVCI neurons. We represent this network organization of HVCRA neurons as a chain of bistable clusters. We see our model as a set of hypotheses, based on experimental data and expressed in quantitative language, from which we can derive predictions to be tested experimentally.
A portion of this work previously appeared in abstract form (Gibb and Abarbanel 2006).
METHODS
We implemented all models in C++ using a neural simulation framework developed by T. Nowotny and extended by L. Gibb, using a Runge–Kutta 6(5) algorithm with a relative error of 10−6, and we performed analyses of model output in MATLAB. We also tested some of the models in Fortran.
Basic spiking model
All neurons in our model are based on a single-compartment Hodgkin–Huxley-type neuron with just Na+, K+, and leak currents (Destexhe and Sejnowski 2001; Destexhe et al. 1998a; Traub and Miles 1991). The membrane potential of this basic spiking model follows the equation
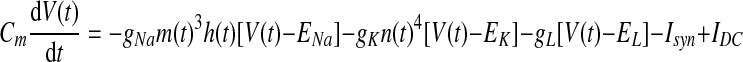 |
where V(t) is the membrane potential; gNa, gK, and gL are the maximal conductances of the Na+, K+, and leak currents; ENa, EK, and EL are the reversal potentials of the Na+, K+, and leak currents; Isyn is the sum of synaptic currents; and IDC is the value of an injected current (IDC = 0, unless otherwise noted). The gating variables X(t) = {m(t), h(t), n(t)} are taken to satisfy the first-order kinetics
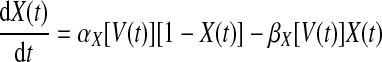 |
where αX(V) and βX(V) are given in Table 1.
TABLE 1.
Rate functions of model neurons
| Basic spiking model (Traub and Miles 1991) | |
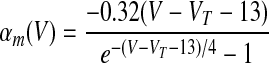
|
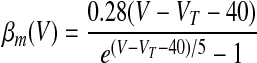 |
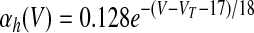
|
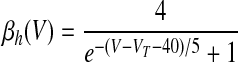 |
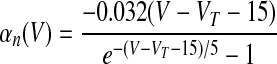
|
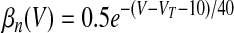 |
| IMs and IMf currents of HVCRA neurons | |
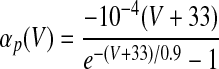
|
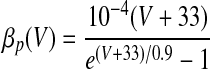 |
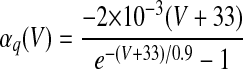
|
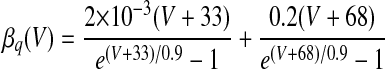 |
| Ih current of HVCI neurons | |
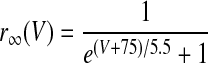
|
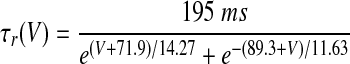 |
V is in millivolts. VT = −53 mV for HVCRA neurons; VT = −63.4 mV for HVCI neurons.
Although the ionic currents of song system neurons have not yet been well characterized, in many cases the responses of neurons to depolarizing and hyperpolarizing current pulses have been recorded in vitro. To match such data from HVCRA and HVCI neurons, we included and modified appropriate currents characterized in mammalian neurons. This level of modeling is appropriate to our long-term goal of spanning cellular, circuit, network, and systems levels of analysis in the song system. As the model develops and new data are obtained, we will replace these neurons with more complex ones where appropriate. We will also explore simplified models to determine which details are essential to the network behavior.
HVCRA neurons
We assumed in the present work that the HVCRA neurons that burst sparsely during singing (Hahnloser et al. 2002; Kozhevnikov and Fee 2007) are of the same short dendrite (SD) class that burst sparsely during bird's own song (BOS) playback (Mooney 2000) and (following Shea 2004) that these are the neurons that spike tonically in response to depolarizing current injection. We fit our model to the physiology described in detail by Kubota and Taniguchi (1998) for their type IIa neurons. There is also a “phasic” HVCRA type, which has been identified with the furry dendrite class (Fortune and Margoliash 1995; Nixdorf et al. 1989; Shea 2004; see discussion). To model Kubota and Taniguchi's HVCRA neurons, we added two voltage-gated K+ currents to the basic spiking model described earlier and modified parameters from those used by Destexhe et al. (1998a) for cortical pyramidal neurons. To better match the in vitro data, we decreased the reversal potential EL of the leak current from −70 to −83 mV and slightly shifted the voltage-dependent rate functions for the Na+ and K+ channels (αm, βm, αh, βh, αn, and βn; see Table 1 and the value of VT) to raise the spike threshold.
The instantaneous spike frequency of these HVCRA neurons declines rapidly over the first few action potentials and then declines much more gradually thereafter during constant-current injection (Kubota and Taniguchi 1998), which suggests that there are two timescales of spike-rate adaptation. Since the currents underlying this behavior have not been characterized in HVCRA neurons, we modified the voltage-gated K+ current IM, described by McCormick et al. (1993) and used by Destexhe et al. (1998a) in their model of cortical pyramidal neurons. The modified current IMs provides the slow component of adaptation and the modified current IMf provides the fast component
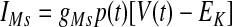 |
and
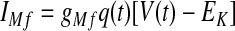 |
where gMs and gMf are the maximal conductances of IMs and IMf. For both IMs and IMf, we shifted the IM rate functions by −3 mV and sharpened them so that small depolarizing current injections trigger very little spike-frequency adaptation, whereas large injections trigger strong adaptation, in accord with the in vitro data. To generate the rate functions of IMf, we multiplied the rate functions of IM by a constant. To prevent a postburst hyperpolarization that is not observed in the data, we modified βq so that IMf rapidly deactivates below about −68 mV (see Table 1).
We adjusted the parameters to match the resting potential and the instantaneous spike frequency as a function of time for different injected currents described by Kubota and Taniguchi (1998; see Supplemental Fig. S1).1 The parameter values for the HVCRA neuron model were gNa = 50 mS/cm2, ENa = 45 mV, gK = 5 mS/cm2, EK = −88 mV, gL = 0.1 mS/cm2, EL = −83 mV, Cm = 1 μF/cm2, VT = −53 mV, gMs = 0.3 mS/cm2, and gMf = 0.8 mS/cm2.
HVCI neurons
To model HVCI neurons, we again built on the basic spiking model described earlier. We modified parameters from those used by Destexhe et al. (1998a) to model cortical interneurons, adjusting the parameters to match the resting potential and the instantaneous spike frequency as a function of time for different injected currents described by Kubota and Taniguchi (1998; see Supplemental Fig. S1). The data reported by Kubota and Taniguchi (1998) and Dutar et al. (1998) demonstrate that HVCI neurons have a depolarizing sag in response to a hyperpolarizing current injection and show little spike-rate adaptation. We represent this in the membrane voltage equation by including Ih but no special K+ currents
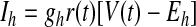 |
where gh and Eh are the maximal conductance and reversal potential of Ih, and
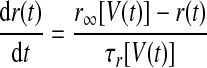 |
where r∞(V) and τr(V) are given in Table 1, based on Huguenard and McCormick (1994). This τr(V) is 20 times smaller than that in the model on which it is based. We made this modification solely to improve the match of the model to the data of Kubota and Taniguchi (1998) and Dutar et al. (1998) by speeding up the sag. This modification can be justified by the broad range of Ih time constants reported in different cortical and subcortical neurons, which depend on the subunit composition of Ih channels (Aponte et al. 2006). The parameter values of the HVCI neuron model were gNa = 50 mS/cm2, ENa = 45 mV, gK = 10 mS/cm2, EK = −85 mV, gL = 0.15 mS/cm2, EL = −64 mV, Cm = 1 μF/cm2, VT = −63.4 mV, gh = 0.07 mS/cm2, and Eh = −40 mV.
Once fit to the in vitro data, the parameters of the neuron models were held fixed; only synaptic strengths and network connectivity were adjusted to achieve the desired network behavior.
Modeling synaptic currents
We modeled both excitatory and inhibitory synaptic currents with the following equations (Destexhe and Sejnowski 2001; Destexhe et al. 1994)
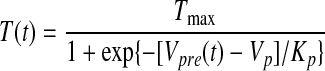 |
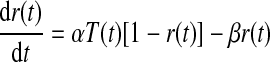 |
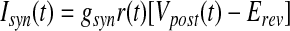 |
where T(t) is the concentration of neurotransmitter in the synaptic cleft, Tmax is the maximal neurotransmitter concentration, Vpre(t) and Vpost(t) are the membrane potentials of the presynaptic and postsynaptic neurons, r(t) is the fraction of the receptors in the open state, gsyn (gAMPA or gGABAA) is the maximal synaptic conductance, and Erev (EAMPA or EGABAA) is the synaptic reversal potential.
The rate constants that we used are based on the time constants of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA)–and γ-aminobutyric acid type A (GABAA)–mediated currents recorded in mammalian neocortical neurons and interneurons in vitro (Destexhe and Sejnowski 2001; Destexhe et al. 1994; Hestrin 1993). AMPA receptors at excitatory synapses onto interneurons are about twice as fast as those onto pyramidal neurons (Destexhe and Sejnowski 2001; Hestrin 1993).
For all synapses in our model, Tmax = 1.5 mM, Vp = 2 mV, and Kp = 5 mV. For excitatory synapses, Erev = 0 mV. For excitatory synapses onto HVCI neurons, α = 2.2 mM−1 ms−1 and β = 0.38 ms−1 (decay time constant 1/β = 2.6 ms). For excitatory synapses onto all other neuron types, α = 1.1 mM−1 ms−1 and β = 0.19 ms−1 (1/β = 5.3 ms). For inhibitory synapses, α = 5.0 mM−1 ms−1 and β = 0.18 ms−1 (1/β = 5.6 ms). In the reduced cluster model and the chain models based on it, Erev = −83 mV for inhibitory synapses. In the models with synapses of physiological strength (see the following text), Erev = −88 mV to give inhibitory postsynaptic potentials (IPSPs) a physiologically correct, nonzero amplitude near the HVCRA resting potential of −83 mV. Mooney and Prather (2005) did not measure Erev experimentally; our assumed value of −88 mV is similar to, and slightly less negative than, the mean GABAA reversal potential measured in neurons of the medial portion of the dorsolateral thalamic nucleus (Person and Perkel 2005). This assumed value is necessarily an informed guess, since Mooney and Prather did not report the resting potentials of the neurons in which they measured IPSP amplitudes. Moreover, in two of the HVCI → HVCRA connections that they reported, the PSP was depolarizing rather than hyperpolarizing. However, we do not expect the precise value of Erev to be critical to the behavior of the model, as long as it is below the spiking threshold.
Modeling temperature dependence of neurons and synapses
In simulations of in vivo activity, as a first approximation of the temperature dependence of neurons and synapses, we scaled all rate functions by a factor of φ(T1) = Q10(T2−T1)/10, assuming a Q10 of 3 for both neuronal and synaptic rates (Collingridge et al. 1984; Hodgkin and Huxley 1952). Here, T2 is the brain temperature in vivo (assumed to be 40°C) and T1 is the approximate temperature at which the measurements were made in vitro. T1 is 32°C for neurons (Kubota and Taniguchi 1998), 31°C for AMPA synapses (Destexhe and Sejnowski 2001; Destexhe et al. 1998b; Xiang et al. 1992), and 34°C for GABAA synapses (Destexhe et al. 1998b, 2001; Otis and Mody 1992a,b).
Synapses of physiological strength
In some simulations, we used synapses of physiological strength, based on the measurements of Mooney and Prather (2005). The values of α and β for these synapses followed the rules described earlier. We list the other parameter values and postsynaptic potential (PSP) peak amplitudes at 40°C here, together with the peak amplitudes measured by Mooney and Prather (abbreviated MP). HVCI → HVCRA: gGABAA = 0.11 mS/cm2, EGABAA = −88 mV, IPSP amplitude −0.9 mV (MP: −0.9 ± 0.2 mV). HVCRA → HVCI: gAMPA = 0.034 mS/cm2, EAMPA = 0 mV, EPSP amplitude 2.0 mV (MP: 2.0 ± 0.4 mV). HVCRA → HVCRA: gAMPA = 0.018 mS/cm2, EAMPA = 0 mV. EPSP amplitude 2.2 mV (MP: 2.2 ± 1.1 mV).
Poisson synapses
In some simulations, we included Poisson synapses to generate spiking of a particular frequency or to mimic background synaptic activity in vivo in a manner similar to that described by Destexhe et al. (1998a). Each Poisson synapse was an AMPA or GABAA synapse receiving 1-ms, 1-mM neurotransmitter pulses as Poisson random events. The values of α and β for these synapses again followed the rules described earlier. To mimic background synaptic activity, each HVCI neuron received 20 Poisson AMPA synapses with a mean rate of 10 Hz and a maximal conductance of 0.02 mS/cm2. This resulted in 8.8-Hz spiking (measured over 100 s), which is consistent with the 8 ± 6 Hz spontaneous firing rate measured by Kozhevnikov and Fee (2007). Similarly, each HVCRA neuron received 20 Poisson AMPA (10 Hz, gAMPA = 0.02 mS/cm2) and 20 Poisson GABAA synapses (10 Hz, gGABAA = 0.1 mS/cm2). This resulted in SD of the membrane potential of 2.8 mV (measured over 100 s). This pattern of Poisson synapses—a combination of AMPA and GABAA for HVCRA neurons and just AMPA for HVCI neurons—mirrors the connectivity of our model: HVCRA → HVCRA, HVCI → HVCRA, HVCRA → HVCI, but no HVCI → HVCI synapses. We acknowledge that there may also be a significant number of HVCI → HVCI connections, but Mooney and Prather (2005) observed only one example. With appropriate parameter adjustments, we would expect to see similar results if Poisson GABAA synapses were included on HVCI neurons.
Sparseness index and spike time
We defined the sparseness of a spike train as S = 1 − D, where D is the fraction of 10-ms bins containing one or more spikes. We defined spike time as the time of the peak depolarization following a crossing of a −15-mV threshold in a positive direction.
Throughout, we report values as means ± SD.
RESULTS
Initial model: dynamic behavior of a single bistable cluster
Our current model has evolved from the simple idea that clusters of HVCRA neurons could act as bistable units in generating the sparse bursting in HVC. The connectivity of our model clusters (Fig. 1C) is based on the HVC microcircuit described by Mooney and Prather (2005), especially their observations of depolarizing HVCRA → HVCRA and HVCRA → HVCI and hyperpolarizing HVCI → HVCRA connections. The recurrent (feedback) excitation of the HVCRA neurons is the basis for the bistability: the HVCRA cluster has two stable states: quiescent and persistently spiking. These states are a fixed point and a stable limit cycle, respectively.
FIG. 1.

Large clusters of HVC neurons with synapses of physiological strength. A: duration of spiking in response to a current pulse injected into 50% of the neurons in clusters of recurrently connected robust nucleus of the arcopallium (RA)–projecting (HVCRA) neurons with synaptic noise (green), no synaptic noise (blue), and no adaptation currents or noise (red). Spiking is truncated at 1 s by the end of the simulations. Each data point is the mean spiking duration of all the neurons in 3 separate simulations. We show error bars only for points at which SD >20 ms. B: bistability of a 160-HVCRA cluster without noise. Membrane potential of a representative HVCRA neuron. Green arrow: time of excitatory current pulse shifting the cluster into its persistently spiking state. Red arrow: time of inhibitory current pulse shifting the cluster back into its quiescent state. C: comparison of 2 distinct modes of inhibition for burst generation in 160-HVCRA, 80-HVCI (HVC interneuron) clusters. Top: schematics of the 2 mechanisms. Arrowheads, excitatory connections; dots, inhibitory connections. Middle: representative HVCRA voltage traces. Bottom: spike time raster plot of 50% of the HVCI neurons.
Despite the existence of synaptic connections from HVCX to HVCRA neurons (Mooney and Prather 2005), we did not give HVCX neurons a role in HVCRA sparse burst generation in our model. The observation that adult song production is not affected by targeted ablation of HVCX neurons, whereas it is often disrupted by ablation of HVCRA neurons (Scharff et al. 2000), suggests that HVCX neurons are a less critical part of the premotor pattern-generating circuit (or possibly that there is much greater redundancy in that part of the circuit with respect to premotor pattern generation).
We created clusters in which each HVCRA neuron sends excitatory synapses of physiological strength (see methods) to a randomly selected set of 50% of the other HVCRA neurons within the same cluster. We chose this degree of connectivity to provide a high level of mutual excitation without implying an excessively large number of reciprocally connected HVCRA neurons within each cluster, since reciprocally connected HVCRA neurons were not observed by Mooney and Prather (2005). We prohibited each HVCRA neuron from making a synapse onto itself or more than one synapse onto the same postsynaptic neuron.
In Fig. 1A, we show the duration of spiking activity evoked by a 3-ms, 40-μA/cm2 DC current pulse injected into 50% of the HVCRA neurons in clusters without inhibitory HVCI neurons, plotted as a function of HVCRA cluster size. The mean spiking duration increases as a function of the number of HVCRA neurons. For intermediate cluster sizes, with the normal adaptation currents (IMs and IMf) present (blue and green traces), the repetitively spiking state of the cluster is only transiently stable. The spiking duration reaches ≥1 s at a cluster size of 160 HVCRA neurons in the absence of synaptic noise (Fig. 1A, blue trace), 190 HVCRA neurons in the presence of synaptic noise (see methods; Fig. 1A, green trace), and 60 HVCRA neurons in the absence of the adaptation currents and noise (Fig. 1A, red trace). These sizes are comparable with the number of HVCRA neurons, about 200, that Fee et al. (2004) estimated are coactive at each time in the song, but we will return to this point in the discussion. Additionally, the spiking duration also increases as a function of the percentage connectivity of the cluster (not shown).
In Fig. 1B, we demonstrate the bistability of a 160-HVCRA cluster without inhibitory HVCI neurons or synaptic noise. At t = 10 ms (green arrow), we shift the cluster into its persistently spiking state by exciting 50% of the HVCRA neurons with a 3-ms, 40-μA/cm2 current pulse. This state persists for ≥1 s in the absence of further inputs (not shown). In Fig. 1B, we shift the cluster back into its quiescent state with a 3-ms, −40-μA/cm2 current pulse into the same 50% of the HVCRA neurons at t = 110 ms (red arrow).
Mechanisms of inhibition
As described in the introduction, HVCI neurons spike and burst densely throughout the song. Given this sustained activity, how could HVCI neurons contribute to sculpting HVCRA activity into sparse bursts? Two hypothetical mechanisms stand out as extremes along a spectrum. At one end of the spectrum (an “inhibitory buildup” mechanism), relatively weak feedback inhibition of HVCRA neurons by HVCI neurons could exert a hyperpolarizing influence throughout the burst, leading to a reduced spiking frequency followed by a failure of spiking and a termination of the burst after several milliseconds (Fig. 1C, left). In this mechanism, relatively small changes in the strength and timescale of the inhibition strongly influence the duration of the burst.
At the other end of the spectrum, the HVCRA burst could occur during a pause in inhibition from HVCI neurons. In this “inhibitory pause” mechanism, HVCRA neurons are disinhibited during the burst and a strong onset of inhibition after several milliseconds abruptly terminates the burst (Fig. 1C, right). Pauses and periods of low-frequency spiking do in fact occur in the midst of the relatively sustained activity of HVCI neurons during singing (Hahnloser et al. 2002; Kozhevnikov and Fee 2007). This mechanism is the basis of our model using globally connected HVCI neurons, described in the following text (Fig. 4). In this mechanism, small changes in the strength and timescale of the inhibition are inconsequential, but the delay between the onset of the burst and the onset of the inhibition controls the duration of the burst. Between the extremes of inhibitory buildup and inhibitory pause is a range of intermediate mechanisms in which the onset of inhibition is delayed but relatively weak, so that a combination of inhibitory strength and inhibitory pause duration controls the duration of the burst.
A second dichotomy between inhibitory mechanisms is that between “local” inhibition, in which each HVCI neuron connects to a set of HVCRA neurons in only one part of the HVCRA network, and “global” inhibition, in which each HVCI neuron connects to a set of HVCRA neurons throughout the HVCRA network.
In the present study, we argue that if inhibition does indeed play a role in HVCRA burst termination, then global inhibition using the inhibitory pause mechanism is a favored candidate (although the true mechanism may be somewhere between the extremes of inhibitory pause and inhibitory buildup). We make this argument along several lines in the following sections.
Our main goals here are to demonstrate that HVCI neurons could participate in the HVCRA burst mechanism, to show a possible mechanism by which they could accomplish this, and to make experimental predictions on the basis of this mechanism.
Computational implementation of inhibitory mechanisms
In this section, we suggest that the inhibitory pause mechanism is a favored candidate for sparse bursting in HVCRA neurons because it is more robust than the inhibitory buildup mechanism to a variety of parameter changes.
To implement these two basic inhibitory mechanisms, we added HVCI neurons to the 160-HVCRA cluster introduced previously, in a proportion based on the literature. Although estimates of the proportions of HVC neuron classes vary substantially (e.g., Alvarez-Buylla et al. 1988; Kirn et al. 1999), one estimate is that 50% of HVC neurons are RA-projecting, 25% are X-projecting, and 25% are interneurons (Nottebohm et al. 1990). Consistent with these proportions, we created a 160-HVCRA, 80-HVCI cluster. To verify the robustness of both of these inhibitory models, we included synaptic noise in the HVCRA and HVCI neurons as described in methods.
In our implementation of the inhibitory buildup mechanism (Fig. 1C, left), each HVCRA neuron in the bistable cluster sends a single synapse to each of nine HVCI neurons and each HVCI neuron sends a single synapse to each of four HVCRA neurons. We initiated the burst with a 3-ms, 40-mS/cm2 DC current pulse into 50% of the HVCRA neurons beginning at t = 20 ms (the simulation began at t = −30 ms). For 10 trials with identical parameters but different random connectivity, the number of spikes per burst was 3.6 ± 1.1 and the burst duration was 6.7 ± 2.3 ms.
In our implementation of the inhibitory pause mechanism (Fig. 1C, right), each HVCI neuron sends a single synapse to each of 70 HVCRA neurons but does not receive synapses from any HVCRA neurons. We excite the bistable HVCRA cluster by a brief current pulse. It is then inhibited by HVCI neurons that we begin to excite 6.4 ms later; this inhibition terminates the burst. To relate this model to our model using globally connected HVCI neurons (see the following text; Fig. 4), we also excite the HVCI neurons until 7 ms before initiating the HVCRA burst; thus the HVCRA burst occurs largely during a pause in the high-frequency presynaptic HVCI spiking. This excitation is a proxy for the input from other HVCRA neurons in the network, which are included in our model using globally connected HVCI neurons. We set the pause to begin 7 ms before burst initiation to give the inhibition sufficient time to decay so that it does not interfere with the burst. As in the inhibitory buildup mechanism, we initiated the burst with a 3-ms, 40-mS/cm2 current pulse into 50% of the HVCRA neurons beginning at t = 20 ms (again, the simulation began at t = −30 ms). We elicited the relatively high-frequency spiking of each HVCI neuron before t = 13 ms and after t = 26.4 ms with a 100-Hz Poisson AMPA synapse with gAMPA = 0.2 mS/cm2 (see methods), together with the synaptic noise. This 89-Hz HVCI spiking frequency (measured over 100 s) is consistent with the 95 ± 40 Hz firing rate of HVCI neurons recorded during singing (Kozhevnikov and Fee 2007). For 10 trials with identical parameters but different random connectivity, the number of spikes per burst was 4.0 ± 0.8 and the burst duration was 5.4 ± 1.4 ms. The HVCI → HVCRA connections are sufficiently strong that in the absence of the pause in the high-frequency HVCI spiking, none of the HVC neurons bursts (10 trials).
Our implementation of the inhibitory buildup mechanism was more sensitive than the inhibitory pause mechanism to a variety of parameter changes. In the inhibitory buildup mechanism, longer bursts tended to be associated with weaker or more rapidly decaying inhibition (Fig. 2, A, B, and D), consistent with a termination of bursting by the hyperpolarizing influence of inhibition during the burst. The burst duration decreased with the inhibitory synaptic decay time constant, 1/β (Fig. 2A; larger values imply longer inhibitory postsynaptic current decay times). Similarly, the burst duration decreased as the number of inhibitory synapses sent to HVCRA neurons by each HVCI neuron increased, either with the normal adaptation currents present (IMs and IMf; Fig. 2B) or with these currents absent (Fig. 2D). In the absence of the adaptation currents, there was an abrupt transition from long to short bursts (Fig. 2D), suggesting an important contribution of these currents to burst termination. Consistent with this observation, the burst duration decreased with increasing strength of the adaptation currents (Fig. 2C). Additionally, the burst duration increased with the number of excitatory HVCRA–HVCRA synapses per HVCRA neuron (Fig. 2E), consistent with a competition between inhibition and excitation in determining the burst duration.
FIG. 2.

Burst duration for the inhibitory buildup mechanism of Fig. 1C as a function of various parameters. Dark gray lines: mean ± SD. Light gray regions: HVCRA burst duration (mean ± SD) measured by Kozhevnikov and Fee (2007). A: inhibitory synaptic decay time constant, 1/β (normal value: 5.6 ms). B: inhibitory synapses per HVCI, normal adaptation currents present (normal number: 9). C: scale factor by which the maximal conductances of both adaptation currents is multiplied (normal value: 1). D: inhibitory synapses per HVCI, adaptation currents absent. E: excitatory synapses per HVCRA (normal number: 80).
For our implementation of the inhibitory buildup mechanism, the mean burst durations fell within 1 SD of the mean recorded in vivo (Fig. 2, light gray regions; Kozhevnikov and Fee 2007) only in a narrow range of values (one to four data points). By contrast, in our implementation of the inhibitory pause mechanism, the mean burst durations fell within this range over a much wider range of values (more than eight data points in every case, for the same spacing; Supplemental Fig. S2). This suggests that the inhibitory pause mechanism is the more robust of the two mechanisms.
Reduced cluster models
Network models of HVC with correct numbers of neurons and synapses are very slow and inefficient to simulate. Therefore we created a reduced cluster model that captures the most essential characteristics of the 160-HVCRA cluster introduced previously, especially its recurrent excitation and bistability. This reduced model contains three HVCRA neurons recurrently connected in a ring by excitatory synapses (Fig. 3A).
FIG. 3.

Chains of clusters with locally connected HVCI neurons produce sparse bursting in HVCRA neurons, but do not produce sustained activity in HVCI neurons beyond their low-frequency spontaneous spiking. A: schematic of the reduced cluster model used in the sparse bursting chain network, consisting of 3 recurrently excitatory HVCRA neurons (light blue) and one local HVCI neuron (dark blue). B: burst duration for the reduced cluster as a function of the inhibitory conductance gGABAA; GABAA, γ-aminobutyric acid type A. Because simulations end at t = 110 ms, the maximum possible duration is 100 ms. For every point, SD <2.1 ms. C: schematic of the sparse bursting chain network. Activity of the network is initiated in the first HVCRA neuron of the first cluster by a DC current pulse. D: raster plot of the spike times of a subset of HVCRA and HVCI neurons in a chain of 250 clusters. Each row represents the spike times of one neuron.
We verified that without the HVCI neuron, the reduced HVCRA cluster is bistable (Supplemental Fig. S3): by exciting a single neuron of the cluster, we shifted the cluster from its quiescent state into its persistently spiking state. By injecting a hyperpolarizing current into the same neuron, we shifted the cluster back into its quiescent state. The resulting plot (Supplemental Fig. S3A) looks almost identical to the corresponding plot for the 160-HVCRA cluster (Fig. 1B). We verified that the persistently spiking state persists for ≥1 s in the absence of further inputs.
Like the 160-HVCRA, 80-HVCI cluster, the 3-HVCRA, 1-HVCI cluster exhibited a burst of appropriate duration for a narrow range of inhibitory coupling. In Fig. 3B, we plot the mean spiking duration of the HVCRA neurons in this cluster as a function of gGABAA. At ≤0.6 mS/cm2, the HVCRA neurons showed persistent activity, whereas at ≥0.9 mS/cm2, the HVCRA neurons spiked only zero to two times. In addition, we found that both two- and four-neuron clusters are also capable of generating brief bursts (not shown). The former may have a smaller parameter regime in which bistability is possible.
A model of HVCRA sparse bursting: the limitations of local inhibition
In this section, we suggest that a chain network using local inhibition and an inhibitory buildup mechanism is not a good model of sparse bursting in HVCRA neurons because it does not reproduce the sustained HVCI activity observed experimentally during singing and is very sensitive to parameter changes.
To create such a chain network, we arranged our reduced clusters in a long chain so that they successively excite each other to generate a burst sequence; we illustrate this in Fig. 3, C and D. To initiate the wave of bursting activity, we set IDC = 30 μA/cm2 in the first neuron of the first chain for 5 ms beginning at t = 0 ms. This “begin song” command may correspond to synaptic input from an afferent nucleus or the end of another chain within HVC. The activity then propagates from cluster to cluster until it reaches the end of the chain. Activity in each cluster is evoked by excitatory input to its HVCRA neuron 1 (Fig. 3A) from the HVCRA neuron 2 of the previous cluster, and terminated by inhibitory input from the local HVCI.
Figure 3D is a raster plot of the spike times of a subset of HVCI neurons from a total of 250 reduced clusters (three HVCRA neurons each) linked in series. For HVCRA → HVCRA synapses within each cluster, gAMPA = 1.95 mS/cm2. For those between clusters, gAMPA = 1.5 mS/cm2. We made the latter weaker to prevent driving the first neuron of each cluster too strongly because it receives a synapse not only from within the cluster but also from the previous cluster (this is an artifact of our reduced model and is not meant to reflect differences in connectivity within HVC). For HVCRA → HVCI synapses, gAMPA = 0.2 mS/cm2. For HVCI → HVCRA synapses, gGABAA = 1.45 mS/cm2. The burst duration was 5.6 ± 1.5 ms, with 3.3 ± 0.9 spikes per burst. We injected a 0.5-μA/cm2 DC current into the HVCI neurons so that they spiked spontaneously at about 8 Hz, consistent with their spontaneous firing rate in awake, nonsinging zebra finches. The HVCI neurons had identical initial conditions and DC current, which accounts for the identical timing of the first spike of most of these neurons.
Significantly, in this model, each HVCI bursts only once as activity propagates along the chain. By contrast, the HVCI neurons described by Hahnloser et al. (2002) and Kozhevnikov and Fee (2007) show an elevated level of spiking and bursting throughout the song. To correct this discrepancy, we developed the model of global inhibition described in the next section. Additionally, this model required fine parameter tuning and was sensitive to changes in the HVCI → HVCRA inhibitory synaptic strength. For example, a decrease from 1.45 to 1.40 mS/cm2 destabilized the burst sequence, resulting in a mean burst duration of 27.0 ± 20.9 ms, with 8.9 ± 6.1 spikes per burst.
A model of HVCRA sparse bursting using global inhibition
In this section, we suggest that a chain network using global inhibition and an inhibitory pause mechanism (i.e., global inhibition with a specific constraint on HVCI–HVCRA connectivity) is a favored candidate for sparse bursting in HVCRA neurons because it generates appropriate HVCRA bursts and reproduces the sustained HVCI activity observed experimentally.
Our current model of sparse bursting is depicted in Fig. 4. As in the previous model, HVCRA neurons are organized into a chain of bistable clusters. However, HVCI neurons are no longer functionally localized to these clusters; rather, they are permitted to receive excitation from, and send inhibition to, any part of the chain. We initiate the burst sequence by setting IDC = 20 μA/cm2 in the first neuron of the first chain for 5 ms beginning at 0 ms. Again, excitation travels through the network via HVCRA → HVCRA synapses. The local excitatory synapses between HVCRA neurons keep each cluster of neurons in an excited state until a subpopulation of global HVCI neurons terminates its activity.
FIG. 4.

Chains of clusters with globally connected HVCI neurons and constrained HVCI–HVCRA connectivity produce sparse bursting in HVCRA neurons and sustained activity in HVCI neurons, consistent with experimental observations. A: schematic illustrating the constraint on HVCI–HVCRA connectivity that permits normal burst propagation. Because the representative HVCI receives an excitatory synapse from the cluster shown, it is not permitted to make inhibitory synapses onto the clusters shown in white. The diagram assumes that the wave of activity propagates from left to right along the HVCRA network. B: raster plot of the spike times of a subset of HVCRA and HVCI neurons from a simulation of 300 HVCI neurons and 200 clusters of 3 HVCRA neurons. C: portion of voltage trace of HVCRA neuron 14 from the same simulation. D: subset of HVCRA neurons in another simulation, in which one of these neurons (magenta) was in a cluster that escaped inhibition to fire a longer burst.
However, without any constraints on HVCI–HVCRA connectivity, it is not possible for the inhibition to sculpt the HVCRA activity into a sequence of appropriate bursts (not shown). With weak inhibition (e.g., gGABAA = 0 or gGABAA = 0.01 mS/cm2), HVCRA bursts failed to terminate, entering a state of persistent activity. With intermediate inhibition (e.g., gGABAA = 0.02 to 0.04 mS/cm2), HVCRA neurons showed some combination of variable-duration bursting, persistent activity, and premature termination of the burst sequence. With strong inhibition (gGABAA = 0.05 to 3.0 mS/cm2), the burst sequence terminated within two clusters of the beginning of the chain: a burst in one cluster caused inhibition in a downstream cluster before that cluster was excited. There was no value of gGABAA for which HVCRA neurons showed bursts with a consistent, correct number of spikes.
A solution to this problem is to constrain each HVCI neuron not to make inhibitory synapses onto any cluster from which it receives an excitatory synapse or onto clusters within some number of clusters downstream of one from which it receives an excitatory synapse (Fig. 4A). We refer to these downstream clusters as an HVCI “downstream connectivity gap.” A downstream connectivity gap of at least one cluster is necessary to allow enough time for the inhibition on HVCRA neurons to decay before excitation arrives from an upstream cluster. When the downstream connectivity gap is zero, only the first cluster spikes. To reduce the influence of residual inhibition on burst duration and propagation speed (see Fig. 5), we used a larger, seven-cluster downstream connectivity gap. The HVCI neuron is constrained not to make excitatory synapses onto any cluster from which it receives an excitatory synapse, so that the HVCRA neurons have time to burst before the inhibition arrives to terminate the bursts. We generated this synaptic connectivity pattern by the following algorithm.
Create a new HVCI neuron.
Select a candidate HVCRA neuron to make an excitatory synapse onto the HVCI neuron. If there exists an inhibitory synapse from the HVCI neuron onto an HVCRA neuron in the candidate neuron's cluster or onto an HVCRA neuron that is within seven clusters downstream of the candidate HVCRA neuron, select a new candidate HVCRA neuron. Repeat this until an appropriate HVCRA neuron is found.
Select a candidate HVCRA neuron to receive an inhibitory synapse from the HVCI neuron. If there exists a synapse onto the HVCI neuron from an HVCRA neuron in the candidate neuron's cluster or from an HVCRA neuron that is within seven clusters downstream of the candidate HVCRA neuron, select a new candidate HVCRA neuron. Repeat this until an appropriate HVCRA neuron is found.
Repeat steps 2 and 3 until all of the synapses onto and from the HVCI have been created.
Repeat steps 1–4 until all of the HVCI neurons have been created.
FIG. 5.

Effects of HVCI–HVCRA connectivity on burst duration and propagation speed. Burst duration increases (A) and burst propagation speed decreases (B) as a function of the HVCI upstream connectivity gap. Burst duration decreases (C) and burst propagation speed increases (D) as a function of the HVCI downstream connectivity gap. Burst propagation terminated prematurely (293 ms) for the simulation with a downstream connectivity gap of 4.
As a result of this constraint on connectivity, the wave of excitation is preceded and followed by inhibition, but not interrupted by it, despite the sustained activity of the HVCI population. The HVCI neurons presynaptic to a given HVCRA neuron are all silent at about the time of the HVCRA burst and shortly before; this is functionally equivalent to the pause in HVCI activity in our implementation of the inhibitory pause mechanism in Fig. 1C.
In addition, we found that neurons in the last cluster of the chain remained persistently active, since there are no clusters downstream of these clusters to inhibit them via the HVCI neurons. To prevent this persistent activity, we added additional connections from the last two HVCRA clusters to the HVCI population.
Figure 4B shows the spike times of a subset of HVCRA and HVCI neurons in a simulation containing a total of 300 HVCI neurons and 200 clusters of 3 HVCRA neurons (i.e., 33% HVCI and 67% HVCRA neurons) with this connectivity pattern. We obtained similar results when the numbers of HVCRA and HVCI neurons were the same. We also obtained similar results with 2 or 4 neurons per cluster. HVCRA2 of each cluster sends an excitatory synapse to HVCRA1 of the next. Each HVCI neuron sends 100 synapses to, and receives 100 synapses from, HVCRA neurons. For HVCRA → HVCRA synapses within each cluster, gAMPA = 1.0 mS/cm2. For those between clusters, gAMPA = 0.5 mS/cm2. We made the latter weaker for the reason described in the previous section. For HVCRA → HVCI synapses, gAMPA = 0.1 mS/cm2. For HVCI → HVCRA synapses, gGABAA = 3.0 mS/cm2.
To prevent persistent activity at the end of the chain, randomly selected HVCRA neurons from the last two clusters make a total of 300 additional synapses (gAMPA = 0.1 mS/cm2) onto randomly selected HVCI neurons. The average burst duration was 4.3 ± 1.4 ms, with 3.2 ± 0.6 spikes per burst. Figure 4C shows a portion of the voltage trace of one of the HVCRA neurons in this simulation.
Because of the element of randomness in generating HVCI → HVCRA connectivity in the model, in some simulations the spiking in one or more HVCRA clusters was not as rapidly terminated by inhibition. In Fig. 4D, we show an example neuron from such a cluster in a simulation containing 90 HVCI neurons and 60 clusters of 2 HVCRA neurons, with 30 synapses to and from each HVCI neuron. The single HVCRA neuron that spikes over an unusually long duration in the experiments of Hahnloser et al. (2002; their Fig. 2B) and Kozhevnikov and Fee (2007; their Fig. 2A) has a lower spiking frequency than this. However, it is sufficiently reminiscent that we suggest the possibility that both cases involve a failure of inhibition.
In this model, the duration of an HVCRA burst is strongly influenced by the timing of inhibition. Most straightforwardly, a larger HVCI upstream connectivity gap (i.e., the number of clusters that an HVCI is not permitted to inhibit upstream of one from which it receives excitation; Fig. 4A) leads to a longer delay before a burst is terminated and therefore a longer burst (Fig. 5A). Longer burst durations also result from smaller downstream connectivity gaps (Fig. 5C). This may be related to the fact that smaller downstream connectivity gaps cause slower burst propagation (Fig. 5D): if the downstream cluster bursts later than usual, then its inhibition of the upstream cluster via HVCI neurons should occur later than usual. The slower burst propagation may be due to residual inhibition delaying the downstream cluster in reaching its spike threshold. Larger upstream connectivity gaps also result in slower burst propagation (Fig. 5B). This may be due to the redistribution of the HVCI → HVCRA synapses: when there are fewer HVCI → upstream HVCRA synapses, there are more HVCI → HVCRA synapses to clusters just downstream of the downstream connectivity gap. This may result in more residual inhibition, which may delay the downstream clusters in reaching their spike threshold.
Sparse bursting without clusters or unidirectional HVCRA chains
The model does not fundamentally rely on a cluster organization nor on the unidirectionality of the HVCRA chain to produce a sequence of sparse bursts. For example, in one set of simulations (not shown), we reduced the clusters in our model to 2 reciprocally connected HVCRA neurons and connected these clusters bidirectionally (gAMPA = 0.75 mS/cm2 for all synapses), effectively creating a chain of 400 bidirectionally connected HVCRA neurons while retaining the HVCRA–HVCI connectivity. Stimulating the first cluster continued to produce a sequence of bursts (burst duration, 3.9 ± 1.3 ms; spikes per burst, 3.0 ± 0.7). Unidirectionality of burst propagation was maintained by the HVCRA–HVCI connectivity: stimulating the last neuron of the chain produced a burst in only the last 2 neurons (not shown). This is expected, since there is no gap in the reverse direction to allow the inhibition of the HVCRA neurons to decay before excitation arrives from an upstream cluster. Thus we expect this unidirectionality of burst propagation to depend on the asymmetry of the upstream and downstream connectivity gaps (Fig. 4A).
Similarly, we created a short chain model of 20 clusters with physiological synaptic strengths, in which the separate identity of the clusters was all but lost by their interconnectivity. Each cluster contained 80 HVCRA neurons, each of which sent AMPA synapses to a set of 40 other HVCRA neurons in its cluster, 40 in the previous cluster, and 40 in the next cluster. Thus the connectivity along the chain had no directionality and the connectivity was less one of a chain of clusters and more one of a network of HVCRA neurons with local random connections. Each of the 1,600 HVCI neurons sent GABAA synapses to a set of 80 HVCRA neurons and received AMPA synapses from a set of 20 HVCRA neurons. The upstream connectivity gap was one cluster and the downstream connectivity gap was 10 clusters. The connectivity of the clusters in this version of the model allowed us to control the burst duration more finely: setting the upstream connectivity gap to 1 rather than 0 caused the HVC neurons to spike 4.3 ± 0.9 times in 5.3 ± 1.3 ms, longer than the bursts in our previous model. To prevent persistent activity at the end of the chain, each HVCRA neuron in the last cluster sent AMPA synapses to a set of 30 HVCI neurons. In every case, we prohibited a neuron from making a synapse onto itself or more than one synapse onto the same postsynaptic neuron. We initiated activity in the first cluster with a 4-ms, 40-μA/cm2 current pulse in 50% of the HVCRA neurons, beginning at t = 0 ms.
Experimental predictions of the sparse bursting model
In Fig. 6, we show experimental predictions of the sparse bursting model. Since GABAA synapses play a critical role in the model, the model predicts that manipulations that influence GABAA synaptic transmission can profoundly affect HVC's behavior. When we blocked inhibitory synapses (i.e., set gGABAA = 0), the bursts of HVCRA neurons began normally but failed to terminate, entering a state of persistent activity. The prediction of normal burst onset will hold only if blocking GABAA synapses does not in itself lead to runaway excitation in HVC before the onset of singing.
FIG. 6.

Sparse bursting model predictions: A: burst duration (top), spikes per burst (middle), and sparseness (bottom) as a function of gGABAA. Solid lines: mean. Dashed lines: mean ± SD. At gGABAA ≥ 0.4 mS/cm2, the burst duration is 5.0 ± 0.3 ms and the number of spikes per burst is 3.6 ± 0.1. B and C: HVCI interspike interval (ISI) distributions may differentiate between connectivity patterns. B, top: ISI distribution of HVCI neurons in the simulation shown in Fig. 4B, connected according to the rule described herein. Bottom: ISI distribution of HVCI neurons receiving completely random synapses from the same set of HVCRA neurons. C: the same ISI distributions, with lower y-axis and higher x-axis limits. Top: ISI distribution of HVCI neurons connected according to the rule described herein shows a characteristic dip. Bottom: ISI distribution of HVCI neurons receiving random synapses lacks this dip.
Figure 6A shows the effect of varying gGABAA on the sparseness of spiking in a version of the model containing 90 HVCI neurons and 60 clusters of 3 HVCRA neurons, illustrated with the burst duration, spikes per burst, and the sparseness index defined in methods. We calculated burst duration and spikes per burst over the time interval from the beginning of current injection into the first neuron (0 ms) to approximately the end of the last burst in the chain in the normal case (230 ms). For very low values of gGABAA, HVCRA neurons entered a state of persistent activity. However, HVCRA neurons showed relatively short, consistent bursts over a wide range of gGABAA values from about 0.1 to ≥3.0 mS/cm2. This robustness is due to the HVCI–HVCRA connectivity pattern described earlier, which results in an inhibitory pause during which bursting can occur.
We also found that the interspike interval (ISI) distribution may contain a signature of the HVCRA → HVCI connectivity pattern, if sufficiently many HVCI spikes are recorded. Since each HVCI is constrained not to receive excitation from any cluster to which it sends inhibition, or from clusters within seven clusters upstream of one to which it sends inhibition, we would expect ISIs corresponding to this nearly eight cluster gap in connectivity, and less, to be underrepresented in the ISI distribution. Since the propagation speed along the chain is 0.26 cluster/ms, we would expect a dip in the ISI distribution below 31 ms. Figure 6B (top) shows the ISI distribution for all spike times of all HVCI neurons in the simulation illustrated in Fig. 4B. Figure 6B (bottom) shows the ISI distribution of another set of HVCI neurons. Each HVCI neuron in this second set was driven by exactly the same set of HVCRA bursts and received exactly the same number of HVCRA → HVCI synapses, but the HVCRA → HVCI connectivity pattern was completely random. These HVCI neurons did not send synapses to HVCRA neurons. Figure 6C shows the same ISI distributions with altered axis limits, to accentuate their differences. Unlike the random case (bottom), the constrained case (top) resulted in a histogram with a prominent dip in ISIs below 30 to 35 ms, corresponding to the gap in HVCRA → HVCI connectivity. Experimentally, such a histogram should include ISIs only from the period corresponding to singing and not from periods of spontaneous activity. The model predicts that both the size of the connectivity gap and the propagation speed of the waves will determine which ISIs are underrepresented in the histogram.
Additional predictions of the inhibitory pause mechanism
The role of inhibition in the sparse bursting model is similar to that in the inhibitory pause mechanism of Fig. 1C. If sleep replay or BOS playback-evoked activity in HVCRA neurons is the result of the same mechanism as singing-related bursting (see discussion), then the inhibitory pause mechanism makes two predictions that could be tested in head-fixed or anesthetized birds. First, under voltage clamp at the AMPA reversal potential (0 mV), an HVCRA neuron will have a background of outward, hyperpolarizing current during the fictive singing, with a dip shortly before and during the time of the burst (Fig. 7A). Second, chloride loading should result in a background of spiking during the fictive singing by making the GABAA synapses functionally excitatory (Fig. 7B).
FIG. 7.

Experimental predictions from the inhibitory pause mechanism of Fig. 1. A: current in a representative HVCRA neuron under voltage clamp at the AMPA reversal potential (0 mV). The neuron showed a background of outward, hyperpolarizing current, with a dip during and before the time of the burst. B: membrane potential of a representative HVCRA neuron from a different simulation, when we set the GABAA reversal potential to −21 mV to simulate chloride loading. The neuron showed a background of spiking, with a dip in membrane potential before the burst. In both A and B, the HVCI pause lasted from t = 23 to 36.4 ms, the depolarizing DC current pulse into HVCRA neurons lasted from t = 30 to 33 ms, and the simulation began at t = −20 ms.
DISCUSSION
We have presented a model of sparse bursting based on inhibition, recurrent excitation, and bistability. This model builds on the observation by Hahnloser et al. (2002) and Kozhevnikov and Fee (2007) of sparse bursting in HVCRA neurons and on their hypothesis that HVCRA neurons form a chainlike organization in which neuronal ensembles burst in sequence at every moment of the song, driving RA neurons to burst in sequence (Fee et al. 2004).
The chainlike organization of our bistable clusters is reminiscent of synfire chains, which have been discussed elsewhere (Abeles 1982, 1991; Diesmann et al. 1999; Hermann et al. 1995). Recently, chain models have been proposed for HVC (Fiete et al. 2005; Jin et al. 2007; Li and Greenside 2006), also building on the work of Fee et al. (2004). Although our model includes chainlike networks, we have focused on the role of bistability and inhibition rather than chains. Unlike other chainlike models, our model postulates a central role for inhibitory interneurons in sparsely bursting telencephalic premotor networks, HVC in particular. This mechanism is related to those of central pattern generators and cortical networks, which make use of inhibition, recurrent excitation, and bistability (McCormick 2005; Shu et al. 2003; Yuste et al. 2005).
Inhibitory interneurons also played a key role in the Drew and Abbott (2003) model of song selectivity and sequence generation in HVC, although this role was not in terminating sparse HVCRA bursts. Their model was limited by its dependence on a long afterhyperpolarization in HVCRA neurons and by the fact that its HVCRA neurons were active only during extrinsically generated excitatory pulses applied every 75 to 100 ms.
Our sparse bursting model is distinguished from other chainlike models by a number of key assumptions and predictions.
Our model assumes that a large proportion of HVCRA neurons participate in a local connectivity of recurrent excitation with other HVCRA neurons that are nearby in the network, which is the basis of the bistability.
Our model assumes that each HVCI neuron is constrained not to make inhibitory synapses onto HVCRA neurons within some distance upstream and downstream of one from which it receives an excitatory synapse (Fig. 4A). This gives the HVCRA neurons time to burst before the inhibition arrives and reduces the influence of residual inhibition on burst duration and propagation speed (Fig. 5).
Our model assumes that an additional mechanism activates HVCI neurons at the end of the HVCRA burst sequence to prevent persistent activity at the end of the chain. In this model, we added additional connections from HVCRA neurons at the end of the chain to the HVCI population. In the model presented in our companion paper (Gibb et al. 2009), which includes neural feedback via nucleus uvaeformis (Uva), these connections are replaced by ones from Uva to HVCI neurons.
As a result of the HVCI–HVCRA connectivity, our model predicts that every HVCRA neuron that participates in sparse bursting receives a background of inhibition throughout the song, with a reduction shortly before and during the time that it bursts (Figs. 1C and 7A). If the mechanism of sparse bursting during sleep or BOS playback is sufficiently similar to that during singing (which is not certain, given the involvement of NIf [nucleus interface of the nidopallium] in the former but not the latter; Cardin et al. 2005; Hahnloser and Fee 2007), then voltage-clamp or chloride loading experiments can be used to test this prediction (Fig. 7). During fictive singing, an HVCRA neuron will have a background of outward current, with a dip shortly before and during the time of the burst (Fig. 7A), and chloride loading of an HVCRA neuron will result in a background of spiking (Fig. 7B).
Our model predicts that, if GABAA synapses are blocked, excited groups of HVCRA neurons will enter a state of persistent activity, whether in vitro or in vivo. How long this activity will last depends on the size of the groups (Fig. 1A), the percentage connectivity of the HVCRA neurons within groups, and the strength of voltage-dependent hyperpolarizing currents (Figs. 1A and 2, B and C) and neurotransmitter-activated hyperpolarizing currents (e.g., GABAB; Dutar et al. 1998). However, this effect will be seen only if blocking GABAA synapses does not itself evoke a high level of song-independent activity. To evoke this persistent activity in vitro, it is necessary to excite a sufficiently large fraction of the HVCRA neurons in a cluster or the functional equivalent of a cluster. In vivo, our model predicts that song-related HVCRA bursts will be initiated normally but fail to be rapidly terminated.
Our model predicts that HVCI ISIs during song will have a distribution resembling Fig. 6C, top (with a dip in the frequency of occurrence of certain ISIs), more than Fig. 6C, bottom. Which ISIs will be underrepresented in the histogram depends on both the burst propagation speed and the size of the HVCI connectivity gap. Since spontaneous activity between song bouts may influence the ISI distribution, the ISIs for such a test should be taken from HVCI neurons only during the period corresponding to singing. Additionally, it is possible that a version of the model could be developed in which the HVCI → HVCRA connectivity pattern is constrained but the HVCRA → HVCI connectivity pattern is random. In such a model, HVCI neurons would not be expected to have this ISI distribution.
Potential role for a Ca2+ current in HVCRA bursting
Under normal conditions, injection of depolarizing current into the soma of an HVCRA neuron does not evoke Ca2+ spikes or any evidence of bursting (Dutar et al. 1998; Kubota and Taniguchi 1998; Long and Fee 2006; Mooney and Prather 2005; Shea 2004). Consequently, we formulated a model in which bursting is not intrinsic to HVCRA neurons but instead arises in the microcircuitry of HVC. However, recent preliminary work shows that Ca2+ spikes, which appear to be of the L-type, are elicited by somatic depolarization of HVCRA neurons in the presence of a Na+-channel blocker (Long and Fee 2006). The same study demonstrates that sleep bursts of HVCRA neurons are augmented by an agonist of the L-type Ca2+ current. These results suggest that an L-type Ca2+ current is present in HVCRA neurons and is capable of influencing bursting. If this current proves to be essential to the bursting mechanism of HVCRA neurons, it may play a complementary role to inhibition and recurrent excitation.
If this current merely serves to enhance the excitation of postsynaptic HVCRA neurons, then we would expect augmenting it to have a similar effect to increasing the number or strength of excitatory synapses onto HVCRA neurons in our model. In this case, the presence of the current may not fundamentally alter our predictions. By contrast, if inactivation of a Ca2+ current, rather than inhibition mediated by HVCI neurons, provided the basis for burst termination, then our predictions would have to be revised. In particular, bursts would continue to terminate even in the absence of inhibition (persistent activity would not be observed). However, it is unlikely that inactivation of an L-type Ca2+ current is responsible for terminating the bursts, since this type of current inactivates slowly (Catterall et al. 2005).
Even if intrinsic currents are identified that provide the basis for burst termination, our proposed inhibitory mechanism could still play important roles: for example, it could enhance the temporal precision of the bursts and prevent excitatory inputs from evoking activity outside the normal burst time.
Cluster size
Fee et al. (2004) estimated that about 200 HVCRA neurons may be coactive at each time in the song motif, which is similar to the number of neurons in our large clusters with physiological synaptic strengths. However, this may be an overestimate, since the study on which they based it (Wang et al. 2002) did not use stereological methods for reconstructing total cell number (JR Kirn, personal communication; West 1999). HVC contains an average of about 40,000 to 50,000 neurons per hemisphere (Burek et al. 1991, 1997; Nordeen and Nordeen 1988, 1989; Ward et al. 1998).
Using the estimate that half of HVC neurons are RA-projecting (Nottebohm et al. 1990) and assuming that only half of the HVCRA neurons participate in syllable networks, we arrive at an estimate of 10,000 to 12,500 participating HVCRA neurons. Taking the motif duration to be about 0.5 to 1 s (Fee et al. 2004; Immelman 1969; Price 1979) and the average burst duration to be 5 ms (Kozhevnikov and Fee 2007), we calculate that about 50 to 125 HVCRA neurons may be coactive at each time in the song motif. In a chain framework, the propagation speed implied by these figures is about 10 to 25 HVCRA neurons per millisecond. Thus if the number of HVCRA neurons per cluster is 160, cluster activations must be staggered by about 6 to 16 ms to achieve such slow propagation. The result presented in Fig. 5D suggests that a smaller HVCI downstream connectivity gap may promote slower propagation. Alternatively, clusters could be smaller and the synaptic response of HVCRA neurons could be enhanced by a dendritic Ca2+ current (Jin et al. 2007; Long and Fee 2006) to achieve an equivalent synaptic effect.
Additionally, our chain model with physiological synaptic strengths suggests that the sparse burst mechanism does not fundamentally rely on a cluster organization; the important elements are recurrent local excitation among HVCRA neurons and patterned inhibition from HVCI neurons.
A possible role for developmental plasticity in the establishment of HVCI → HVCRA connectivity
What developmental mechanism could generate the pattern of HVCI → HVCRA connectivity shown in Fig. 4A? We suggest that a novel form of timing-dependent synaptic plasticity at inhibitory synapses could serve this role. In this proposed model, early in development, HVCRA → HVCI and HVCI → HVCRA synapses are weak or absent and sustained (nonsparse) spiking activity propagates along chains of recurrently excitatory HVCRA neurons, terminated by slow hyperpolarization. HVCRA → HVCI synapses are gradually added and/or strengthened with time. As they strengthen, subsets of them become strong enough that concurrent spiking in the presynaptic HVCRA neurons can elicit spiking in the postsynaptic HVCI neuron.
Additionally, HVCI → HVCRA synapses begin weak and follow a specific timing-dependent synaptic plasticity rule: if the presynaptic HVCI neuron spikes within a few tens of milliseconds before or a few milliseconds after the postsynaptic HVCRA neuron, the inhibitory synapse is weakened. If the HVCI neuron spikes outside this time window, the synapse is strengthened. For some period of time after the induction of plasticity, the synapse is resistant to further induction, so that only the first spikes of the propagating activity are involved in triggering plasticity. We suggest that over many bouts of propagation, a plasticity mechanism like this could generate the HVCI → HVCRA connectivity required by our model. Our proposed plasticity rule is similar to, although distinct from, various forms of timing-dependent plasticity that have been observed at excitatory and inhibitory synapses (Dan and Poo 2006; Haas et al. 2006; Holmgren and Zilberter 2001; Woodin et al. 2003).
GRANTS
This work was partially supported by National Science Foundation (NSF) Grant NSF PHY0097134 and Multidisciplinary University Research Initiative Contract ONR N00014-07-1-0741 to H. D. I. Abarbanel. H. D. I. Abarbanel also acknowledges partial support through the NSF-sponsored Center for Theoretical Biological Physics at University of California, San Diego (UCSD). D. L. Gibb received support from the NSF Integrative Graduate Education and Research Traineeship program through the UCSD Computational Neurobiology Graduate Program and a predoctoral fellowship from the Training Program in Cognitive Neuroscience of the Institute for Neural Computation at UCSD, supported by National Institute of Mental Health Grant 2 T32 MH-20002.
Supplementary Material
Acknowledgments
We thank R. Mooney for discussions and ideas for experimental tests of the sparse bursting model; T. Nowotny for providing the basic C++ neural simulation framework and programming advice; R. Ashmore, M. Bazhenov, M. Brainard, M. Coleman, A. Doupe, M. Fee, R. Hahnloser, J. Kirn, M. Long, D. Margoliash, D. Perkel, M. Rabinovich, C. Scharff, T. Sejnowski, S. Shea, M. Solis, M. Schmidt, D. Vicario, E. Vu, and M. Wild for helpful discussions and communications; and two anonymous reviewers, whose comments significantly improved this manuscript.
Footnotes
The online version of this article contains supplemental data.
REFERENCES
- Abarbanel et al. 2004a.Abarbanel HD, Gibb L, Mindlin GB, Rabinovich MI, Talathi S. Spike timing and synaptic plasticity in the premotor pathway of birdsong. Biol Cybern 91: 159–167, 2004a. [DOI] [PubMed] [Google Scholar]
- Abarbanel et al. 2004b.Abarbanel HD, Gibb L, Mindlin GB, Talathi SS. Mapping neural architectures onto acoustic features of birdsong. J Neurophysiol 92: 96–110, 2004b. [DOI] [PubMed] [Google Scholar]
- Abarbanel et al. 2004c.Abarbanel HD, Talathi SS, Mindlin G, Rabinovich M, Gibb L. Dynamical model of birdsong maintenance and control. Phys Rev E Stat Nonlin Soft Matter Phys 70: 051911, 2004c. [DOI] [PubMed] [Google Scholar]
- Abeles 1982.Abeles M. Local Cortical Circuits: An Electrophysiological Study. Berlin: Springer-Verlag, 1982.
- Abeles 1991.Abeles M. Corticonics, Neural Circuits of the Cerebral Cortex. Cambridge, UK: Cambridge Univ. Press, 1991.
- Alvarez-Buylla et al. 1988.Alvarez-Buylla A, Theelen M, Nottebohm F. Birth of projection neurons in the higher vocal center of the canary forebrain before, during, and after song learning. Proc Natl Acad Sci USA 85: 8722–8726, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aponte et al. 2006.Aponte Y, Lien CC, Reisinger E, Jonas P. Hyperpolarization-activated cation channels in fast-spiking interneurons of rat hippocampus. J Physiol 574: 229–243, 2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bottjer et al. 1984.Bottjer SW, Miesner EA, Arnold AP. Forebrain lesions disrupt development but not maintenance of song in passerine birds. Science 224: 901–903, 1984. [DOI] [PubMed] [Google Scholar]
- Brainard and Doupe 2000.Brainard MS, Doupe AJ. Interruption of a basal ganglia–forebrain circuit prevents plasticity of learned vocalizations. Nature 404: 762–766, 2000. [DOI] [PubMed] [Google Scholar]
- Burek et al. 1991.Burek MJ, Nordeen KW, Nordeen EJ. Neuron loss and addition in developing zebra finch song nuclei are independent of auditory experience during song learning. J Neurobiol 22: 215–223, 1991. [DOI] [PubMed] [Google Scholar]
- Burek et al. 1997.Burek MJ, Nordeen KW, Nordeen EJ. Sexually dimorphic neuron addition to an avian song-control region is not accounted for by sex differences in cell death. J Neurobiol 33: 61–71, 1997. [PubMed] [Google Scholar]
- Cardin et al. 2005.Cardin JA, Raksin JN, Schmidt MF. Sensorimotor nucleus NIf is necessary for auditory processing but not vocal motor output in the avian song system. J Neurophysiol 93: 2157–2166, 2005. [DOI] [PubMed] [Google Scholar]
- Catterall et al. 2005.Catterall WA, Perez-Reyes E, Snutch TP, Striessnig J. International Union of Pharmacology. XLVIII. Nomenclature and structure–function relationships of voltage-gated calcium channels. Pharmacol Rev 57: 411–425, 2005. [DOI] [PubMed] [Google Scholar]
- Collingridge et al. 1984.Collingridge GL, Gage PW, Robertson B. Inhibitory post-synaptic currents in rat hippocampal CA1 neurones. J Physiol 356: 551–564, 1984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dan and Poo 2006.Dan Y, Poo MM. Spike timing-dependent plasticity: from synapse to perception. Physiol Rev 86: 1033–1048, 2006. [DOI] [PubMed] [Google Scholar]
- Destexhe et al. 1998a.Destexhe A, Contreras D, Steriade M. Mechanisms underlying the synchronizing action of corticothalamic feedback through inhibition of thalamic relay cells. J Neurophysiol 79: 999–1016, 1998a. [DOI] [PubMed] [Google Scholar]
- Destexhe et al. 1994.Destexhe A, Mainen ZF, Sejnowski TJ. Synthesis of models for excitable membranes, synaptic transmission and neuromodulation using a common kinetic formalism. J Comput Neurosci 1: 195–230, 1994. [DOI] [PubMed] [Google Scholar]
- Destexhe et al. 1998b.Destexhe A, Mainen M, Sejnowski TJ. Kinetic models of synaptic transmission. In: Methods in Neuronal Modeling (2nd ed.), edited by Koch C, Segev I. Cambridge, MA: MIT Press, 1998b, p. 1–26.
- Destexhe and Sejnowski 2001.Destexhe A, Sejnowski TJ. Thalamocortical Assemblies. Oxford, UK: Oxford Univ. Press, 2001.
- Diesmann et al. 1999.Diesmann M, Gewaltig MO, Aertsen A. Stable propagation of synchronous spiking in cortical neural networks. Nature 402: 529–533, 1999. [DOI] [PubMed] [Google Scholar]
- Drew and Abbott 2003.Drew PJ, Abbott LF. Model of song selectivity and sequence generation in area HVc of the songbird. J Neurophysiol 89: 2697–2706, 2003. [DOI] [PubMed] [Google Scholar]
- Dutar et al. 1998.Dutar P, Vu HM, Perkel DJ. Multiple cell types distinguished by physiological, pharmacological, and anatomic properties in nucleus HVc of the adult zebra finch. J Neurophysiol 80: 1828–1838, 1998. [DOI] [PubMed] [Google Scholar]
- Fee et al. 2004.Fee MS, Kozhevnikov AA, Hahnloser RH. Neural mechanisms of vocal sequence generation in the songbird. Ann NY Acad Sci 1016: 153–170, 2004. [DOI] [PubMed] [Google Scholar]
- Fiete et al. 2005.Fiete IR, Burger L, Senn W, Hahnloser RHR. A biophysical network model for the emergence of ultrasparse sequences in HVC of the songbird. Soc Neurosci Abstr 79.12, 2005.
- Fiete et al. 2007.Fiete IR, Fee MS, Seung HS. Model of birdsong learning based on gradient estimation by dynamic perturbation of neural conductances. J Neurophysiol 98: 2038–2057, 2007. [DOI] [PubMed] [Google Scholar]
- Fiete et al. 2004.Fiete IR, Hahnloser RH, Fee MS, Seung HS. Temporal sparseness of the premotor drive is important for rapid learning in a neural network model of birdsong. J Neurophysiol 92: 2274–2282, 2004. [DOI] [PubMed] [Google Scholar]
- Fortune and Margoliash 1995.Fortune ES, Margoliash D. Parallel pathways and convergence onto HVc and adjacent neostriatum of adult zebra finches (Taeniopygia guttata). J Comp Neurol 360: 413–441, 1995. [DOI] [PubMed] [Google Scholar]
- Gibb and Abarbanel 2006.Gibb L, Abarbanel HD. Multifunctional interneurons and brainstem feedback in a computational model of birdsong. Soc Neurosci Abstr 44.4, 2006.
- Gibb et al. 2009.Gibb L, Gentner TQ, Abarbanel HD. Brain stem feedback in a computational model of birdsong sequencing. J Neurophysiol 102: 1763–1778, 2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haas et al. 2006.Haas JS, Nowotny T, Abarbanel HD. Spike-timing-dependent plasticity of inhibitory synapses in the entorhinal cortex. J Neurophysiol 96: 3305–3313, 2006. [DOI] [PubMed] [Google Scholar]
- Hahnloser and Fee 2007.Hahnloser RH, Fee MS. Sleep-related spike bursts in HVC are driven by the nucleus interface of the nidopallium. J Neurophysiol 97: 423–435, 2007. [DOI] [PubMed] [Google Scholar]
- Hahnloser et al. 2002.Hahnloser RH, Kozhevnikov AA, Fee MS. An ultra-sparse code underlies the generation of neural sequences in a songbird. Nature 419: 65–70, 2002. [DOI] [PubMed] [Google Scholar]
- Hahnloser et al. 2006.Hahnloser RH, Kozhevnikov AA, Fee MS. Sleep-related neural activity in a premotor and a basal–ganglia pathway of the songbird. J Neurophysiol 96: 794–812, 2006. [DOI] [PubMed] [Google Scholar]
- Hermann et al. 1995.Hermann M, Herz JA, Pruegel-Bennett A. Analysis of synfire chains. Network Comput Neural Syst 6: 403–414, 1995. [Google Scholar]
- Hestrin 1993.Hestrin S. Different glutamate receptor channels mediate fast excitatory synaptic currents in inhibitory and excitatory cortical neurons. Neuron 11: 1083–1091, 1993. [DOI] [PubMed] [Google Scholar]
- Hodgkin and Huxley 1952.Hodgkin AL, Huxley AF. A quantitative description of membrane current and its application to conduction and excitation in nerve. J Physiol 117: 500–544, 1952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holmgren and Zilberter 2001.Holmgren CD, Zilberter Y. Coincident spiking activity induces long-term changes in inhibition of neocortical pyramidal cells. J Neurosci 21: 8270–8277, 2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huguenard and McCormick 1994.Huguenard JR, McCormick DA. Electrophysiology of the Neuron. New York: Oxford Univ. Press, 1994.
- Jin et al. 2007.Jin DZ, Ramazanoglu FM, Seung HS. Intrinsic bursting enhances the robustness of a neural network model of sequence generation by avian brain area HVC. J Comput Neurosci 23: 283–299, 2007. [DOI] [PubMed] [Google Scholar]
- Kirn et al. 1999.Kirn JR, Fishman Y, Sasportas K, Alvarez-Buylla A, Nottebohm F. Fate of new neurons in adult canary high vocal center during the first 30 days after their formation. J Comp Neurol 411: 487–494, 1999. [PubMed] [Google Scholar]
- Kozhevnikov and Fee 2007.Kozhevnikov A, Fee MS. Singing-related activity of identified HVC neurons in the zebra finch. J Neurophysiol 97: 4271–4283, 2007. [DOI] [PubMed] [Google Scholar]
- Kubota and Taniguchi 1998.Kubota M, Taniguchi I. Electrophysiological characteristics of classes of neuron in the HVc of the zebra finch. J Neurophysiol 80: 914–923, 1998. [DOI] [PubMed] [Google Scholar]
- Long and Fee 2006.Long M, Fee MS. Mechanisms of burst generation within HVC of the zebra finch. Soc Neurosci Abstr 44.7, 2006.
- McCormick 2005.McCormick DA. Neuronal networks: flip-flops in the brain. Curr Biol 15: R294–R296, 2005. [DOI] [PubMed] [Google Scholar]
- McCormick Taniguchi1993.McCormick DA, Wang Z, Huguenard J. Neurotransmitter control of neocortical neuronal activity and excitability. Cereb Cortex 3: 387–398, 1993. [DOI] [PubMed] [Google Scholar]
- Mooney 2000.Mooney R. Different subthreshold mechanisms underlie song selectivity in identified HVc neurons of the zebra finch. J Neurosci 20: 5420–5436, 2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mooney and Prather 2005.Mooney R, Prather J. The HVC microcircuit: the synaptic basis for interactions between song motor and vocal plasticity pathways. J Neurosci 25: 1952–1964, 2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nixdorf et al. 1989.Nixdorf BE, Davis SS, DeVoogd TJ. Morphology of Golgi-impregnated neurons in hyperstriatum ventralis, pars caudalis in adult male and female canaries. J Comp Neurol 284: 337–349, 1989. [DOI] [PubMed] [Google Scholar]
- Nordeen and Nordeen 1988.Nordeen EJ, Nordeen KW. Sex and regional differences in the incorporation of neurons born during song learning in zebra finches. J Neurosci 8: 2869–2874, 1988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nordeen and Nordeen 1989.Nordeen EJ, Nordeen KW. Estrogen stimulates the incorporation of new neurons into avian song nuclei during adolescence. Brain Res Dev Brain Res 49: 27–32, 1989. [DOI] [PubMed] [Google Scholar]
- Nottebohm et al. 1990.Nottebohm F, Alvarez-Buylla A, Cynx J, Kirn J, Ling CY, Nottebohm M, Suter R, Tolles A, Williams H. Song learning in birds: the relation between perception and production. Philos Trans R Soc Lond B Biol Sci 329: 115–124, 1990. [DOI] [PubMed] [Google Scholar]
- Otis and Mody 1992a.Otis TS, Mody I. Modulation of decay kinetics and frequency of GABAA receptor-mediated spontaneous inhibitory postsynaptic currents in hippocampal neurons. Neuroscience 49: 13–32, 1992a. [DOI] [PubMed] [Google Scholar]
- Otis and Mody 1992b.Otis TS, Mody I. Differential activation of GABAA and GABAB receptors by spontaneously released transmitter. J Neurophysiol 67: 227–235, 1992b. [DOI] [PubMed] [Google Scholar]
- Perkel 2004.Perkel DJ. Origin of the anterior forebrain pathway. Ann NY Acad Sci 1016: 736–748, 2004. [DOI] [PubMed] [Google Scholar]
- Person and Perkel 2005.Person AL, Perkel DJ. Unitary IPSPs drive precise thalamic spiking in a circuit required for learning. Neuron 46: 129–140, 2005. [DOI] [PubMed] [Google Scholar]
- Price 1979.Price PH. Developmental determinants of structure in zebra finch song. J Comp Physiol Psychol 93: 260–277, 1979. [Google Scholar]
- Rauske et al. 2003.Rauske PL, Shea SD, Margoliash D. State and neuronal class-dependent reconfiguration in the avian song system. J Neurophysiol 89: 1688–1701, 2003. [DOI] [PubMed] [Google Scholar]
- Reiner et al. 2004.Reiner A, Perkel DJ, Bruce LL, Butler AB, Csillag A, Kuenzel W, Medina L, Paxinos G, Shimizu T, Striedter G, Wild M, Ball GF, Durand S, Gunturkun O, Lee DW, Mello CV, Powers A, White SA, Hough G, Kubikova L, Smulders TV, Wada K, Dugas-Ford J, Husband S, Yamamoto K, Yu J, Siang C, Jarvis ED for the Avian Brain Nomenclature Forum. Revised nomenclature for avian telencephalon and some related brainstem nuclei. J Comp Neurol 473: 377–414, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharff et al. 2000.Scharff C, Kirn JR, Grossman M, Macklis JD, Nottebohm F. Targeted neuronal death affects neuronal replacement and vocal behavior in adult songbirds. Neuron 25: 481–492, 2000. [DOI] [PubMed] [Google Scholar]
- Scharff and Nottebohm 1991.Scharff C, Nottebohm F. A comparative study of the behavioral deficits following lesions of various parts of the zebra finch song system: implications for vocal learning. J Neurosci 11: 2896–2913, 1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shea 2004.Shea SD. Basal Forebrain Control of Sensorimotor Activity (PhD Dissertation). Chicago, IL: Division of Biological Sciences and Pritzker School of Medicine, Univ. of Chicago, 2004.
- Shu et al. 2003.Shu Y, Hasenstaub A, McCormick DA. Turning on and off recurrent balanced cortical activity. Nature 423: 288–293, 2003. [DOI] [PubMed] [Google Scholar]
- Sohrabji et al. 1990.Sohrabji F, Nordeen EJ, Nordeen KW. Selective impairment of song learning following lesions of a forebrain nucleus in the juvenile zebra finch. Behav Neural Biol 53: 51–63, 1990. [DOI] [PubMed] [Google Scholar]
- Traub and Miles 1991.Traub RD, Miles R. Neuronal Networks of the Hippocampus. Cambridge, UK: Cambridge Univ. Press, 1991.
- Wang et al. 2002.Wang N, Hurley P, Pytte C, Kirn JR. Vocal control neuron incorporation decreases with age in the adult zebra finch. J Neurosci 22: 10864–10870, 2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward et al. 1998.Ward BC, Nordeen EJ, Nordeen KW. Individual variation in neuron number predicts differences in the propensity for avian vocal imitation. Proc Natl Acad Sci USA 95: 1277–1282, 1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- West 1999.West MJ. Stereological methods for estimating the total number of neurons and synapses: issues of precision and bias. Trends Neurosci 22: 51–61, 1999. [DOI] [PubMed] [Google Scholar]
- Woodin et al. 2003.Woodin MA, Ganguly K, Poo MM. Coincident pre- and postsynaptic activity modifies GABAergic synapses by postsynaptic changes in Cl− transporter activity. Neuron 39: 807–820, 2003. [DOI] [PubMed] [Google Scholar]
- Xiang et al. 1992.Xiang Z, Greenwood AC, Brown T. Measurement and analysis of hippocampal mossy-fiber synapses. Soc Neurosci Abstr 18: 1350, 1992. [Google Scholar]
- Yuste et al. 2005.Yuste R, MacLean JN, Smith J, Lansner A. The cortex as a central pattern generator. Nat Rev Neurosci 6: 477–483, 2005. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.